

Background: The TRPV1 receptor is an ionotropic receptor implicated in a variety of pain and inflammatory disorders.
Results: β-Arrestin-2 scaffolds phosphodiesterase PDE4D5 to TRPV1 to regulate receptor phosphorylation and activity.
Conclusion: β-Arrestin-2 functions as a scaffold protein to mediate TRPV1 desensitization in multiple cell models.
Significance: Our findings presented herein provide compelling support for the contribution of β-arrestins as scaffolding proteins in the regulation of ligand-gated ion channels.
Keywords: Arrestin, Neurons, Pain, Protein Kinase A (PKA), TRP Channels, TRPV1, cAMP, Desensitization
Abstract
Transient receptor potential vanilloid 1 (TRPV1) is a nonselective cation channel activated by multiple stimuli and is implicated in a variety of pain disorders. Dynamic sensitization of TRPV1 activity by A-kinase anchoring protein 150 demonstrates a critical role for scaffolding proteins in nociception, yet few studies have investigated scaffolding proteins capable of mediating receptor desensitization. In this study, we identify β-arrestin-2 as a scaffolding protein that regulates TRPV1 receptor activity. We report β-arrestin-2 association with TRPV1 in multiple cell models. Moreover, siRNA-mediated knockdown of β-arrestin-2 in primary cultures resulted in a significant increase in both initial and repeated responses to capsaicin. Electrophysiological analysis further revealed significant deficits in TRPV1 desensitization in primary cultures from β-arrestin-2 knock-out mice compared with wild type. In addition, we found that β-arrestin-2 scaffolding of phosphodiesterase PDE4D5 to the plasma membrane was required for TRPV1 desensitization. Importantly, inhibition of PDE4D5 activity reversed β-arrestin-2 desensitization of TRPV1. Together, these results identify a new endogenous scaffolding mechanism that regulates TRPV1 ligand binding and activation.
Introduction
TRPV1 is a nonselective cation channel that is activated by multiple stimuli, including noxious heat (>42 °C), acidic pH, and capsaicin (1, 2). TRPV1 sensitivity to many chemical agonists is due to post-translational modifications of the receptor, including phosphorylation, which functionally reduces the threshold for receptor activation (3, 4). Recent research indicates that TRPV1 phosphorylation by protein kinases A and C is mediated by the scaffolding protein PKA-anchoring protein 150 (AKAP150) (5–8). Conversely, de-phosphorylation of TRPV1 affects receptor activity by increasing the threshold for receptor activation (4, 9) in a calcium-dependent manner (10). The calcium-dependent serine/threonine phosphatase calcineurin (protein phosphatase 2B or PP2B) de-phosphorylates and subsequently desensitizes TRPV1 (11, 12), whereas calmodulin binds to both termini of TRPV1 to influence receptor desensitization (13–15). However, to date no one has identified a scaffolding protein that endogenously restricts either tonic or stimulation-induced TRPV1 phosphorylation. In this study, we identify β-arrestin-2 as a scaffolding protein capable of preventing PKA phosphorylation of TRPV1, thereby reducing receptor response to agonist-mediated stimulation.
β-Arrestin proteins, initially identified for their role in G-protein-coupled receptor (GPCR)2 desensitization, have recently been ascribed with new roles as scaffolding and signaling molecules that promote multiprotein complex formation (16, 17). GPCR desensitization theory contends that G-protein-coupled receptor kinase-mediated phosphorylation of agonist-stimulated receptors drives β-arrestin association and subsequent internalization of the receptor for recycling or degradation (18, 19). Following agonist binding, kinases, including PKA, PKC, and G-protein-coupled receptor kinase, phosphorylate the receptor and attract β-arrestins (20–23), although several studies have demonstrated that β-arrestins can associate with GPCRs prior to agonist-mediated phosphorylation (24, 25). Recent reports identify β-arrestins as significant regulators of several unrelated ionotropic receptors, albeit through variable signaling mediators and pathways, via their ability to function as scaffold proteins (26, 28, 29).
It is now well accepted that β-arrestins can form scaffolding complexes with various proteins to modulate the strength and duration of multiple signaling pathways, including Src kinase, phosphodiesterase 4D (PDE4D), and Mdm2 (26, 30–32). Of particular interest, β-arrestins selectively scaffold phosphodiesterase 4D (PDE4D) to locally regulate subcellular cyclic AMP availability and subsequent PKA activity (32–34). Perry et al. (34) have demonstrated the contribution of β-arrestins to ligand-activated β2-adrenergic receptors by scaffolding PDE4D isoforms that hydrolyze cAMP to regulate PKA activity and subsequent receptor sensitivity (34). Additionally, the ability of β-arrestins to regulate metabotropic receptor activity is supported by many studies; however, their ability to modulate the activity of ionotropic receptors and downstream signaling pathways is an emerging area of research. A recent study demonstrates a scaffolding role for β-arrestin in the regulation of the TRPV4 receptor (29), a member of the TRP family with ∼40% homology to TRPV1 (36, 37). Therefore, we sought to determine whether β-arrestins also act to modulate TRPV1.
In this study, we demonstrate the association between TRPV1 and β-arrestin-2 in primary sensory neurons. Furthermore, we provide support that β-arrestin-2-mediated scaffolding of the phosphodiesterase PDE4D5 contributes to TRPV1 desensitization. De-phosphorylation of TRPV1 is known to mediate desensitization of the receptor, yet only calcineurin (calcineurin/PP2B) has been characterized to regulate this process (9, 11). Our present data demonstrate a novel endogenous mechanism underlying the down-regulation of TRPV1 activity, by which β-arrestin-2 scaffolds PDE4D5 at the plasma membrane to control the phosphorylation status of TRPV1.
EXPERIMENTAL PROCEDURES
Reagents
Capsaicin and dimethyl sulfoxide (DMSO) were purchased from Sigma. Rolipram was obtained from Cayman Chemical (Ann Arbor, MI). Stock solutions were made as follows. Capsaicin was dissolved in ethanol for a 50 nm treatment concentration and rolipram in DMSO for a 20 mm treatment concentration.
Tissue Culture
All procedures utilizing animals were approved by the Institutional Animal Care and Use Committee of University of Texas Health Science Center, San Antonio, and were conducted in accordance with policies for the ethical treatment of animals established by the National Institutes of Health. Trigeminal ganglia (TG) were dissected bilaterally from male Sprague-Dawley rats (200–250 g; Charles River Laboratories, Wilmington, MA) and disassociated by treatment with collagenase (Worthington) for 30 min, followed by treatment with trypsin (Sigma) for 15 min. Cells were centrifuged, aspirated, and resuspended in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 ng/ml nerve growth factor (Harlan Laboratories, Indianapolis, IN), 1% penicillin/streptomycin (Invitrogen), and 1% glutamine (Sigma) and then placed on poly-d-lysine-coated plates. Cultures were maintained at 37 °C and 5% CO2 and grown for 5–7 days. Briefly, experiments conducted with TG dissected bilaterally from C57BL/6 wild-type and knock-out β-arrestin-2 mice (Duke University, Durham, NC) were disassociated by treatment with collagenase (Worthington) and dispase (Sigma). Next, cells were centrifuged, aspirated, and resuspended in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 ng/ml nerve growth factor (Harlan Laboratories, Indianapolis, IN), 1% penicillin/streptomycin (Invitrogen), and 1% glutamine (Sigma), plated on poly-d-lysine/laminin coverslips (BD Biosciences), and used the next day for electrophysiology experiments. Chinese hamster ovarian (CHO) cells were utilized for heterologous expression of cDNA constructs. CHO cells were maintained at 37 °C and 5% CO2 and transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions.
siRNA Transfection
FITC-labeled siRNA duplexes custom-designed to target β-arrestin-2 were generated by Qiagen (Valencia, CA). The sense strand of β-arrestin-2 siRNA was 5′-GGAAAGTGTTTGTGACCCTTT-3′ and antisense strand of β-arrestin-2 siRNA was 5′-UUCUCCUCCAGGUUCUCAGUU-3′. TG sensory neurons were transfected with scrambled siRNA (Silencer-1, Ambion, Austin, TX), no siRNA (mock), or β-arrestin-2 FITC-labeled siRNA (20 μg of siRNA/10-cm plate) (38) using HiPerFect (Qiagen), following the manufacturer's directions. Following 18 h of transfection incubation, cells were replenished with complete media, and total cell lysates were harvested 3 h post-media change. Western blot analysis was conducted with antibodies specific for TRPV1 (R-130, Santa Cruz Biotechnology, Santa Cruz, CA), β-arrestin-1/2 (A-1, Santa Cruz Biotechnology), β-arrestin-2 (H-9, Santa Cruz Biotechnology), and β-actin (Sigma).
Immunoprecipitation and Western Blot Analysis
For each experimental condition, cells were treated with the indicated compounds and harvested as described previously (11). Protein quantification of crude plasma membrane homogenates was performed using the Bradford assay (39) (Sigma) following the manufacturer's directions. Samples (200 μg) were immunoprecipitated with 2 μg of anti-TRPV1 (R-130, Santa Cruz Biotechnology) or β-arrestin-2 antiserum (H-9, Santa Cruz Biotechnology). Next, immunoprecipitates were resolved via 15% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). Western blots were blocked in 5% nonfat milk in Tris-buffered saline/Tween 20 and visualized using anti-TRPV1, anti-β-arrestin-2, or anti-PDE4D primary antibodies (H-69, Santa Cruz Biotechnology) followed by the appropriate horseradish peroxidase-conjugated secondary antisera (GE Healthcare) and enhanced chemiluminescence detection following the manufacturer's instructions (GE Healthcare). To study TRPV1 phosphorylation, transfected CHO cells in 6-cm plates were incubated with 250 mCi of [32P]orthophosphate (PerkinElmer Life Sciences) for 4 h at 37 °C in phosphate-free DMEM (Invitrogen) on the day of experimentation. Crude plasma membranes were prepared for immunoprecipitation, SDS-PAGE separation, and transfer following University of Texas Health Science Center, San Antonio, radiation safety protocols. Autoradiographs were developed after an 18-h exposure to blots at −80 °C. Densitometry measurements were determined using Image 1.62 (National Institutes of Health), with the reported pixel density = (band density) − (lane background density).
Biotinylation
Cultured CHO cells transfected with mock, TRPV1, or TRPV1 and β-arrestin-2/GFP cDNA (3 μg/6-cm plate) were treated with vehicle or CAP (100 nm; 30 s) and rinsed three times with PBS. Next, cells were biotinylated with EZ-Link biotin (0.5 mg/ml; Pierce), harvested, and precipitated as described previously (40). To determine expression of TRPV1, SDS-PAGE was performed followed by transfer to a PVDF membrane and Western blotting with antibodies specific for TRPV1 (R-130, Santa Cruz Biotechnology) and β1-integrin (M-106, Santa Cruz Biotechnology).
Crude Membrane Preparation
Following homogenization by 20 strokes in a Potter-Elvehjem homogenizer in a hypotonic homogenization buffer (25 mm HEPES, 25 mm sucrose, 1.5 mm MgCl2, 50 mm NaCl, pH 7.2), the cell extract was incubated on ice for 15 min and then centrifuged at 1000 × g for 1 min at 4 °C to remove nuclei and unlysed cells from the homogenate. The resulting supernatant was centrifuged at 16,000 × g for 30 min at 4 °C, separating cytosolic proteins from cell membrane proteins. The pellet (crude membrane fraction) was then resuspended in 400 μl of homogenization buffer containing 1% Triton X-100 and utilized for co-immunoprecipitation experiments.
Calcium Imaging
To measure intracellular Ca2+ levels, Fura-2 AM (2 μm; Molecular Probes, Carlsbad, CA) was incubated with cells for 30 min at 37 °C in the presence of 0.05% Pluronic (Calbiochem). Fluorescence was detected with a Nikon Eclipse Ti-U microscope fitted with a ×20/0.8 NA Fluor objective. Fluorescence images from 340 and 380 nm excitation wavelengths were collected and analyzed with MetaFluor software (MetaMorph, Web Universal Imaging Corp., Downingtown, PA). Transfected CHO cells plated on poly-d-lysine coverslips (BD Biosciences) were identified by their GFP fluorescence. To assess Ca2+ accumulation following TRPV1 activation CAP (50 nm) was administered for 30 s followed by a 3-min washout with standard extracellular solution (SES) buffer (140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm d-(+)-glucose, 10 mm HEPES, pH 7.4). For analysis of TRPV1 desensitization, repeated CAP (50 nm; 30 s) applications were interrupted by SES washout for 3 min. Ratiometric data were converted to [Ca2+]i by using the following equation: [Ca2+]i = K·(R − Rmin)/(Rmax − R), where R is the 340/380 nm fluorescence ratio. For TG neurons, cells were plated on poly-d-lysine/laminin coverslips (BD Biosciences) and transfected in a mock fashion or with β-arrestin-2 siRNA (0.5 μg of siRNA/coverslip) using HiPerFect (Qiagen), following the manufacturer's directions. Next, TRPV1 activity was assessed following repeat application of CAP (50 nm; 30 s) in the same manner as transfected CHO cells.
Electrophysiology
All recordings were made in a whole-cell patch voltage clamp configuration at a holding potential (Vh) of −60 mV. Recordings and following analysis were carried out at 22–24 °C with TG sensory neurons from β-arrestin-2 wild-type and knock-out mice (5-week-old littermates) using an Axopatch 200B amplifier and pCLAMP 9 software (Axon Instruments, Molecular Devices, Sunnyvale, CA). Briefly, TG neurons were harvested and plated on poly-d-lysine/laminin coverslips (BD Biosciences) and assessed for TRPV1 activity the following day. Data were filtered at 0.5 kHz and sampled at 2 kHz. Borosilicate pipettes (Sutter Instrument Co., Novato, CA) were polished to resistances of Mr 4–6 in standard pipette solution. If necessary, access resistance (Rs) was compensated by 40–80% to Mr <10–12. All recordings were made in the presence of 2 mm Ca2+ in external solution. Drugs were applied using a computer-controlled pressure-driven eight-channel system (ValveLink8; AutoMate Scientific, Berkeley, CA).
Total Internal Reflection Fluorescence-Forster Resonance Energy Transfer (TIRF-FRET) Microscopy
Fluorescence emissions from enhanced cyan fluorescent protein (CFP)-tagged TRPV1 (41) or enhanced yellow fluorescent protein (YFP)-tagged PDE4D5 were collected at room temperature using TIRF microscopy. All TIRF experiments were performed in the total internal reflection fluorescence microscopy core facility housed within the Department of Physiology at the University of Texas Health Science Center, San Antonio. Fluorescence emissions were collected using an inverted Nikon TE2000 microscope with through-the-lens (prismless) TIRF imaging (Nikon). This system was equipped with a vibration isolation system (Technical Manufacturing, Peabody, MA) to minimize drift and noise. Samples were viewed through a plan-Apo TIRF ×60 oil immersion high resolution (1.45 numerical aperture) TIRF objective. Coupled to the microscope was a laser light-delivery system (Prairie Technologies, Middleton, WI) consisting of a 40-milliwatt argon laser outputting 488- and 514-nm lines and a 442-nm diode-pumped solid state laser. The excitation light was selected with an acoustic optical tunable filter, controlled by MetaMorph software. CFP and YFP emissions were simultaneously collected using the Dual-View chip splitter (Optical Insights, Photometrics, Tucson, AZ), equipped with a filter cube containing HQ470-nm/30-m and HQ550-nm/30-m emission filters for CFP and YFP emission, respectively, and a 505-nm dichroic mirror for separation of emission wavelengths. In this configuration, the microscope uses only a dual bandpass TIRF dichroic mirror to separate the excitation and emission light, with no excitation filters in the microscope cube. The TIRF angle was adjusted by eye to give the signature TIRF illumination to the experimental chamber. Fluorescence images were collected and processed with a 16-bit, cooled charge-coupled device camera (Cascade 512F; Roper Scientific, Tucson, AZ) interfaced to a PC running MetaMorph software. This camera uses a front-illuminated electron-multiplying charge-coupled device with on-chip multiplication gain. Images were collected (300-ms exposure time, focused to best exploit the dynamic range of the camera without pixel saturation) immediately before and after photobleaching. Images were not binned or filtered, with pixel size corresponding to a square of 122 × 122 nm.
Radioligand Binding
Saturation binding assays using [3H]RTX (PerkinElmer Life Sciences) with cell membrane preparations were conducted as described by Szallasi and Blumberg (42, 43). Membranes were prepared by repeated trituration of cells through a 1-ml tuberculin syringe with 26-gauge needle in ice-cold buffer (10 mm HEPES, 2 mm MgCl2, 0.75 mm CaCl2, 5 mm KCl and 58 mm NaCl, pH 7.4) and transferred to a 50-ml centrifuge tube containing 35 ml of ice-cold buffer. The homogenate was centrifuged (39,000 × g, 4 °C, 10 min), and the pellet was resuspended in 4.5 ml of buffer, vortexed and sonicated. Membrane protein content was measured with the Bradford assay (Pierce). Aliquots (100 μl containing 40–50 μg of protein) of membrane suspension were incubated with multiple concentrations of radioligand in triplicate over a 4-log unit range (final volume 200 μl) in 96-well multiscreen filtration plates (Millipore) previously soaked with buffer containing 10 mg/ml BSA. Incubations were carried out for 1 h at 37 °C followed by rapid filtration, followed by 10 washes of cold buffer. Nonspecific binding was determined in the presence of 1 μm cold RTX.
Statistical Analysis
If not stated otherwise, one-way ANOVA with Bonferroni post hoc corrections were used for multiple comparisons (GraphPad Prism software, La Jolla, CA). Student's t test was utilized for comparison of normalized CAP responses for calcium imaging data. All values refer to means ± S.E.; n indicates the sample number; p denotes the significance (**, p < 0.01; ***, p < 0.001), and NS indicates not significant. For receptor saturation binding experiments, the data were fit with nonlinear regression to Equation 1 to provide estimates of Bmax and Kd,
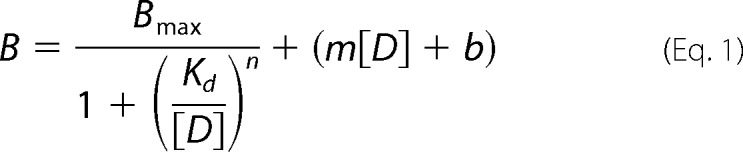 |
where B is the measured amount of radioligand bound (fmol/mg of protein) in the presence of various concentrations of radioligand ([D]); Bmax is the maximal amount of radioligand bound; Kd is the concentration of radioligand producing half-maximal binding; n is the slope factor; m is the slope, and b is the intercept of the linear regression line for nonspecific binding.
RESULTS
The coordinated organization and targeting of specific enzymes within close proximity to TRPV1 leads to efficient control of receptor activity. Recently, β-arrestins were identified as novel scaffolding proteins (32, 44) and have been implicated in the regulation of ionotropic receptors, including TRPV4 (16, 26, 29). Thus, we sought to determine whether β-arrestins participate in the regulation of TRPV1, a receptor that shares 40% homology with TRPV4. Co-immunoprecipitation analyses of primary cultures of trigeminal peripheral sensory neurons reveal association of β-arrestin-2 (over β-arrestin-1) with TRPV1 in crude plasma membrane preparations (Fig. 1A). To confirm specificity of the β-arrestin-1/2 antibody, CHO cells were mock-transfected with empty vector or β-arrestin-2 cDNA and immunoprecipitated with β-arrestin-1/2 antibody. A specific band was observed in cells selectively transfected with β-arrestin-2 but not in mock (Fig. 1B). Moreover, CHO cells immunoprecipitated with IgG did not yield nonspecific immunoreactivity in cells transfected with β-arrestin-2 (Fig. 1B, right panel). To further confirm the association between β-arrestin-2 and TRPV1 in our heterologous CHO cell model, β-arrestin-2 and TRPV1 cDNA were transfected into CHO cells, and association was observed at the plasma membrane (Fig. 1C).
FIGURE 1.

β-Arrestin-2 associates with TRPV1 in primary sensory neurons. A, primary cultures of TG sensory neurons were homogenized and differentially centrifuged to isolate crude plasma membrane fractions. Next, co-immunoprecipitation (IP) was conducted with antibodies specific for β-arrestin-1/2 (β-arr1/2) and TRPV1 followed by Western blotting. CL, cell lysate (50 μg). Molecular sizes of proteins are indicated on the left side of immunoblots (β-arr1; 50 kDa, β-arr2; 48 kDa). WB, Western blot. B, CHO cells mock-transfected or transfected with β-arrestin-2/GFP, followed by co-immunoprecipitation with a β-arrestin-1/2 antibody (left panel) or with IgG (right panel). Red arrow indicates position of β-arrestin-2/GFP, and blue arrow indicates mouse IgG. C, co-immunoprecipitates and crude plasma membrane homogenate samples of CHO cells transfected with the indicated cDNAs. D, CHO cells transfected with the indicated cDNAs were surface-biotinylated to identify plasma membrane expression of TRPV1 and GFP. E, CHO cells were transfected with TRPV1 alone or with β-arrestin-2 and assessed for TRPV1 plasma membrane expression via surface biotinylation following vehicle or CAP (100 nm; 1 min) treatment. β1 integrin expression was used as a positive control for crude plasma membrane protein expression. Results are representative of four independent experiments. F, CHO cells transfected with TRPV1 alone or with β-arrestin-2 were assessed for TRPV1 activity via calcium imaging. Following acquisition of base line, CAP (50 nm) was administered for 30 s, and cells were rinsed with SES buffer for 3 min prior to a second CAP challenge. To assess desensitization, a second application of CAP was administered for 30 s after the SES buffer washout. Accumulation of Ca2+ was calculated as the change in [Ca2+]i, n = 144–206 cells per transfection group. Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001. G, normalized second CAP response was calculated as percent of initial CAP response. t test was for normalized CAP response; ***, p < 0.001.
Because β-arrestins are well characterized for their role in directing GPCR desensitization and internalization (28, 45, 46), we next investigated whether β-arrestin-2 alters plasma membrane expression of TRPV1. Cells co-transfected with TRPV1 and β-arrestin-2 demonstrate no difference in plasma membrane expression of TRPV1 compared with cells expressing TRPV1 alone, as determined by surface biotinylation (Fig. 1D). Because agonist stimulation of GPCRs stimulates β-arrestin-mediated internalization, we questioned whether CAP-mediated stimulation of TRPV1 would result in β-arrestin-dependent internalization. Cells co-transfected with β-arrestin-2 and TRPV1 demonstrated no difference in plasma membrane expression of TRPV1 following vehicle or CAP treatment (Fig. 1E). These data together indicate that TRPV1 associates with β-arrestin-2 in multiple cell types, but it does not affect TRPV1 receptor expression on the plasma membrane. We next investigated whether β-arrestin-2 co-expression alters TRPV1 response to CAP, a receptor-specific agonist. Calcium imaging analyses revealed that repeated CAP (50 nm; 30 s) treatments of CHO cells transfected with TRPV1 and β-arrestin-2 resulted in a significant increase in receptor desensitization, compared with cells expressing TRPV1 alone (Fig. 1, F and G).
To explore the regulatory contribution of β-arrestin-2 to TRPV1 in peripheral sensory neurons, we designed siRNA to selectively knock down β-arrestin-2 protein expression. Transfection with β-arrestin-2 siRNA reduced β-arrestin-2 expression 65%, compared with mock and scrambled siRNA-transfected cultures (Fig. 2, A and B). To confirm that selective knockdown of β-arrestin-2 with siRNA does not alter plasma membrane expression of TRPV1, we conducted surface biotinylation experiments with mock-transfected and β-arrestin-2 siRNA-transfected cultures. Importantly, no differences in surface expression of TRPV1 were observed between mock and siRNA-transfected neurons (Fig. 2C). Following siRNA validation, we investigated the TRPV1 response to CAP following siRNA-mediated knockdown of β-arrestin-2 expression. β-Arrestin-2 siRNA knockdown resulted in a significant increase in both initial and tachyphylactic responses to CAP, as well as response rates, compared with mock-transfected cultures (Fig. 2, D and E). Specifically, siRNA-mediated knockdown of β-arrestin-2 resulted in a significant decrease in TRPV1 desensitization, indicated by an increase in the normalized second CAP response (62%) as compared with mock-transfected neurons (28%) (Fig. 2F). Because the magnitudes of calcium influx can be influenced by channel kinetics, localization of channels on internal membranes, as well as sarcoplasmic/endoplasmic reticulum calcium functions and un-clamped voltage-gated calcium channels, we next evaluated the effects of β-arrestin2 siRNA using voltage clamp whole-cell recording (Fig. 3). Importantly, cumulative results between the two approaches yielded similar results, demonstrating reduced TRPV1 desensitization following β-arrestin2 knockdown.
FIGURE 2.

β-Arrestin-2 knockdown decreases TRPV1 desensitization. A, primary cultures of TG neurons were transfected with siRNA directed against β-arrestin-2, silencer negative siRNA (Scr), or no siRNA (mock) and assessed for β-arrestin-2 expression by Western blotting. B, quantification of β-arrestin-2 expression calculated as percent of mock-transfected cells. C, TG neurons were mock-transfected or transfected with β-arrestin-2 siRNA. Next, cells were surface-biotinylated to assess plasma membrane expression of TRPV1. Molecular sizes of proteins are indicated on left side of immunoblots. D, TRPV1 activity was assessed via calcium imaging in TG neurons following mock transfection or transfection with scrambled or β-arrestin-2 siRNA. Following acquisition of base line, CAP (50 nm; representative traces (lines)) was administered for 30 s, and cells were rinsed with SES buffer for 3 min prior to second CAP challenge. E, CAP-evoked Ca2+ influx in TG neurons mock-transfected or transfected with scrambled or β-arrestin-2 siRNA, n = 84–130 cells per transfection group. Accumulation of Ca2+ was calculated from the change in [Ca2+]i. Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001. F, normalized second CAP response was calculated as percent of initial CAP response. t test was for normalized CAP response; ***, p < 0.001. NS, not significant. Results are representative of four independent experiments.
FIGURE 3.

β-Arrestin-2 knockdown affects TRPV1-desensitized current. A–C, representative current traces of TG sensory neurons transfected in a mock fashion or with scrambled or β-arrestin-2 siRNA, following repeat applications of CAP (100 nm; 30 s; representative traces (lines)). D, normalized second CAP response was calculated as a percent of the initial CAP response. One-way ANOVA was for normalized CAP response with Bonferroni post hoc correction; **, p < 0.01.
β-Arrestin isoforms 1 and 2 are co-expressed in TG neurons and contain high amino acid sequence homology (19). However, co-immunoprecipitation results confirm preferential association of TRPV1 with β-arrestin-2 (Fig. 1). The selective role of β-arrestin-2, but not β-arrestin-1, in TRPV1 regulation was further confirmed from mouse embryonic fibroblasts, which selectively express β-arrestin-1 or β-arrestin-2 (18) (data not shown). Our findings from molecular and functional analyses indicate a fundamental role for β-arrestin-2 in the regulation of TRPV1 activation, namely receptor desensitization (Figs. 1G, 2F, and 3D). Genetic ablation of β-arrestin-2 is known to significantly enhance and prolong anti-nociception in an animal model. Specifically, β-arrestin-2 KO mice demonstrate potentiated morphine analgesia as compared with their WT littermates (47, 48). To further confirm the contribution of β-arrestin-2 to TRPV1 activity, we conducted patch clamp electrophysiology on TG sensory neurons harvested from β-arrestin-2 KO and WT animals. Following repeat application of CAP (100 nm; 30 s), β-arrestin-2 KO neurons displayed a significant reduction in receptor desensitization (19%) as compared with WT neurons (50%) (Fig. 4C). Unlike our siRNA model, we did not observe significant differences in CAP-induced initial currents between β-arrestin-2 KO and WT neurons (Fig. 4D). Yet both siRNA and KO models implicate β-arrestin-2 in the regulation of TRPV1 receptor desensitization.
FIGURE 4.

Genetic ablation of β-arrestin-2 decreases TRPV1 desensitization. A, representative current traces of TG sensory neurons cultured from β-arrestin-2 WT mice, following a repeat application of CAP (100 nm; 30 s; representative traces (lines)). B, representative current traces from TG sensory neurons cultured from β-arrestin-2 KO mice. C, normalized second CAP response was calculated as a percent of the initial CAP response. t test was for normalized CAP response;' **, p < 0.01. D, initial current amplitude (pA) of β-arrestin-2 KO and WT neurons. NS, not significant.
Recent studies demonstrate a scaffolding role of β-arrestins in the regulation of both metabotropic and ionotropic receptors (29, 32, 44). Based on calcium imaging and electrophysiology results, sensory neurons transfected with β-arrestin-2-specific siRNA (Figs. 2 and 3) or harvested from β-arrestin-2 KO animals (Fig. 4) both demonstrate deficits in receptor desensitization. Because β-arrestin-2 does not influence TRPV1 localization to the plasma membrane (Fig. 1D), we sought to identify the mechanism underlying β-arrestin-2- regulation of receptor activity. Although β-arrestin scaffolding of the E3 ubiquitin ligase, AIP4, controls TRPV4 down-regulation (29), no differences in TRPV1 ubiquitination were observed in CHO cells co-expressing β-arrestin-2 (data not shown). However, several studies have identified β-arrestin-2-scaffolded PDE4D in the regulation of metabotropic receptors (33, 34). Additionally, two specific regions in the C-domain of β-arrestin-2 confer binding specificity for the PDE4D5 isoform (49). Because PKA acts both to phosphorylate/sensitize TRPV1 (4) and to reverse receptor desensitization (9), we investigated whether a PDE4D isoform could be the β-arrestin-2-scaffolded protein involved in mediating TRPV1 activity.
In cultured TG neurons, we observed a greater association of β-arrestin-2 with PDE4D5 (over PDE4D3) at the level of the plasma membrane (Fig. 5A). To substantiate PDE4D5 as the specific isoform involved in β-arrestin-2-mediated desensitization of TRPV1, we performed calcium imaging on CHO cells transfected with TRPV1 and β-arrestin-2 with PDE4D3, PDE4D5, or the catalytically inactive mutant PDE4D5-D556A (33). Co-expression of PDE4D5, but not PDE4D3 or PDE4D5-D556A, significantly increased TRPV1 desensitization following multiple CAP applications, as indicated by diminished calcium accumulation (Fig. 5, B and C). Conversely, co-transfection with the PDE4D5-D556A mutant resulted in a significant decrease in receptor desensitization compared with cells expressing PDE4D5 (Fig. 5C). These data indicate that co-expressed PDE4D5 plays a role in TRPV1 desensitization.
FIGURE 5.

β-Arrestin-2 preferentially associates with the phosphodiesterase PDE4D5. A, primary cultures of TG neurons were immunoprecipitated (IP) with the indicated antibodies followed by Western blot analysis. Cell lysate (CL; 50 μg) was used as a loading control. Molecular sizes of proteins indicated on left side of immunoblots (PDE4D5; 105 kDa, PDE4D3; 95 kDa). WB, Western blot. B and C, TRPV1 activity from CHO cells transfected with the indicated PDE4D cDNA was assessed via calcium imaging following repeat applications of CAP (50 nm, 30 s; bars) from CHO cells transfected with the indicated PDE4D cDNA. D, accumulation of Ca2+ was calculated as the change in [Ca2+]i, n = 121–135 cells per transfection group. Two-way ANOVA with Bonferroni post hoc correction is shown; **, p < 0.01; ***, p < 0.001. E, normalized second CAP response was calculated as percent of initial CAP response. One-way ANOVA was for normalized CAP response; ***, p < 0.001.
Next, we sought to determine whether β-arrestin-2 is required for PDE4D5 to modulate the TRPV1 agonist response by first investigating β-arrestin-2-dependent proximity between PDE4D5 and TRPV1. We employed TIRF-FRET microscopy to quantify PDE4D5-TRPV1 interaction at the plasma membrane in the presence or absence of β-arrestin-2 expression. For this experiment, CHO cells were transfected with PDE4D5-YFP and TRPV1-CFP to establish FRET, in the presence of either empty vector or β-arrestin-2. As shown in Fig. 6, transfection of β-arrestin-2 significantly increased CFP emission in CHO cells expressing PDE4D5-YFP and TRPV1-CFP, and this increase was comparable with emission generated by the Rho-pYC positive control (plasma membrane-targeted CFP-YFP tandem proteins) (Fig. 6, A and B) (50). Although these results indicate β-arrestin-2-dependent subcellular co-localization of PDE4D5 to TRPV1, we sought to identify the functional relationship between the phosphodiesterase and receptor. To determine this point, we employed rolipram, a selective inhibitor of the PDE4D family of phosphodiesterases. Although rolipram (20 μm, 10 min) failed to alter the primary CAP response in TG neurons (Fig. 6C), it significantly decreased TRPV1 desensitization (Fig. 6, E and F). Importantly, neurons transfected with β-arrestin-2-specific siRNA exhibited a decrease in CAP/CAP desensitization compared with vehicle, which was not further potentiated following rolipram (Fig. 6F). Therefore, the reduction in pharmacological desensitization of TRPV1 by rolipram was blocked by β-arrestin-2 knockdown, suggesting that PDE4D5-dependent modulation of TRPV1 is dependent upon β-arrestin-2 expression.
FIGURE 6.

PDE4D5 effects on TRPV1 require β-arrestin-2. A, images of CHOs under TIRF illumination expressing CFP-tagged TRPV1, YFP-tagged PDE4D5, and either empty vector or β-arrestin-2 under 442- and 514-nm laser lines. Images of CFP (left, rainbow pseudocolor) and YFP (right, yellow pseudocolor) emissions are shown before and after YFP photobleaching. B, percentage increase in CFP emission after YFP photobleaching for groups in A, including membrane-targeted CFP-YFP tandem proteins (Rho-pYC, positive control). One-way ANOVA was with Bonferroni post hoc correction; ***, p < 0.001. C and D, representative traces of CAP-induced calcium accumulation in TG neurons following vehicle (Veh) or rolipram (PDE4D inhibitor, 20 μm, 10 min) transfected in a mock fashion or with β-arrestin-2-specific siRNA. TRPV1 activity was assessed via calcium imaging following repeat applications of CAP (50 nm, 30 s; bars). Accumulation of Ca2+ was from the change in [Ca2+]ii. E, quantified peaks of calcium accumulation from C and D. Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001. F, normalized second CAP response was calculated as percent of initial CAP response. One-way ANOVA was for normalized CAP response, n = 75–120 neurons for each treatment paradigm. ***, p < 0.001; NS, no significance.
β-Arrestin-mediated scaffolding of phosphodiesterases to a variety of receptors leads to localized down-regulation of cAMP concentrations and subsequent downstream signaling cascades (33, 51, 52). Thus, we investigated the role of β-arrestin-2 in PKA-mediated phosphorylation of TRPV1. Serum-starved CHO cells co-expressing TRPV1 and either empty vector or β-arrestin-2 were metabolically labeled with [32P]orthophosphate and treated with vehicle or forskolin (adenylyl cyclase activator, 10 μm, 5 min). As shown in Fig. 7A, β-arrestin-2 overexpression blocked forskolin-stimulated incorporation of [32P]orthophosphate into TRPV1. Post-translational modifications of TRPV1, including phosphorylation and de-phosphorylation, are implicated in the sensitization and desensitization of the receptor, respectively (5, 6, 53, 54). Importantly, specific residues of TRPV1, including Ser-116, Thr-144, and Thr-370, are selectively phosphorylated by PKA and are important for receptor activity (4, 9, 53). With these PKA phosphorylation site mutants of TRPV1, we demonstrate that β-arrestin-2 co-expression reduces normalized TRPV1 desensitization in wild-type and the TRPV1 T144A mutant cells only, although no changes were observed in the desensitized responses of TRPV1 mutants S116A and T370A (Fig. 7, B and C). Importantly, pretreatment with the phosphodiesterase inhibitor rolipram (20 μm; 30 min) reversed the effects on TRPV1 desensitization in cells transfected with the T370A mutant (Fig. 7, D and E). These data identify TRPV1 Ser-116 and to a greater extent Thr-370 as residues in which PKA phosphorylation is decreased by β-arrestin-2-scaffolded PDE4D5 and are critical to reversing TRPV1 desensitization. Because TRPV1 de-phosphorylation reduces agonist affinity (55), we also sought to determine whether the presence of β-arrestin-2 or inhibition of PDE4D5 would affect the pharmacological profile for the TRPV1-specific agonist RTX. Full saturation of [3H]RTX binding curves were generated, yielding Kd and Bmax values for CHO cells transfected with TRPV1 alone, TRPV1, and β-arrestin-2 or TRPV1 and β-arrestin-2 treated with rolipram (Table 1). Interestingly, the co-expression of β-arrestin-2 with TRPV1 significantly increased the Bmax more than 3-fold over TRPV1 alone and in a rolipram-sensitive manner. Furthermore, Kd values for [3H]RTX binding to CHO cells expressing both TRPV1 and β-arrestin-2 were lower than those for cells expressing TRPV1 alone or following rolipram treatment. These data indicate that β-arrestin-2 functions to selectively regulate PKA-mediated phosphorylation at residues Ser-116 and Thr-370 within TRPV1, thereby reducing agonist affinity for TRPV1 in a rolipram-sensitive manner.
FIGURE 7.

β-Arrestin-2 modulates TRPV1 phosphorylation. A, autoradiographic detection (top) and quantification (n = 3, bottom) of [32P]orthophosphate uptake by TRPV1 in CHO cells when co-expressed with β-arrestin-2 and treated with forskolin (10 μm, 5 min). Molecular sizes of proteins are indicated on left side of immunoblots. Results are representative of three independent experiments. One-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001. IP, immunoprecipitation; WB, Western blot. B, multiple CAP-stimulated Ca2+ accumulation measurements from CHO cells expressing WT TRPV1 and the PKA phosphorylation mutants TRPV1 S116A, T144A, T370A, and either empty vector or β-arrestin-2/GFP. Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001. C, second CAP response from B is normalized to the first and depicted per TRPV1 species in the presence/absence of β-arrestin-2/GFP; n = 71–86 cells for each paradigm. Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001; NS, not significant. D, multiple CAP-stimulated Ca2+ accumulation measurements from CHO cells expressing TRPV1 WT and the PKA phosphorylation mutants TRPV1 S116A, T370A, and treated with vehicle or rolipram (20 mm, 30 min). Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001. E, second CAP response from D is normalized to the first and depicted per TRPV1 species in the presence/absence of rolipram. Two-way ANOVA with Bonferroni post hoc correction is shown; ***, p < 0.001; NS = no significance.
TABLE 1.
Co-expression of β-arrestin-2 with TRPV1 increases Bmax but reduces the Kd value for [3H]RTX
Saturation binding assays were done with CHO cells transiently expressing either TRPV1 alone or with β-arrestin-2 (β-arr2). Cells were then treated (trtd) with vehicle or the PDE4D phosphodiesterase inhibitor rolipram, as described under “Experimental Procedures.” Data represent the means ± S.E. of five independent experiments.
| Treatment conditions | pKd | Bmax |
|---|---|---|
| nm | fmol/mg protein | |
| TRPV1 | 9.87 ± 0.06 (0.136) | 1076 ± 292 |
| TRPV1 + β-arr2 | 8.88 ± 0.25 (0.741)* | 3949 ± 533** |
| TRPV1 + β-arr2 (rolipram-trtd) | 9.76 ± 0.31 (0.174) | 1308 ± 284 |
a One-way ANOVA, p < 0.05.
b p < 0.01 versus TRPV1 expression alone.
DISCUSSION
β-Arrestins are well characterized for their role in the regulation of a variety of GPCRs (45, 56). To date, extensive research by numerous groups has revealed the contributions of β-arrestins to multiple physiological functions and processes (44, 57, 58). β-Arrestins are now ascribed as novel scaffolding proteins, which associate with a variety of proteins to effectively regulate receptor activity and downstream signaling pathways (32). Here, we provide the first evidence of a scaffolding role for β-arrestin-2 in the regulation of the TRPV1 receptor, which is critically implicated in a variety of inflammatory and pain conditions. We demonstrate a novel association between β-arrestin-2 and the ionotropic TRPV1 receptor at the level of the plasma membrane both in sensory neurons and transfected heterologous cells, where this association enhances the pharmacological desensitization of TRPV1 (Fig. 1). In particular, both siRNA-mediated knockdown and genetic ablation of β-arrestin-2 significantly reduce TRPV1 desensitization (Figs. 2–4). Previous studies have demonstrated preferential association of β-arrestin-2 with PDE4D5, via its ability to bind to specific regions within both the N and C termini of β-arrestin-2 (49). Here, we demonstrate that β-arrestin-2 expression is required to localize the phosphodiesterase PDE4D5 to within close spatial proximity to TRPV1 at the plasma membrane (Fig. 6A). This action mediates the phosphorylation state of TRPV1 and contributes to the effective regulation of receptor desensitization (Fig. 7). Furthermore, CAP-induced Ca2+ accumulation measurements following rolipram treatment demonstrate a reduced return to base line, providing further evidence that β-arrestins may play roles in TRPV1 acute desensitization other than those associated with tachyphylactic agonist responses, as reported here.
Pharmacological inhibition of PDE4D, with the selective inhibitor rolipram leads to a significant reduction in TRPV1 pharmacological desensitization in sensory TG neurons following a repeat application of CAP (Fig. 6C). Inhibition of localized concentrations of PDE4D increases subcellular cAMP (59), enhancing PKA-mediated phosphorylation of TRPV1, leading to augmented receptor function and diminished desensitization (4, 9, 53). In support of this model, β-arrestin-2 expression abolished forskolin-mediated phosphorylation of TRPV1 (Fig. 7A). The contribution of β-arrestin-2 to the modulation of TRPV1 activity is further supported in [3H]RTX radioligand experiments, in which β-arrestin-2 co-expression reduced agonist affinity for TRPV1 in a rolipram-sensitive manner as indicated by increases in Bmax (Table 1). Analysis of β-arrestin-2-mediated desensitization revealed two PKA residues within the N terminus of TRPV1 (Ser-116 and Thr-370) that are potentially modulated by β-arrestin-2 (Fig. 7, B and C). Taken together, our findings indicate a role for β-arrestin-2-mediated scaffolding of PDE4D5 in the regulation of TRPV1 by modulating receptor phosphorylation. It is possible that β-arrestin-2 scaffolding of PDE4D5 may also play a role in the removal of calcium from TG neurons. As indicated in Fig. 6D, in which the effect of rolipram is lost in β-arrestin-2 siRNA-treated TG neurons, one could speculate that another channel or mechanism may be modified by this scaffolding mechanism.
The contribution of β-arrestin-2 to the modulation of TRPV1 activity was further supported in [3H]RTX radioligand binding experiments, in which β-arrestin-2 co-expression reduced the agonist affinity for TRPV1 in a rolipram-sensitive manner as indicated by more than a 3-fold increase in Bmax over cells expressing TRPV1 alone (Table 1). TRPV1 phosphorylation is suggested to lead to an increased probability of receptor activation in response to a specific agonist by inducing a conformational change favoring agonist binding (60). Furthermore, phosphorylation of TRPV1 has been shown to increase vanilloid binding in heterologous cells (55). As such, β-arrestin-2 scaffolding of PDE4D5 could serve to effectively control agonist affinities for TRPV1. As indicated by [32P]orthophosphate incorporation, co-expression of β-arrestin-2 abolished forskolin-mediated phosphorylation of TRPV1 (Fig. 7A). The localized regulation of cAMP bioavailability by β-arrestin-2-scaffolded PDE4D5 appears to control the phosphorylation status of TRPV1 and subsequent activity of the receptor. Scaffold proteins have been previously identified for their critical role in the sensitization of the TRPV1 receptor via their ability to localize specific kinases (5–7). Thus, our present findings demonstrate a novel scaffolding mechanism that effectively regulates the phosphorylation of TRPV1 and hence agonist binding. Consequently, aberrations in scaffolding mechanisms could lead to pathological conditions in which the phosphorylation status of TRPV1 is dysregulated.
TRPV1 receptor activity is modulated by phosphorylation at key serine/threonine residues located in both the N and C termini of the receptor (4, 9, 61). Phosphorylation of specific amino acids within TRPV1 are critical to activation, desensitization, and permeability of the receptor (35). Previously, the Ser-116 and Thr-370 residues of TRPV1 were found to play a critical role in the PKA-dependent reduction of desensitization of CAP-activated currents (9, 53). In agreement with these findings, we have identified potential sites on TRPV1 (Ser-116 and Thr-370) where the effects of phosphorylation are inhibited by overexpression of β-arrestin-2 (Fig. 7, B and C). Specifically, rolipram treatment abolished β-arrestin-2-mediated effects on cells transfected with the TRPV1 Thr-370 mutant (Fig. 7E). Therefore, β-arrestin-2 functions as a scaffolding protein by regulating TRPV1 receptor activity through specific PKA residues, providing critical insights into complex yet coordinated regulatory mechanisms of desensitization/resensitization.
The findings presented herein implicate β-arrestin-2 as a scaffolding protein that modulates TRPV1 desensitization through targeted phosphorylation events. Our results complement the myriad of molecules and proteins implicated in the strategic regulation of the TRPV1 receptor and emphasize the fundamental role of scaffolding proteins in the regulation of TRPV1. Further investigation of residues within TRPV1 that can undergo phosphorylation/dephosphorylation may provide additional insights into mechanisms underlying β-arrestin-2 recruitment and association with the receptor. Collectively, our findings reveal the significant contribution of β-arrestin-2 in controlling the phosphorylation status of TRPV1 and, consequently, receptor responses to agonist-mediated stimulation.
Acknowledgments
We thank Drs. David Julius (University of California, San Francisco), Miles Houslay (University of Glasgow), and Jeffrey Benovic (Thomas Jefferson University) for kindly providing cDNAs. We also thank Teresa Sanchez for expert technical assistance with radioligand binding experiments.
This work was supported, in whole or in part, by National Institutes of Health Grants T32 HL07446-29 (to S. M. B.), R21 NS055835 (to K. A. B.), F31 DE022517-01 (to E. D. P), and RO1 NS061884 (to N. A. J.).
- GPCR
- G-protein-coupled receptor
- ANOVA
- analysis of variance
- RTX
- resiniferatoxin
- TG
- trigeminal ganglia
- TIRF
- total internal reflection fluorescence
- CAP
- capsaicin
- CFP
- cyan fluorescent protein.
REFERENCES
- 1. Caterina M. J., Schumacher M. A., Tominaga M., Rosen T. A., Levine J. D., Julius D. (1997) The capsaicin receptor. A heat-activated ion channel in the pain pathway. Nature 389, 816–824 [DOI] [PubMed] [Google Scholar]
- 2. Tominaga M., Caterina M. J., Malmberg A. B., Rosen T. A., Gilbert H., Skinner K., Raumann B. E., Basbaum A. I., Julius D. (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 [DOI] [PubMed] [Google Scholar]
- 3. Bhave G., Hu H. J., Glauner K. S., Zhu W., Wang H., Brasier D. J., Oxford G. S., Gereau R. W., 4th (2003) Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1). Proc. Natl. Acad. Sci. U.S.A. 100, 12480–12485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bhave G., Zhu W., Wang H., Brasier D. J., Oxford G. S., Gereau R. W., 4th (2002) cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35, 721–731 [DOI] [PubMed] [Google Scholar]
- 5. Jeske N. A., Diogenes A., Ruparel N. B., Fehrenbacher J. C., Henry M., Akopian A. N., Hargreaves K. M. (2008) A-kinase anchoring protein mediates TRPV1 thermal hyperalgesia through PKA phosphorylation of TRPV1. Pain 138, 604–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeske N. A., Patwardhan A. M., Ruparel N. B., Akopian A. N., Shapiro M. S., Henry M. A. (2009) A-kinase anchoring protein 150 controls protein kinase C-mediated phosphorylation and sensitization of TRPV1. Pain 146, 301–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schnizler K., Shutov L. P., Van Kanegan M. J., Merrill M. A., Nichols B., McKnight G. S., Strack S., Hell J. W., Usachev Y. M. (2008) Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J. Neurosci. 28, 4904–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang X., Li L., McNaughton P. A. (2008) Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59, 450–461 [DOI] [PubMed] [Google Scholar]
- 9. Mohapatra D. P., Nau C. (2003) Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J. Biol. Chem. 278, 50080–50090 [DOI] [PubMed] [Google Scholar]
- 10. Docherty R. J., Yeats J. C., Bevan S., Boddeke H. W. (1996) Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 431, 828–837 [DOI] [PubMed] [Google Scholar]
- 11. Jeske N. A., Patwardhan A. M., Gamper N., Price T. J., Akopian A. N., Hargreaves K. M. (2006) Cannabinoid WIN 55,212-2 regulates TRPV1 phosphorylation in sensory neurons. J. Biol. Chem. 281, 32879–32890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patwardhan A. M., Jeske N. A., Price T. J., Gamper N., Akopian A. N., Hargreaves K. M. (2006) The cannabinoid WIN 55,212-2 inhibits transient receptor potential vanilloid 1 (TRPV1) and evokes peripheral antihyperalgesia via calcineurin. Proc. Natl. Acad. Sci. U.S.A. 103, 11393–11398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lishko P. V., Procko E., Jin X., Phelps C. B., Gaudet R. (2007) The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54, 905–918 [DOI] [PubMed] [Google Scholar]
- 14. Numazaki M., Tominaga T., Takeuchi K., Murayama N., Toyooka H., Tominaga M. (2003) Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc. Natl. Acad. Sci. U.S.A. 100, 8002–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tominaga M., Numazaki M., Iida T., Moriyama T., Togashi K., Higashi T., Murayama N., Tominaga T. (2004) Regulation mechanisms of vanilloid receptors. Novartis Found Symp. 261, 4–12 [PubMed] [Google Scholar]
- 16. Luttrell L. M., Ferguson S. S., Daaka Y., Miller W. E., Maudsley S., Della Rocca G. J., Lin F., Kawakatsu H., Owada K., Luttrell D. K., Caron M. G., Lefkowitz R. J. (1999) β-Arrestin-dependent formation of β2-adrenergic receptor-Src protein kinase complexes. Science 283, 655–661 [DOI] [PubMed] [Google Scholar]
- 17. Luttrell L. M., van Biesen T., Hawes B. E., Koch W. J., Krueger K. M., Touhara K., Lefkowitz R. J. (1997) G-protein-coupled receptors and their regulation. Activation of the MAP kinase signaling pathway by G-protein-coupled receptors. Adv. Second Messenger Phosphoprotein Res. 31, 263–277 [PubMed] [Google Scholar]
- 18. Kohout T. A., Lin F. S., Perry S. J., Conner D. A., Lefkowitz R. J. (2001) β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. U.S.A. 98, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krupnick J. G., Benovic J. L. (1998) The role of receptor kinases and arrestins in G-protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 38, 289–319 [DOI] [PubMed] [Google Scholar]
- 20. Gurevich V. V., Pals-Rylaarsdam R., Benovic J. L., Hosey M. M., Onorato J. J. (1997) Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J. Biol. Chem. 272, 28849–28852 [DOI] [PubMed] [Google Scholar]
- 21. Kohout T. A., Nicholas S. L., Perry S. J., Reinhart G., Junger S., Struthers R. S. (2004) Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 279, 23214–23222 [DOI] [PubMed] [Google Scholar]
- 22. Lohse M. J., Andexinger S., Pitcher J., Trukawinski S., Codina J., Faure J. P., Caron M. G., Lefkowitz R. J. (1992) Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of β-arrestin and arrestin in the β2-adrenergic receptor and rhodopsin systems. J. Biol. Chem. 267, 8558–8564 [PubMed] [Google Scholar]
- 23. Violin J. D., Ren X. R., Lefkowitz R. J. (2006) G-protein-coupled receptor kinase specificity for β-arrestin recruitment to the β2-adrenergic receptor revealed by fluorescence resonance energy transfer. J. Biol. Chem. 281, 20577–20588 [DOI] [PubMed] [Google Scholar]
- 24. Jala V. R., Shao W. H., Haribabu B. (2005) Phosphorylation-independent β-arrestin translocation and internalization of leukotriene B4 receptors. J. Biol. Chem. 280, 4880–4887 [DOI] [PubMed] [Google Scholar]
- 25. Key T. A., Foutz T. D., Gurevich V. V., Sklar L. A., Prossnitz E. R. (2003) N-Formyl peptide receptor phosphorylation domains differentially regulate arrestin and agonist affinity. J. Biol. Chem. 278, 4041–4047 [DOI] [PubMed] [Google Scholar]
- 26. Dasgupta P., Rastogi S., Pillai S., Ordonez-Ercan D., Morris M., Haura E., Chellappan S. (2006) Nicotine induces cell proliferation by β-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Invest. 116, 2208–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deleted in proof
- 28. Lin F. T., Daaka Y., Lefkowitz R. J. (1998) β-Arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J. Biol. Chem. 273, 31640–31643 [DOI] [PubMed] [Google Scholar]
- 29. Shukla A. K., Kim J., Ahn S., Xiao K., Shenoy S. K., Liedtke W., Lefkowitz R. J. (2010) Arresting a transient receptor potential (TRP) channel. β-Arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4. J. Biol. Chem. 285, 30115–30125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Houslay M. D., Baillie G. S. (2005) β-Arrestin-recruited phosphodiesterase-4 desensitizes the AKAP79/PKA-mediated switching of β2-adrenoceptor signalling to activation of ERK. Biochem. Soc. Trans. 33, 1333–1336 [DOI] [PubMed] [Google Scholar]
- 31. Li X., Baillie G. S., Houslay M. D. (2009) Mdm2 directs the ubiquitination of β-arrestin-sequestered cAMP phosphodiesterase-4D5. J. Biol. Chem. 284, 16170–16182 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Xiao K., McClatchy D. B., Shukla A. K., Zhao Y., Chen M., Shenoy S. K., Yates J. R., 3rd, Lefkowitz R. J. (2007) Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 12011–12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baillie G. S., Sood A., McPhee I., Gall I., Perry S. J., Lefkowitz R. J., Houslay M. D. (2003) β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. U.S.A. 100, 940–945 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Perry S. J., Baillie G. S., Kohout T. A., McPhee I., Magiera M. M., Ang K. L., Miller W. E., McLean A. J., Conti M., Houslay M. D., Lefkowitz R. J. (2002) Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science 298, 834–836 [DOI] [PubMed] [Google Scholar]
- 35. Tominaga M., Tominaga T. (2005) Structure and function of TRPV1. Pflugers Arch. 451, 143–150 [DOI] [PubMed] [Google Scholar]
- 36. Liedtke W., Choe Y., Martí-Renom M. A., Bell A. M., Denis C. S., Sali A., Hudspeth A. J., Friedman J. M., Heller S. (2000) Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 103, 525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strotmann R., Harteneck C., Nunnenmacher K., Schultz G., Plant T. D. (2000) OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2, 695–702 [DOI] [PubMed] [Google Scholar]
- 38. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2-adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 39. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 40. Jeske N. A., Patwardhan A. M., Henry M. A., Milam S. B. (2009) Fibronectin stimulates TRPV1 translocation in primary sensory neurons. J. Neurochem. 108, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ghilardi J. R., Röhrich H., Lindsay T. H., Sevcik M. A., Schwei M. J., Kubota K., Halvorson K. G., Poblete J., Chaplan S. R., Dubin A. E., Carruthers N. I., Swanson D., Kuskowski M., Flores C. M., Julius D., Mantyh P. W. (2005) Selective blockade of the capsaicin receptor TRPV1 attenuates bone cancer pain. J. Neurosci. 25, 3126–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Szallasi A., Blumberg P. M. (1990) Specific binding of resiniferatoxin, an ultrapotent capsaicin analog, by dorsal root ganglion membranes. Brain Res. 524, 106–111 [DOI] [PubMed] [Google Scholar]
- 43. Szallasi A., Blumberg P. M. (1990) Resiniferatoxin and its analogs provide novel insights into the pharmacology of the vanilloid (capsaicin) receptor. Life Sci. 47, 1399–1408 [DOI] [PubMed] [Google Scholar]
- 44. Lefkowitz R. J., Rajagopal K., Whalen E. J. (2006) New roles for β-arrestins in cell signaling. Not just for seven-transmembrane receptors. Mol. Cell 24, 643–652 [DOI] [PubMed] [Google Scholar]
- 45. Daaka Y., Luttrell L. M., Ahn S., Della Rocca G. J., Ferguson S. S., Caron M. G., Lefkowitz R. J. (1998) Essential role for G-protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J. Biol. Chem. 273, 685–688 [DOI] [PubMed] [Google Scholar]
- 46. Vögler O., Nolte B., Voss M., Schmidt M., Jakobs K. H., van Koppen C. J. (1999) Regulation of muscarinic acetylcholine receptor sequestration and function by β-arrestin. J. Biol. Chem. 274, 12333–12338 [DOI] [PubMed] [Google Scholar]
- 47. Bohn L. M., Lefkowitz R. J., Caron M. G. (2002) Differential mechanisms of morphine antinociceptive tolerance revealed in β-arrestin-2 knock-out mice. J. Neurosci. 22, 10494–10500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., Lin F. T. (1999) Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 49. Baillie G. S., Adams D. R., Bhari N., Houslay T. M., Vadrevu S., Meng D., Li X., Dunlop A., Milligan G., Bolger G. B., Klussmann E., Houslay M. D. (2007) Mapping binding sites for the PDE4D5 cAMP-specific phosphodiesterase to the N- and C-domains of β-arrestin using spot-immobilized peptide arrays. Biochem. J. 404, 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sevcik M. A., Ghilardi J. R., Peters C. M., Lindsay T. H., Halvorson K. G., Jonas B. M., Kubota K., Kuskowski M. A., Boustany L., Shelton D. L., Mantyh P. W. (2005) Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain 115, 128–141 [DOI] [PubMed] [Google Scholar]
- 51. Houslay M. D., Adams D. R. (2003) PDE4 cAMP phosphodiesterases. Modular enzymes that orchestrate signalling cross-talk, desensitization, and compartmentalization. Biochem. J. 370, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li X., Huston E., Lynch M. J., Houslay M. D., Baillie G. S. (2006) Phosphodiesterase-4 influences the PKA phosphorylation status and membrane translocation of G-protein receptor kinase 2 (GRK2) in HEK-293beta2 cells and cardiac myocytes. Biochem. J. 394, 427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mohapatra D. P., Nau C. (2005) Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J. Biol. Chem. 280, 13424–13432 [DOI] [PubMed] [Google Scholar]
- 54. Mohapatra D. P., Wang S. Y., Wang G. K., Nau C. (2003) A tyrosine residue in TM6 of the vanilloid receptor TRPV1 involved in desensitization and calcium permeability of capsaicin-activated currents. Mol. Cell. Neurosci. 23, 314–324 [DOI] [PubMed] [Google Scholar]
- 55. Jung J., Shin J. S., Lee S. Y., Hwang S. W., Koo J., Cho H., Oh U. (2004) Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J. Biol. Chem. 279, 7048–7054 [DOI] [PubMed] [Google Scholar]
- 56. Mundell S. J., Benovic J. L. (2000) Selective regulation of endogenous G protein-coupled receptors by arrestins in HEK293 cells. J. Biol. Chem. 275, 12900–12908 [DOI] [PubMed] [Google Scholar]
- 57. Kovacs J. J., Hara M. R., Davenport C. L., Kim J., Lefkowitz R. J. (2009) Arrestin development. Emerging roles for β-arrestins in developmental signaling pathways. Dev. Cell 17, 443–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. DeFea K. A. (2011) β-Arrestins as regulators of signal termination and transduction. How do they determine what to scaffold? Cell. Signal. 23, 621–629 [DOI] [PubMed] [Google Scholar]
- 59. Jin S. L., Bushnik T., Lan L., Conti M. (1998) Subcellular localization of rolipram-sensitive, cAMP-specific phosphodiesterases. Differential targeting and activation of the splicing variants derived from the PDE4D gene. J. Biol. Chem. 273, 19672–19678 [DOI] [PubMed] [Google Scholar]
- 60. Studer M., McNaughton P. A. (2010) Modulation of single-channel properties of TRPV1 by phosphorylation. J. Physiol. 588, 3743–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mandadi S., Numazaki M., Tominaga M., Bhat M. B., Armati P. J., Roufogalis B. D. (2004) Activation of protein kinase C reverses capsaicin-induced calcium-dependent desensitization of TRPV1 ion channels. Cell Calcium 35, 471–478 [DOI] [PubMed] [Google Scholar]