

Background: GLUT4 translocation in cardiomyocytes is impaired during insulin resistance leading to insufficient glucose supply and eventually heart failure.
Results: Cardiomyocytes overexpressing VAMP3 maintain full insulin-stimulated GLUT4 translocation and do not accumulate intramyocellular lipids.
Conclusion: Overexpression of VAMP3 protects cardiac glucose metabolism under conditions of impaired insulin sensitivity.
Significance: These data indicate a mechanism how contraction signaling improves insulin-dependent GLUT4 translocation.
Keywords: Cardiac Metabolism, Fatty Acid Synthase, GLUT4, Insulin Resistance, Trafficking
Abstract
Cardiac glucose utilization is regulated by reversible translocation of the glucose transporter GLUT4 from intracellular stores to the plasma membrane. During the onset of diet-induced insulin resistance, elevated lipid levels in the circulation interfere with insulin-stimulated GLUT4 translocation, leading to impaired glucose utilization. Recently, we identified vesicle-associated membrane protein (VAMP) 2 and 3 to be required for insulin- and contraction-stimulated GLUT4 translocation, respectively, in cardiomyocytes. Here, we investigated whether overexpression of VAMP2 and/or VAMP3 could protect insulin-stimulated GLUT4 translocation under conditions of insulin resistance. HL-1 atrial cardiomyocytes transiently overexpressing either VAMP2 or VAMP3 were cultured for 16 h with elevated concentrations of palmitate and insulin. Upon subsequent acute stimulation with insulin, we measured GLUT4 translocation, plasmalemmal presence of the fatty acid transporter CD36, and myocellular lipid accumulation. Overexpression of VAMP3, but not VAMP2, completely prevented lipid-induced inhibition of insulin-stimulated GLUT4 translocation. Furthermore, the plasmalemmal presence of CD36 and intracellular lipid levels remained normal in cells overexpressing VAMP3. However, insulin signaling was not retained, indicating an effect of VAMP3 overexpression downstream of PKB/Akt. Furthermore, we revealed that endogenous VAMP3 is bound by the contraction-activated protein kinase D (PKD), and contraction and VAMP3 overexpression protect insulin-stimulated GLUT4 translocation via a common mechanism. These observations indicate that PKD activates GLUT4 translocation via a VAMP3-dependent trafficking step, which pathway might be valuable to rescue constrained glucose utilization in the insulin-resistant heart.
Introduction
Obesity is hallmarked by deposition of excess lipids in nonadipose tissue, like heart and skeletal muscle. These excess lipids interfere with insulin signaling (1, 2) and urge the β-cells of the pancreas to compensate by increasing the secretion of insulin. Still, the elevated lipid levels in this insulin-resistant state induce mitochondrial dysfunction (3) and endoplasmic reticulum stress (4). In the heart, levels of intramyocellular triacylglycerol (IMTG)2 correlate negatively with systolic function (5) and positively with the risk for heart failure and stroke (6). The increase in IMTG is associated with the elevated presence of the fatty acid transporter CD36 at the plasma membrane of cardiomyocytes, leading to an influx of fatty acids which exceeds the capacity to oxidize these (7, 8). Furthermore, an association between insulin resistance and increased sarcolemmal CD36 has also been found in human skeletal muscle (9).
Cardiac IMTG itself is biologically rather inert; yet it is in equilibrium with biologically active lipid species like diacylglycerol and ceramides, which impair the activity of insulin receptor substrate proteins and protein kinase B/Akt (Akt) (10–12). As a consequence, insulin-stimulated translocation of the glucose transporter GLUT4 to the plasma membrane is impaired, leading to insufficient supply of the cardiomyocytes with glucose at early stages of diet-induced insulin resistance (13, 14). In search for physiological pathways to restore insulin sensitivity and GLUT4 translocation in insulin-resistant cardiomyocytes, activation of contraction signaling with its key enzymes AMP-activated protein kinase (AMPK) and protein kinase D (PKD) is a promising target (15–17). Interestingly, contraction increases insulin sensitivity not by increasing the activity of the insulin receptor kinase or its downstream signaling enzymes (18, 19). Rather, activation of contraction signaling increases (i) the availability of GLUT4 vesicles, e.g. by inhibition of AS160 (20, 21) and (ii) the capacity of the myocytes to oxidize lipids (22, 23). Additionally to activation of contraction signaling, manipulation of proteins that regulate the intracellular distribution of GLUT4 could form the basis to protect GLUT4 translocation under insulin-resistant conditions (24).
One family of proteins that regulates intracellular GLUT4 distribution is the family of soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), which constitute the mechanistic core complexes of membrane fusion (25, 26). Depending on the conserved amino acid residue within the SNARE motif they are classified as Q-SNAREs (glutamine) or R-SNAREs (arginine). R-SNAREs are typically present on transport vesicles that shuttle GLUT4 (or CD36) between the different intracellular subcompartments and are clustered in the family of vesicle-associated membrane proteins (VAMPs). Recently, we identified VAMP3 to be involved in contraction-stimulated translocation of both GLUT4 and CD36 (27).
Because VAMP3 links contraction signaling to GLUT4 mobilization, we speculated that overexpression of VAMP3 would be beneficial to protect insulin-stimulated GLUT4 translocation under conditions of insulin resistance, similar to the effects observed after acute exercise. Furthermore, we intended to compare the effects of VAMP3 overexpression with VAMP2, which mediates insulin-stimulated translocation of both GLUT4 and CD36 (27). For this, we established a model of insulin-resistant cardiomyocytes and measured the effects of VAMP2 and VAMP3 overexpression, respectively, on insulin-stimulated GLUT4 translocation, plasmalemmal CD36 presence, and lipid accumulation. In addition, we investigated the interaction of both VAMP2 and VAMP3 with key enzymes of the insulin- and the contraction-signaling pathways and compared the protective effects of VAMP3 overexpression with low intensity contraction in insulin-resistant cardiomyocytes. The data presented in this study show that VAMP3, but not VAMP2, is able to protect insulin-stimulated GLUT4 translocation during early stages of diet-induced insulin resistance and preserves normal CD36 distribution.
EXPERIMENTAL PROCEDURES
Antibodies, siRNAs, and Plasmids
Antibodies against VAMP2 and VAMP3 were from Abcam and against GLUT4 from Santa Cruz Biotechnology. Antibodies against the Myc epitope, PKB/Akt, phospho-PKB/Akt (Ser-473), phospho-ACC (Ser-79) and phospho-AMPKα (Thr-172) were from Cell Signaling, and against mouse CD36 from Chemicon. The plasmid coding for GLUT4myc was kindly provided by Dr. J. Eckel (German Diabetes Center, Düsseldorf, Germany). pEGFP-N3 was from Clontech. Lipofectamine 2000 was from Invitrogen. Ortho-phenylenediamine-H2O2 fizzing tablets were from AbD Serotec (Kidlington, UK).
Cell Culture of HL-1 Atrial Cardiomyocytes
HL-1 cells were kindly provided by Dr. W. Claycomb (Louisiana State University, New Orleans, LA), cultured in Claycomb medium (supplemented with 10% FBS, 0.1 mm norepinephrine, 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin) at 37 °C and 5% CO2. Twice a week cells were split after reaching confluence.
GLUT4myc Cell Surface Staining
Plasmalemmal GLUT4 was detected using a GLUT4 variant carrying a myc tag on its first extracellular epitope, as described previously (27). Cells expressing GLUT4myc were incubated in serum-free depletion medium or serum-free depletion medium containing 20 μm palmitate and 50 nm insulin for 16 h at 37 °C and 5% CO2. Translocation of GLUT4myc was induced by incubation with 200 nm insulin for 30 min at 37 °C and 5% CO2. After fixation, the nonpermeabilized cells were incubated with an anti-myc antibody for 30 min at room temperature and subsequently washed extensively. Next, cells were incubated with a HRP-linked secondary antibody for 30 min at room temperature. After extensive washing, an ortho-phenylenediamine-H2O2 solution was added as a substrate for the bound HRP. The reaction was carried out at room temperature and stopped after 30 min by addition of 1 m H2SO4. Color development, representative for the amount of GLUT4myc present at the plasma membrane, was quantified by measurement of the absorbance at 490 nm. The background signal of the controls (1: incubation without primary antibody, 2: nontransfected cells) was subtracted from the raw data.
CD36 Cell Surface Staining
Prior to measuring CD36, cells were incubated in serum-free depletion medium or serum-free depletion medium containing 20 μm palmitate and 50 nm insulin for 16 h at 37 °C and 5% CO2. Translocation of CD36 was induced by incubation with 200 nm insulin for 30 min at 37 °C and 5% CO2. The staining of plasmalemmal CD36 in the nonpermeabilized HL-1 cells was done with a primary antibody against murine CD36 (Chemicon) and an appropriate HRP-linked secondary antibody. Aside from these details, the CD36 surface staining was homologous to the method of GLUT4myc cell surface staining. The background signal of the controls (incubation without primary antibody) was subtracted from the raw data.
Lipid Extraction and HP-TLC
Upon scraping from the plate, cells were lysed by repeated freeze-thaw cycles. Equal amounts of protein (300 μg) per sample were dissolved in 800 μl of H2O and mixed with 3 ml of methanol/chloroform (2:1). Next, 500 μl of chloroform and 2 μg of internal standard (cholesterol acetate) were added. Upon addition of 1 ml of H2O, the suspension was centrifuged and the chloroform phase transferred to a new tube. The samples were dried with a N2 airflow in a 50 °C water bath and resuspended in 50 μl of chloroform. Lipids were separated on a silica HP-TLC plate preactivated with methanol/chloroform (2:1) and 100 mm NaOH. Afterward, lipids were stained and imaged on a flatbed scanner. Quantification was done with the Quantity One Software (Bio-Rad).
Electric Field Stimulation
HL-1 cells were electrically paced using the C-Pace Stimulator and 6-well C-Dish Culture Dish Electrodes from IonOptix. Prior to electric field stimulation, medium was changed to depletion medium (DMEM low glucose, supplemented with 2 mm l-glutamine, 100 μm non-essential amino acids, 100 units/ml penicillin, and 100 μg/ml streptomycin) or depletion medium containing 20 μm palmitate and 50 nm insulin. Electric field stimulation was performed for 16 h at 40 V with a frequency of 0.3 Hz and a pulse duration of 5 ms, at 37 °C and 5% CO2.
Generation of VAMP2- and VAMP3-Plasmids
Total mRNA was isolated from mouse heart tissue using the RNeasy Mini Kit from Qiagen and transcribed to cDNA using the cDNA Synthesis Kit from Roche Applied Science. VAMP2-cDNA was flanked by XhoI and BamHI restriction sites, VAMP3-cDNA was flanked by BglII and BamHI restriction sites, and inserted into the pEGFP-N3 plasmid from Clontech.
Transfection of HL-1 Cells
Cells were transfected at 60–70% confluence, 24 h after seeding. Transfection of plasmid DNA was done with 0.25 μg of DNA/cm2 and 0.75 μl of Lipofectamine 2000/cm2, in antibiotic- and norepinephrine-free culture medium. After 6 h, medium was changed to regular culture medium. 48 h after transfection, cell lysis or functional assays were performed.
Isolation of Primary Rat Cardiomyocytes
Cardiac myocytes were isolated from male Lewis rats (200–250 g) using a Langendorff perfusion system and a Krebs-Henseleit bicarbonate medium equilibrated with a 95% O2/5% CO2 gas phase at 37 °C, as described previously (28).
Immunoprecipitation
Upon isolation, primary rat cardiomyocytes were resuspended in a modified Krebs-Ringer buffer, containing 1 mm CaCl2 and 0.45% BSA. Afterward, cells were stimulated with 10 nm insulin and 5 μm oligomycin, respectively, for 20 min. The reaction was stopped by addition of ice-cold radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% (v/v) Igepal CA-630, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, supplemented with the protease-inhibitor Complete (Roche Applied Science), and the phosphatase inhibitor PhosSTOP (Roche Applied Science)). 2 μg of anti-VAMP2 and 1 μg of anti-VAMP3, respectively, were added to 1 mg of protein lysate in 1 ml of radioimmune precipitation assay buffer and incubated on a rotating wheel at 4 °C for 1 h. Washed protein G-Sepharose 4 Fast Flow (GE Healthcare) was added with a bed volume of 25 μl per condition and incubated on a rotating wheel at 4 °C overnight. After three washing steps, 2× Laemmli buffer was added to the Sepharose beads and incubated at 95 °C for 5 min. The eluate was collected with a syringe and either directly used for SDS-PAGE or stored at −20 °C.
Cell Lysis and Western Blotting
Cell lysis and Western blotting were performed as described earlier (29).
Confocal Microscopy
For confocal microcopy cells were seeded on glass slides. Upon stimulation, cells were fixed with 3.7% formalin for 10 min. Upon permeabilization with 0.1% Triton X-100 cells were incubated with the α-myc antibody. During the secondary antibody step DAPI was added to stain the nuclei. After washing, the glass slides were mounted on object plates and imaged using a Bio-Rad MRC600 confocal microscope (Bio-Rad) equipped with an air-cooled argon-krypton mixed gas laser and mounted onto an Axiophote microscope (Zeiss), using oil-immersion objectives. Quantification of the degree of overlap between GFP and myc staining has been carried out with ImageJ software (National Institutes of Health, Bethesda, MD).
Statistics
All data are presented as means ± S.E. Statistical analysis was performed by using Student's t test or ANOVA (including Newman-Keuls Multiple Comparison Test) and statistical analysis software Prism 4 (GraphPad Software, Inc.). A p value of <0.05 was considered statistically significant.
RESULTS
In Vitro Model for Lipid-induced Insulin Resistance in the Heart
In patients, diet-induced insulin resistance is hallmarked by elevated plasma levels of fatty acids and insulin (30). Incubation with various concentrations of palmitate for 6–18 h is an established approach to study insulin resistance in skeletal muscle cell lines like C2C12 (31–33), L6 (34, 35), but also in human skeletal muscle myotubes (31). To establish a similar model to study diet-induced insulin resistance in the heart, we incubated HL-1 cardiomyocytes overnight (16 h) with a medium containing 20 μm palmitate (palmitate:BSA ratio 6:1) and 50 nm insulin (palm/ins medium). Compared with the standard culture conditions, this is an 8-fold elevation of free fatty acids and an 18-fold increase in insulin. Under these conditions, HL-1 cells accumulated mono-, di-, and triacylglycerols (Fig. 1A). Furthermore, insulin-stimulated Akt phosphorylation was largely inhibited, although the basal levels were elevated (Fig. 1B). As observed in several human studies, in the presence of elevated palmitate, CD36 accumulated at the plasma membrane and lost its insulin responsiveness (Fig. 1C). Furthermore, basal GLUT4 levels at the plasma membrane remained low, and insulin-stimulated GLUT4 translocation was completely inhibited (Fig. 1C). Taken together, these data indicate that incubation with 20 μm palmitate and 50 nm insulin for 16 h is sufficient to induce insulin resistance in HL-1 cardiomyocytes.
FIGURE 1.

Palmitate-induced insulin resistance in HL-1 cardiomyocytes. Cells were incubated with 20 μm palmitate and 50 nm insulin (palm/ins) overnight to induce insulin resistance. A, lipids were extracted from cells treated with control (white bars) or palm/ins medium (black bars). Intracellular levels of mono-, di-, and triacylglycerol were determined using HP-TLC. B, upon treatment with palm/ins medium, HL-1 cells were acutely stimulated with insulin for 30 min. Lysates of these cells were subsequently blotted against pAkt. C, cells treated with control (white bars) or palm/ins medium (black bars) were acutely stimulated with insulin for 30 min and subsequently stained for sarcolemmal CD36 and GLUT4myc, respectively. Data are mean values ± S.E. (error bars) of at least three independent experiments (n = 3), with duplicate measurements for each condition. *, statistically different from corresponding basal value (p < 0.05); **, statistically different from corresponding basal value (p < 0.01); #, statistically different from corresponding value in control group (p < 0.05).
Overexpression of VAMP3 Restores Insulin-stimulated GLUT4 Translocation
Previously, we showed that VAMP2 is involved in insulin-stimulated GLUT4 and CD36 translocation, and VAMP3 in contraction-mediated GLUT4 and CD36 translocation (27). Here, we investigated whether overexpression of these VAMPs would prevent the inhibition of insulin-stimulated GLUT4 translocation in insulin-resistant cardiomyocytes. Overexpression of GFP-VAMP2 and GFP-VAMP3 did not influence basal GLUT4myc presence at the plasma membrane under normal culture conditions (Fig. 2A). In the control cells overexpressing GFP, insulin increased the presence of GLUT4myc at the plasma membrane by 46 ± 9% (Fig. 2A). Overexpression of GFP-VAMP2 or GFP-VAMP3 did not significantly affect insulin-stimulated GLUT4 translocation. In HL-1 cardiomyocytes incubated overnight with palm/ins medium, insulin-stimulated GLUT4 translocation is completely inhibited (Fig. 2B). Cells overexpressing GFP-VAMP2 under these conditions did not display a significant increase in GLUT4myc translocation upon insulin stimulation (increase of 28 ± 12%). However, in cells overexpressing GFP-VAMP3, insulin-stimulated GLUT4myc translocation is retained (increase of 43 ± 8%).
FIGURE 2.
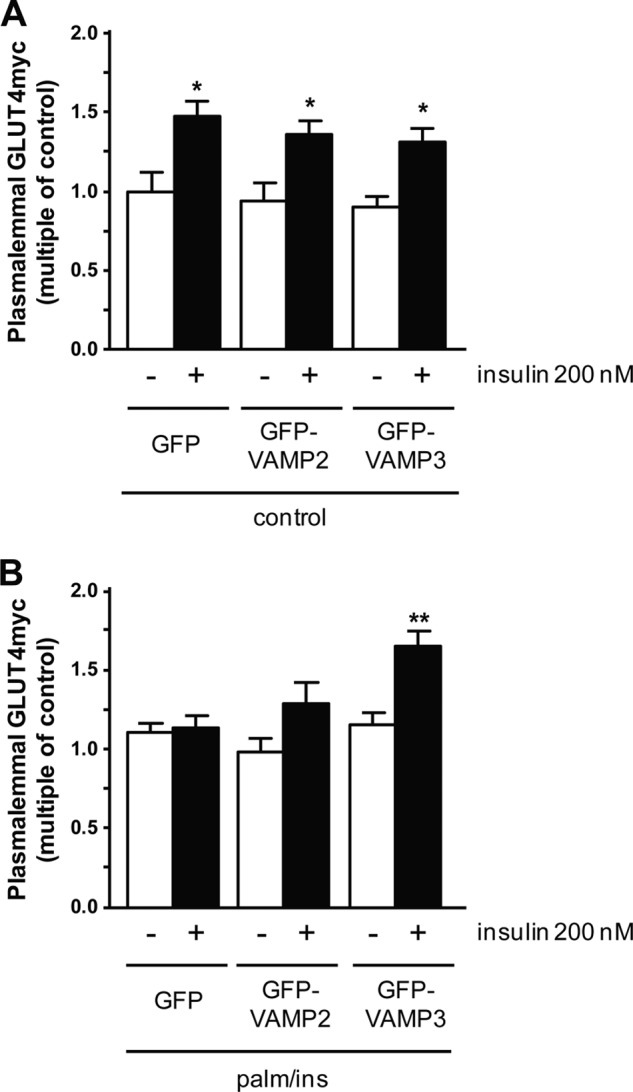
Effect of VAMP overexpression on GLUT4myc translocation. Cells were either transfected with GLUT4myc and with GFP as a control, or GFP-VAMP2 and GFP-VAMP3, respectively. Afterward, cells were either cultured with depletion medium (control) (A) or medium containing 20 μm palmitate and 50 nm insulin (palm/ins) (B) for 16 h. To induce GLUT4myc translocation, cells were acutely stimulated with insulin for 30 min. After fixation of the cells, sarcolemmal GLUT4myc was stained and quantified. Sarcolemmal GLUT4myc under basal conditions (white bars) is compared with sarcolemmal GLUT4myc after acute insulin stimulation (black bars). Data are mean values ± S.E. (error bars) of at least three independent experiments (n = 3), with triplicate measurements for each condition. *, statistically different from corresponding basal value with p < 0.05; **, statistically different from corresponding basal value with p < 0.01.
Next, we measured the effect of GFP-VAMP2 and GFP-VAMP3 overexpression, respectively, on insulin-stimulated Akt phosphorylation under both culture conditions. With GFP alone, overnight incubation with palm/ins medium inhibits acute insulin-stimulated Akt phosphorylation (Fig. 3A). Neither overexpression of GFP-VAMP2 nor overexpression of GFP-VAMP3 had an effect on insulin-stimulated Akt phosphorylation under any culture condition (Fig. 3A). Furthermore, overexpression of GLUT4myc was neither altered by culture condition (control versus palm/ins) nor by overexpression of GFP-VAMP2 and GFP-VAMP3, respectively (Fig. 3B). In insulin-stimulated cells, GLUT4myc and GFP-VAMP2 show a near complete colocalization at both intracellular compartments and the plasma membrane (94 ± 1%, n = 3). Interestingly, such strong colocalization is also observed for GLUT4myc and GFP-VAMP3 (94 ± 2%, n = 4) (Fig. 3C). Taken together, overexpression of VAMP3 prevents the loss of insulin-stimulated translocation of GLUT4 to the plasma membrane under lipotoxic culture conditions, without affecting GLUT4 expression or insulin signaling.
FIGURE 3.

Effect of VAMP overexpression on insulin signaling and GLUT4myc expression. Cells were either transfected with GFP as a control, or GFP-VAMP2 and GFP-VAMP3, respectively. Afterward, cells were either cultured with depletion medium (control) or medium containing 20 μm palmitate and 50 nm insulin (palm/ins) for 16 h. A and B, afterward, HL-1 cells were acutely stimulated with insulin for 30 min. Lysates of these cells were subsequently blotted against caveolin-3 (loading control), pAkt, GLUT4myc, and VAMP2 and VAMP3, respectively. Representative Western blots from at least three different experiments (n = 3) are shown. C, after acute stimulation with insulin, control cells were fixed and stained for confocal microscopy. DAPI was used to stain the nuclei, myc represents GLUT4myc, GFP represents GFP-VAMP2 and GFP-VAMP3, respectively. Representative confocal pictures from at least three different experiments (n = 3) are shown.
Intramyocellular Lipid Accumulation Is Prevented by VAMP3 Overexpression
Besides the protective effects on GLUT4 translocation, overexpression of VAMP3 could also affect intracellular CD36 distribution and therefore lipid accumulation in HL-1 cells. Under control conditions with GFP alone, insulin stimulated CD36 translocation to the plasma membrane by 51 ± 14% (Fig. 4A). Overexpression of GFP-VAMP2 increased the basal presence of CD36 at the plasma membrane by 40 ± 15%, and acute stimulation with insulin did not further increase CD36 translocation. In contrast, overexpression of GFP-VAMP3 did not affect plasmalemmal CD36 presence under normal culture conditions. Consistent with the data from Fig. 1C, the presence of CD36 at the plasma membrane was elevated by 33 ± 11% already in the basal state upon induction of insulin resistance, and acute stimulation with insulin was without effect (Fig. 4A). This increased presence of CD36 at the plasma membrane was not altered by overexpression of GFP-VAMP2. However, overexpression of GFP-VAMP3 normalized plasmalemmal CD36 levels under nonstimulated conditions. Still, acute insulin stimulation did not increase plasmalemmal CD36 under these conditions.
FIGURE 4.

Effect of VAMP overexpression on CD36 translocation and lipid accumulation. Cells were either transfected with GFP as a control, or GFP-VAMP2 and GFP-VAMP3, respectively. Afterward, cells were either cultured with depletion medium (control) or medium containing 20 μm palmitate and 50 nm insulin (palm/ins) for 16 h. A, to induce CD36 translocation, cells were acutely stimulated with insulin for 30 min. After fixation of the cells, sarcolemmal CD36 was stained and quantified. Sarcolemmal CD36 under basal conditions (white bars) is compared with sarcolemmal CD36 after acute insulin stimulation (black bars). B–D, lipids were extracted from cells treated with normal depletion medium (control) or palm/ins medium, and quantified using HP-TLC. Levels of mono-, di-, and triacylglycerols were compared between cells overexpressing GFP (white bars), GFP-VAMP2 (gray bars), and GFP-VAMP3 (black bars). Data are mean values ± S.E. (error bars) of at least three independent experiments (n = 3), with triplicate measurements for each condition. *, statistically different from corresponding basal value with p < 0.05; **, statistically different from corresponding basal value with p < 0.01; $, statistically different from corresponding value of the GFP transfection with p < 0.05; #, statistically different from corresponding value of control group with p < 0.05.
It has been well established that CD36 is rate-limiting for uptake and utilization of fatty acids into cardiac myocytes (7, 36, 37). Therefore, we investigated whether the altered CD36 distribution of the GFP-VAMP-transfected cells would affect lipid deposition. Overexpression of GFP-VAMP2 did not prevent the accumulation of monoacylglycerol, whereas GFP-VAMP3 overexpression completely prevented the accumulation of this lipid species under lipotoxic conditions (Fig. 4B). The accumulation of diacylglycerol under lipotoxic conditions (54 ± 8%) was reduced upon overexpression of GFP-VAMP2 (28 ± 2%) and prevented upon overexpression of GFP-VAMP3 (9 ± 11%) compared with the control situation (Fig. 4C). Triacylglycerol levels increased by 45 ± 8% upon GFP-VAMP2 overexpression during insulin resistance (Fig. 4D). In the cells overexpressing GFP-VAMP3, triacylglycerol levels did not increase.
Taken together, overexpression of VAMP2 and VAMP3 has opposite effects on plasmalemmal CD36 presence and intramyocellular lipid accumulation. Whereas overexpression of VAMP2 increases CD36 at the plasma membrane already under normal culture conditions, overexpression of VAMP3 keeps plasmalemmal CD36 amounts unaltered even under conditions of elevated palmitate and insulin levels in the culture medium.
VAMP3 Is Linked to Contraction Signaling via PKD
Insulin-stimulated GLUT4 translocation in myocytes relies on activation of Akt and PKCζ (38), whereas contraction-mediated GLUT4 translocation is dependent on AMPK and PKD activation (17). To investigate whether GLUT4 mobilization by these protein kinases is mediated by a direct interaction with the corresponding VAMP proteins (VAMP2 in insulin-, VAMP3 in contraction signaling) we performed immunoprecipitation experiments of endogenous VAMP2 and VAMP3 from rat cardiomyocytes. We did not detect either Akt or PKCζ in the VAMP2 precipitate from cardiomyocytes incubated with or without insulin (Fig. 5A), indicating that there is no physical interaction between any of these kinases with VAMP2. To investigate binding of contraction-activated kinases to VAMP3, we blotted the VAMP3 precipitates of cardiomyocytes stimulated with the contraction-mimicking agent oligomycin (17, 39) against AMPK and PKD. Although we did not detect any AMPK in the VAMP3 precipitate, we found a strong binding of PKD to basal VAMP3, which was further enhanced under oligomycin-stimulated conditions (Fig. 5B). Blotting the same samples against the phospho-specific PKD antibody revealed an enrichment of phosphorylated PKD in the immunoprecipitate of both basal and oligomycin-stimulated cardiomyocytes (Fig. 5B), indicating a preferential interaction of VAMP3 with the activated form of this kinase.
FIGURE 5.
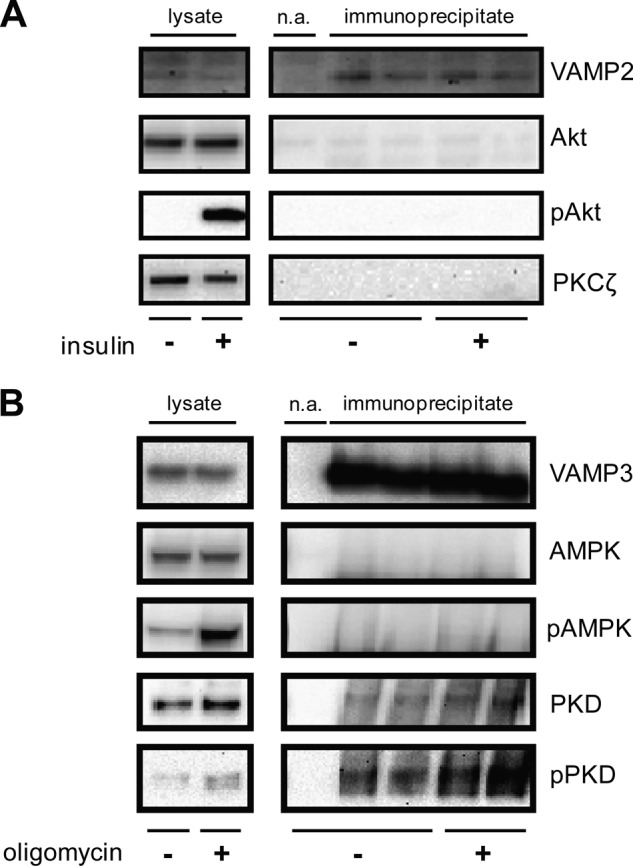
Immunoprecipitation of VAMP2 and VAMP3. Freshly isolated rat cardiomyocytes were either treated with insulin or oligomycin for 15 min. A, upon lysis, VAMP2 was immunoprecipitated from insulin-treated cardiomyocytes. Subsequently, the immunoprecipitate was blotted against VAMP2, Akt, pAkt, and PKCζ. B, upon lysis, VAMP3 was immunoprecipitated from oligomycin-treated cardiomyocytes. Subsequently, the immunoprecipitate was blotted against VAMP3, AMPK, pAMPK, PKD, and pPKD. Representative Western blots from at least three different experiments (n = 3) are shown. n.a., no antibody during immunoprecipitation (negative control).
PKD selectively stimulates translocation of GLUT4 to the plasma membrane during contraction, and VAMP3 is the vesicle-associated SNARE protein regulating trafficking under these conditions (17, 27). Therefore, the increased binding of activated PKD to VAMP3 suggests that PKD initiates GLUT4 movement to the plasma membrane via an interaction with VAMP3, located at intracellular stores of GLUT4 (40). If VAMP3 overexpression and contraction signal via the same pathway to GLUT4 mobilization, both interventions should not have additive effects on GLUT4 translocation if either PKD activation or VAMP3 is rate-limiting. To test this hypothesis, we first investigated whether low frequency contraction would protect insulin-stimulated GLUT4 translocation in insulin-resistant HL-1 cardiomyocytes, and second, whether contraction and VAMP3 overexpression have additive effects on insulin-stimulated GLUT4 translocation under lipotoxic conditions. In quiescent HL-1 cardiomyocytes, insulin increased plasmalemmal GLUT4 by 54 ± 17% (Fig. 6A). Under the same resting conditions, insulin-stimulated GLUT4 translocation is inhibited upon incubation with palm/ins medium and protected when cells were additionally overexpressing VAMP3 (increase of 66 ± 10%). Pacing the cells with 0.3 Hz during the time of serum depletion increased basal GLUT4 by 38 ± 15%, acute stimulation with insulin increased the plasmalemmal GLUT4 to a magnitude similar as in the quiescent cells (70 ± 5%) (Fig. 6B). When cells were paced in palm/ins medium we observed an increased plasmalemmal GLUT4 under basal conditions compared with the cells cultured in normal depletion medium (increase of 46 ± 18%). Importantly, the inhibition of insulin-stimulated GLUT4 translocation by palm/ins medium (Fig. 6A) was prevented when cells were paced with 0.3 Hz (Fig. 6B). Furthermore, a combination of 0.3-Hz pacing and overexpression of GFP-VAMP3 had no additional effect on basal or insulin-stimulated GLUT4 translocation (Fig. 6B). Taken together, these data indicate that both interventions, low frequency contraction and VAMP3 overexpression, enhanced the plasmalemmal GLUT4 via a common mechanism.
FIGURE 6.
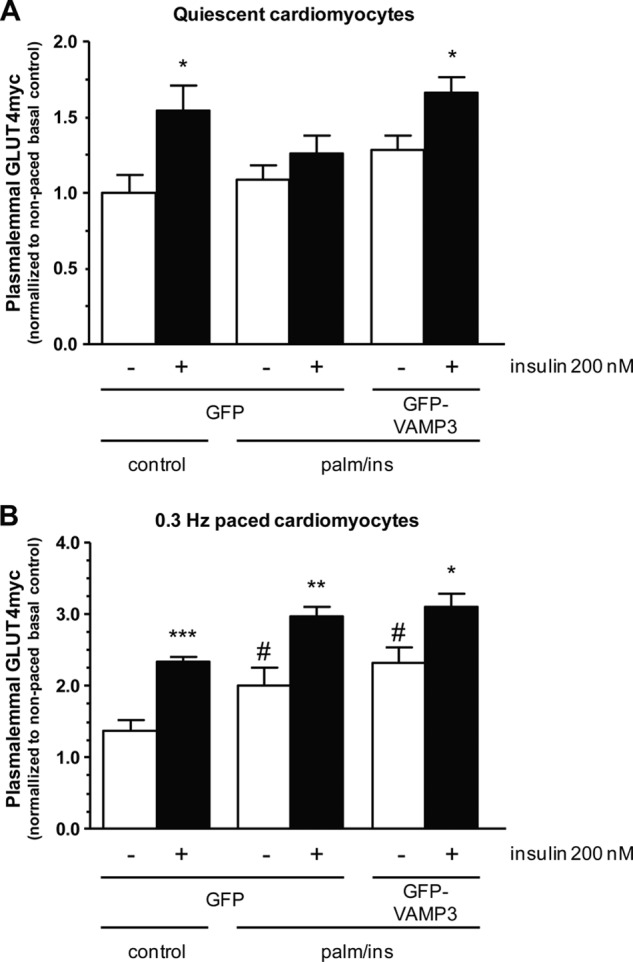
Effect of contraction and VAMP3 overexpression on GLUT4myc translocation under lipotoxic conditions. Cells were either transfected with GLUT4myc plus GFP as a control, or with GLUT4myc plus GFP-VAMP3, respectively. After addition of depletion medium (control) or medium containing 20 μm palmitate and 50 nm insulin (palm/ins), the cells were either left quiescent (A) or paced with 0.3 Hz (B) for 16 h. To induce GLUT4myc translocation, cells were acutely stimulated with insulin for 30 min. After fixation of the cells, sarcolemmal GLUT4myc was stained and quantified. Sarcolemmal GLUT4myc under basal conditions (white bars) is compared with sarcolemmal GLUT4myc after acute insulin stimulation (black bars). Data are mean values ± S.E. (error bars) of at least three independent experiments (n = 3), with triplicate measurements for each condition. *, statistically different from corresponding basal value with p < 0.05; **, statistically different from corresponding basal value with p < 0.01; ***, statistically different from corresponding basal value with p < 0.001; #, statistically different from corresponding value of control group with p < 0.05.
DISCUSSION
Lipids are important sources of energy for the heart. However, under insulin-resistant conditions an oversupply of lipids impairs cardiac function as lipid metabolites interfere with key enzymes of the insulin signaling cascade and subsequently impair insulin-stimulated GLUT4 translocation and glucose utilization (10–12). During early insulin resistance, the β-cells compensate for this insulin desensitizing by increasing insulin disposal. Still, with progression of this nutrient oversupply the accumulation of lipids in cardiomyocyte will impair cardiac glucose utilization and function. Here, we investigated the potential of vesicle-associated membrane receptors VAMP2 and VAMP3 to protect insulin-stimulated GLUT4 translocation in cardiomyocytes under culture conditions mimicking diet-induced insulin resistance. However, caution should be taken to translate findings obtained in this in vitro model of overnight insulin resistance to the in vivo chronic insulin resistance in humans. Overexpression of VAMP2 marginally preserved GLUT4 translocation in insulin-resistant cardiomyocytes. However, overexpression of the contraction-regulated VAMP3 not only prevented the loss of insulin-stimulated GLUT4 translocation, but also preserved normal CD36 distribution and lipid levels during the same adverse culture conditions. Recently, we established that PKD selectively enhances GLUT4 translocation during cardiomyocyte contraction without affecting CD36 distribution or fatty acid uptake (41). The binding of VAMP3 to phosphorylated PKD could serve as an explanation of how PKD selectively targets GLUT4 translocation and how exercise improves insulin-stimulated GLUT4 translocation under conditions of impaired insulin signaling.
Given the role of VAMP2 in insulin-stimulated GLUT4 translocation (26), one would assume that overexpression of this protein might be beneficial for hindered GLUT4 translocation. Therefore, we were surprised to observe that overexpression of VAMP2 in lipotoxic cardiomyocytes was without significant effect on GLUT4 translocation, whereas overexpression of VAMP3 completely protected insulin-stimulated GLUT4 translocation under these conditions. An earlier study in L6 myoblasts, in which all VAMP proteins were ablated using tetanus toxins, showed that transient reexpression of VAMP2, but not VAMP3, restored insulin-stimulated GLUT4 translocation (42), supporting the common view that insulin-stimulated GLUT4 translocation is dependent on VAMP2. In a previous study, we also showed that the presence of VAMP2 is crucial for proper insulin-stimulated GLUT4 translocation (27). However, overexpression of the cytoplasmic domains of both VAMP2 and VAMP3 inhibited insulin-stimulated GLUT4 translocation in another study (43), indicating a potential involvement of VAMP3 in this process, too. Taken together, the elevation of insulin-stimulated GLUT4 translocation by VAMP3 is dependent on the continued presence of VAMP2. However, the mechanism behind this observation needs further investigation. Most likely, VAMP3 overexpression alters the kinetics of intracellular GLUT4 trafficking which makes GLUT4 more available for the VAMP2-dependent translocation upon an insulin signal. This is supported by our finding that VAMP3 has a strong colocalization with intracellular GLUT4, but additionally also at the plasma membrane upon insulin stimulation.
Cardiomyocytes overexpressing VAMP2 displayed an increased presence of CD36 at the plasma membrane. Interestingly, VAMP2 has been found to be up-regulated in skeletal muscle of Zucker diabetic fatty rats compared with their lean controls (44). In the same model, CD36 expression and presence at the sarcolemma of skeletal muscle cells and cardiomyocytes is elevated (7, 45, 46). Therefore, our observation that overexpression of VAMP2 in cardiomyocytes leads to an increased content of sarcolemmal CD36 links both observations in the Zucker diabetic fatty rat to a common mechanism, suggesting a novel function for VAMP2 in the accumulation of CD36 at the plasma membrane in the insulin-resistant heart.
Under normal culture conditions, overexpression of VAMP3 did not affect insulin-stimulated GLUT4 translocation. This is in line with the observation that knockdown of VAMP3 is without effect on insulin-stimulated GLUT4 translocation and supports the conclusion that VAMP3 is not required for insulin-stimulated GLUT4 translocation under normal conditions (27). However, in the elevated presence of free fatty acids and insulin, VAMP3 reveals beneficial effects toward both GLUT4 and CD36 distribution, independent of full maintenance of insulin signaling. Here, as mentioned above, insulin-stimulated GLUT4 translocation is protected in cells overexpressing VAMP3, whereas CD36 accumulation at the plasma membrane is prevented. The repressive effect of VAMP3 on CD36, the predominant fatty acid transporter also in HL-1 cells (27), is reflected by the low levels of intramyocellular lipids under any culture condition. Because myocellular lipid accumulation is known to inhibit insulin-stimulated GLUT4 translocation due to impairment of downstream insulin signaling (10–12), the maintenance of low intracellular levels of tri- and diacylglycerol in lipid-exposed cells should preserve insulin-stimulated GLUT4 translocation. Still, maintaining the low lipid levels was not sufficient to protect insulin sensitivity of Akt phosphorylation, most likely due to the high insulin levels of the culture medium mimicking conditions of insulin resistance. However, studies in transgenic mice are needed to investigate whether the improved glucose utilization by insulin-sensitive tissues would also normalize the circulating insulin levels.
What is the mechanism that selectively preserves GLUT4 translocation in cardiomyocytes under conditions inducing insulin resistance? In our earlier study on VAMP proteins we identified VAMP3 to be involved in contraction-stimulated translocation of both GLUT4 and CD36 to the plasma membrane (27). Beneficial effects of muscular contraction on myocellular insulin sensitivity have been observed in several studies (15, 47, 48). Hence, the beneficial effects of VAMP3 overexpression on insulin-stimulated GLUT4 translocation might be mediated by elements of the contraction signaling pathway. As we demonstrate here, this is indeed the case, with VAMP3 overexpression and contraction having similar and nonadditive effects on preserving insulin-stimulated GLUT4 translocation. It is worth mentioning here that both interventions have different effects on basal GLUT4 presence at the plasma membrane. Whereas VAMP3 overexpression did not affect basal GLUT4, low frequency contraction strongly did. This suggests that components of the contraction signaling cascade, but not VAMP3, are rate-limiting for basal GLUT4 distribution. Possible candidates could be kinases like AMPK and PKD, which activity is low in the basal state independent of VAMP3, but increased during contraction. This is supported by our finding that induction of contraction signaling further enhanced PKD phosphorylation and its binding to VAMP3, suggesting a higher affinity of VAMP3 to the activated kinase. In contrast to PKD, AMPK does not bind to VAMP3 in cardiomyocytes. Interestingly, PKD has already been linked to contraction-stimulated GLUT4 translocation independent of AMPK (17). Furthermore, we recently showed that PKD selectively increased GLUT4 translocation during contraction without increasing plasmalemmal CD36 (41). Therefore, our data indicate that PKD selectively increases GLUT4 translocation through a pathway involving VAMP3. This novel pathway might be responsible for the preservation of insulin-stimulated GLUT4 translocation in cardiomyocytes exposed to elevated levels of palmitate and insulin, either overexpressing VAMP3 or being contracted. Whether and how this putative PKD-VAMP3 axis succeeds in refraining CD36 from relocalizing to the plasma membrane in lipid-exposed cells deserves further investigation.
In conclusion, our data indicate that VAMP3, downstream of PKD, is involved in the contraction-mediated improvement of insulin-stimulated GLUT4 translocation under conditions of impaired insulin signaling. Furthermore, we provide a novel function for VAMP2, which overexpression appears to be detrimental, as it does not improve insulin-stimulated GLUT4 translocation substantially, but rather shifts CD36 toward the plasma membrane. Taken together, pharmacological up-regulation of VAMP3, but not VAMP2, might be beneficial for the insulin-resistant heart, as it would normalize both GLUT4 and CD36 distribution and therefore is expected also to normalize substrate utilization.
This work was supported by the Transnational University Limburg. Part of this research was performed within the framework of CTMM, the Center for Translational Molecular Medicine, project PREDICCt, Grant 01C-104, and supported by the Netherlands Heart Foundation, Dutch Diabetes Research Foundation Grant 2006.00.044, and the Dutch Kidney Foundation.
- IMTG
- intramyocellular triacylglycerol
- AMPK
- AMP-activated protein kinase
- GLUT4
- glucose transporter 4
- palm/ins medium
- palmitate/insulin medium
- PKD
- protein kinase D
- VAMP
- vesicle-associated membrane protein.
REFERENCES
- 1. Bachmann O. P., Dahl D. B., Brechtel K., Machann J., Haap M., Maier T., Loviscach M., Stumvoll M., Claussen C. D., Schick F., Häring H. U., Jacob S. (2001) Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes 50, 2579–2584 [DOI] [PubMed] [Google Scholar]
- 2. Griffin M. E., Marcucci M. J., Cline G. W., Bell K., Barucci N., Lee D., Goodyear L. J., Kraegen E. W., White M. F., Shulman G. I. (1999) Free fatty acid-induced insulin resistance is associated with activation of protein kinase Cθ and alterations in the insulin signaling cascade. Diabetes 48, 1270–1274 [DOI] [PubMed] [Google Scholar]
- 3. Schrauwen P., Schrauwen-Hinderling V., Hoeks J., Hesselink M. K. (2010) Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 1801, 266–271 [DOI] [PubMed] [Google Scholar]
- 4. Borradaile N. M., Han X., Harp J. D., Gale S. E., Ory D. S., Schaffer J. E. (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 47, 2726–2737 [DOI] [PubMed] [Google Scholar]
- 5. Szczepaniak L. S., Dobbins R. L., Metzger G. J., Sartoni-D'Ambrosia G., Arbique D., Vongpatanasin W., Unger R., Victor R. G. (2003) Myocardial triglycerides and systolic function in humans: in vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 49, 417–423 [DOI] [PubMed] [Google Scholar]
- 6. Abel E. D., Litwin S. E., Sweeney G. (2008) Cardiac remodeling in obesity. Physiol. Rev. 88, 389–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coort S. L., Hasselbaink D. M., Koonen D. P., Willems J., Coumans W. A., Chabowski A., van der Vusse G. J., Bonen A., Glatz J. F., Luiken J. J. (2004) Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese Zucker rats. Diabetes 53, 1655–1663 [DOI] [PubMed] [Google Scholar]
- 8. Ouwens D. M., Diamant M., Fodor M., Habets D. D., Pelsers M. M., El Hasnaoui M., Dang Z. C., van den Brom C. E., Vlasblom R., Rietdijk A., Boer C., Coort S. L., Glatz J. F., Luiken J. J. (2007) Cardiac contractile dysfunction in insulin-resistant rats fed a high fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 50, 1938–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonen A., Parolin M. L., Steinberg G. R., Calles-Escandon J., Tandon N. N., Glatz J. F., Luiken J. J., Heigenhauser G. J., Dyck D. J. (2004) Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. 18, 1144–1146 [DOI] [PubMed] [Google Scholar]
- 10. Itani S. I., Zhou Q., Pories W. J., MacDonald K. G., Dohm G. L. (2000) Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes 49, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 11. Muoio D. M. (2010) Intramuscular triacylglycerol and insulin resistance: guilty as charged or wrongly accused? Biochim. Biophys. Acta 1801, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paz K., Hemi R., LeRoith D., Karasik A., Elhanany E., Kanety H., Zick Y. (1997) A molecular basis for insulin resistance: elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J. Biol. Chem. 272, 29911–29918 [DOI] [PubMed] [Google Scholar]
- 13. Uphues I., Kolter T., Goud B., Eckel J. (1995) Failure of insulin-regulated recruitment of the glucose transporter GLUT4 in cardiac muscle of obese Zucker rats is associated with alterations of small-molecular-mass GTP-binding proteins. Biochem. J. 311, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolter T., Uphues I., Eckel J. (1997) Molecular analysis of insulin resistance in isolated ventricular cardiomyocytes of obese Zucker rats. Am. J. Physiol. 273, E59–67 [DOI] [PubMed] [Google Scholar]
- 15. Frøsig C., Rose A. J., Treebak J. T., Kiens B., Richter E. A., Wojtaszewski J. F. (2007) Effects of endurance exercise training on insulin signaling in human skeletal muscle: interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes 56, 2093–2102 [DOI] [PubMed] [Google Scholar]
- 16. Hawley J. A., Lessard S. J. (2008) Exercise-training-induced improvements in insulin action. Acta Physiol. 192, 127–135 [DOI] [PubMed] [Google Scholar]
- 17. Luiken J. J., Vertommen D., Coort S. L., Habets D. D., El Hasnaoui M., Pelsers M. M., Viollet B., Bonen A., Hue L., Rider M. H., Glatz J. F. (2008) Identification of protein kinase D as a novel contraction-activated kinase linked to GLUT4-mediated glucose uptake, independent of AMPK. Cell. Signal. 20, 543–556 [DOI] [PubMed] [Google Scholar]
- 18. Treadway J. L., James D. E., Burcel E., Ruderman N. B. (1989) Effect of exercise on insulin receptor binding and kinase activity in skeletal muscle. Am. J. Physiol. 256, E138–144 [DOI] [PubMed] [Google Scholar]
- 19. Wojtaszewski J. F., Hansen B. F., Gade, Kiens B., Markuns J. F., Goodyear L. J., Richter E. A. (2000) Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49, 325–331 [DOI] [PubMed] [Google Scholar]
- 20. Treebak J. T., Glund S., Deshmukh A., Klein D. K., Long Y. C., Jensen T. E., Jørgensen S. B., Viollet B., Andersson L., Neumann D., Wallimann T., Richter E. A., Chibalin A. V., Zierath J. R., Wojtaszewski J. F. (2006) AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 55, 2051–2058 [DOI] [PubMed] [Google Scholar]
- 21. Kramer H. F., Witczak C. A., Fujii N., Jessen N., Taylor E. B., Arnolds D. E., Sakamoto K., Hirshman M. F., Goodyear L. J. (2006) Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55, 2067–2076 [DOI] [PubMed] [Google Scholar]
- 22. Bergman B. C., Tsvetkova T., Lowes B., Wolfel E. E. (2009) Myocardial FFA metabolism during rest and atrial pacing in humans. Am. J. Physiol. Endocrinol. Metab. 296, E358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eisele J. C., Schaefer I. M., Randel Nyengaard J., Post H., Liebetanz D., Brüel A., Mühlfeld C. (2008) Effect of voluntary exercise on number and volume of cardiomyocytes and their mitochondria in the mouse left ventricle. Basic Res. Cardiol. 103, 12–21 [DOI] [PubMed] [Google Scholar]
- 24. Glatz J. F., Luiken J. J., Bonen A. (2010) Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 90, 367–417 [DOI] [PubMed] [Google Scholar]
- 25. Hong W. (2005) SNAREs and traffic. Biochim. Biophys. Acta 1744, 493–517 [PubMed] [Google Scholar]
- 26. Kawanishi M., Tamori Y., Okazawa H., Araki S., Shinoda H., Kasuga M. (2000) Role of SNAP23 in insulin-induced translocation of GLUT4 in 3T3-L1 adipocytes. Mediation of complex formation between syntaxin4 and VAMP2. J. Biol. Chem. 275, 8240–8247 [DOI] [PubMed] [Google Scholar]
- 27. Schwenk R. W., Dirkx E., Coumans W. A., Bonen A., Klip A., Glatz J. F., Luiken J. J. (2010) Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 53, 2209–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luiken J. J., van Nieuwenhoven F. A., America G., van der Vusse G. J., Glatz J. F. (1997) Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J. Lipid Res. 38, 745–758 [PubMed] [Google Scholar]
- 29. van Oort M. M., van Doorn J. M., Bonen A., Glatz J. F., van der Horst D. J., Rodenburg K. W., Luiken J. J. (2008) Insulin-induced translocation of CD36 to the plasma membrane is reversible and shows similarity to that of GLUT4. Biochim. Biophys. Acta 1781, 61–71 [DOI] [PubMed] [Google Scholar]
- 30. Alberti K. G., Zimmet P., Shaw J. (2005) The metabolic syndrome: a new worldwide definition. Lancet 366, 1059–1062 [DOI] [PubMed] [Google Scholar]
- 31. Hage Hassan R., Hainault I., Vilquin J. T., Samama C., Lasnier F., Ferré P., Foufelle F., Hajduch E. (2012) Endoplasmic reticulum stress does not mediate palmitate-induced insulin resistance in mouse and human muscle cells. Diabetologia 55, 204–214 [DOI] [PubMed] [Google Scholar]
- 32. Coll T., Alvarez-Guardia D., Barroso E., Gómez-Foix A. M., Palomer X., Laguna J. C., Vázquez-Carrera M. (2010) Activation of peroxisome proliferator-activated receptor-δ by GW501516 prevents fatty acid-induced nuclear factor-κB activation and insulin resistance in skeletal muscle cells. Endocrinology 151, 1560–1569 [DOI] [PubMed] [Google Scholar]
- 33. Wang C., Liu M., Riojas R. A., Xin X., Gao Z., Zeng R., Wu J., Dong L. Q., Liu F. (2009) Protein kinase Cθ (PKCθ)-dependent phosphorylation of PDK1 at Ser-504 and Ser-532 contributes to palmitate-induced insulin resistance. J. Biol. Chem. 284, 2038–2044 [DOI] [PubMed] [Google Scholar]
- 34. Watson M. L., Coghlan M., Hundal H. S. (2009) Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem. J. 417, 791–801 [DOI] [PubMed] [Google Scholar]
- 35. Sinha S., Perdomo G., Brown N. F., O'Doherty R. M. (2004) Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor κB. J. Biol. Chem. 279, 41294–41301 [DOI] [PubMed] [Google Scholar]
- 36. Coort S. L., Willems J., Coumans W. A., van der Vusse G. J., Bonen A., Glatz J. F., Luiken J. J. (2002) Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. Mol. Cell. Biochem. 239, 213–219 [PubMed] [Google Scholar]
- 37. Luiken J. J., Niessen H. E., Coort S. L., Hoebers N., Coumans W. A., Schwenk R. W., Bonen A., Glatz J. F. (2009) Etomoxir-induced partial carnitine palmitoyltransferase-I (CPT-I) inhibition in vivo does not alter cardiac long chain fatty acid uptake and oxidation rates. Biochem. J. 419, 447–455 [DOI] [PubMed] [Google Scholar]
- 38. Chang L., Chiang S. H., Saltiel A. R. (2004) Insulin signaling and the regulation of glucose transport. Mol. Med. 10, 65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hawley S. A., Ross F. A., Chevtzoff C., Green K. A., Evans A., Fogarty S., Towler M. C., Brown L. J., Ogunbayo O. A., Evans A. M., Hardie D. G. (2010) Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rose A. J., Jeppesen J., Kiens B., Richter E. A. (2009) Effects of contraction on localization of GLUT4 and vSNARE isoforms in rat skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1228–1237 [DOI] [PubMed] [Google Scholar]
- 41. Dirkx E., Schwenk R. W., Coumans W. A., Hoebers N., Angin Y., Viollet B., Bonen A., van Eys G. J., Glatz J. F., Luiken J. J. (2012) Protein kinase D1 is essential for contraction-induced glucose uptake but is not involved in fatty acid uptake into cardiomyocytes. J. Biol. Chem. 287, 5871–5881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Randhawa V. K., Bilan P. J., Khayat Z. A., Daneman N., Liu Z., Ramlal T., Volchuk A., Peng X. R., Coppola T., Regazzi R., Trimble W. S., Klip A. (2000) VAMP2, but not VAMP3/cellubrevin, mediates insulin-dependent incorporation of GLUT4 into the plasma membrane of L6 myoblasts. Mol. Biol. Cell 11, 2403–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olson A. L., Knight J. B., Pessin J. E. (1997) Syntaxin 4, VAMP2, and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol. 17, 2425–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maier V. H., Melvin D. R., Lister C. A., Chapman H., Gould G. W., Murphy G. J. (2000) v- and t-SNARE protein expression in models of insulin resistance: normalization of glycemia by rosiglitazone treatment corrects overexpression of cellubrevin, vesicle-associated membrane protein-2, and syntaxin 4 in skeletal muscle of Zucker diabetic fatty rats. Diabetes 49, 618–625 [DOI] [PubMed] [Google Scholar]
- 45. Coort S. L., Luiken J. J., van der Vusse G. J., Bonen A., Glatz J. F. (2004) Increased FAT (fatty acid translocase)/CD36-mediated long-chain fatty acid uptake in cardiac myocytes from obese Zucker rats. Biochem. Soc. Trans. 32, 83–85 [DOI] [PubMed] [Google Scholar]
- 46. Bonen A., Holloway G. P., Tandon N. N., Han X. X., McFarlan J., Glatz J. F., Luiken J. J. (2009) Cardiac and skeletal muscle fatty acid transport and transporters and triacylglycerol and fatty acid oxidation in lean and Zucker diabetic fatty rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1202–1212 [DOI] [PubMed] [Google Scholar]
- 47. Dela F., Larsen J. J., Mikines K. J., Ploug T., Petersen L. N., Galbo H. (1995) Insulin-stimulated muscle glucose clearance in patients with NIDDM: effects of one-legged physical training. Diabetes 44, 1010–1020 [DOI] [PubMed] [Google Scholar]
- 48. Bertrand L., Ginion A., Beauloye C., Hebert A. D., Guigas B., Hue L., Vanoverschelde J. L. (2006) AMPK activation restores the stimulation of glucose uptake in an in vitro model of insulin-resistant cardiomyocytes via the activation of protein kinase B. Am. J. Physiol. Heart Circ. Physiol. 291, H239–250 [DOI] [PubMed] [Google Scholar]