

Abstract
During brain development, neurogenesis is precisely regulated by the concerted action of intrinsic factors and extracellular signalling systems that provide the necessary niche information to proliferating and differentiating cells. A number of recent studies have revealed a previously unknown role for the endocannabinoid (ECB) system in the control of embryonic neuronal development and maturation. Thus, the CB1 cannabinoid receptor in concert with locally produced ECBs regulates neural progenitor (NP) proliferation, pyramidal specification and axonal navigation. In addition, subcellularly restricted ECB production acts as an axonal growth cone signal to regulate interneuron morphogenesis. These findings provide the rationale for understanding better the consequences of prenatal cannabinoid exposure, and emphasize a novel role of ECBs as neurogenic instructive cues involved in cortical development. In this review the implications of altered CB1-receptor-mediated signalling in developmental disorders and particularly in epileptogenesis are briefly discussed.
Keywords: cortical progenitor, neurogenesis, endocannabinoid signalling
1. Introduction
The developing nervous system is characterized by highly active and dynamically regulated cellular processes involving cell generation and differentiation, migration to their final destination, neuronal maturation and establishment of appropriate neuronal connectivity [1,2]. The precise regulation of these processes is achieved by a complex network of intrinsic molecular determinants and intracellular signalling pathways that are in turn modulated by surrounding information from the neurogenic niche [3]. Numerous studies have begun to delineate some of these determinants and signalling pathways involved in neural cell fate decisions, such as the regulatory switch responsible for neuronal versus glial differentiation [4] and the specification of dorsal (pallial) versus ventral (subpallial) neurons [3]. Developmental neurobiology studies and advances in stem cell research have allowed the identification of some of the molecular mechanisms involved in the specification and differentiation of specific neuronal lineages with different neurotransmitter phenotypes (e.g. glutamatergic, GABAergic, dopaminergic, etc.) [5]. However, the precise extracellular signalling pathways that modulate the acquisition of the diversity of developing neuronal populations and guarantee their appropriate integration are still only partially understood. Exposure of the developing and maturing nervous system to marijuana-derived cannabinoids exerts a significant impact on behavioural aspects, particularly regarding the control of emotions and cognitive responses. The implications of cannabinoid exposure in human neuropsychiatric disorders (see other reviews in this special issue and [6,7]) have driven the investigation on the mechanism of action and neurobiological substrate underlying developmental action of cannabinoids. Endocannabinoids (ECBs) have recently been underscored as neurodevelopmental signalling cues that, by targeting the CB1 cannabinoid receptor, exert a regulatory role on the molecular and cellular mechanisms involved in brain development. Here, we review the experimental evidence supporting the functional role of the ECB system during cortical development, as derived from genetic and pharmacological manipulation studies. The CB1 receptor has emerged as a novel signalling platform that drives neuronal generation and specification, thereby modulating brain maturation and connectivity. We also discuss the potential implications of these findings in proper neuronal activity of the adult brain.
2. The endocannabinoid system in the developing brain
The expression pattern of the ECB system elements (including receptors and enzymes of synthesis and degradation) in the developing brain has been addressed, revealing the presence of diverse ECB-metabolizing enzymes with restricted subcellular and spatio-temporal distribution. This complexity and the rapid rates of ECB synthesis/degradation reveal the existence of a dynamically regulated ECB tone during active neurogenesis. We will focus here on the expression pattern of the CB1 receptor as the most important molecular target of the ECB tone [8]. The CB1 receptor is expressed from very early stages of embryonic development, even before the appearance of the neural tube and neuroectoderm development. CB1 is present in trophoblast stem cells and its deletion results in reduced cell proliferation and differentiation that is followed by aberrant placentation and compromised embryo implantation [9]. In addition to CB1 receptor expression in the blastocyst stage, the other G-protein-coupled cannabinoid receptor, the CB2 receptor, is also present in the inner cell mass, and has been proposed to be involved in embryonic stem-derived haematopoietic cell proliferation and lineage differentiation [10]. Cannabinoid administration during chick gastrulation results in alterations of neural tube formation and patterning, thus revealing the early sensitivity of the developing nervous system to cannabinoid signalling interference [11].
In mammals, CB1 receptor expression during neural development is characterized by its abundant levels in white matter areas, with their levels progressively increasing from prenatal stages to adulthood in grey matter areas [12]. This atypical distribution of CB1 receptor expression during development occurs while active neurogenesis and axonal migration occurs and prior to synaptic maturation and neuronal activity. Therefore, neurodevelopmental CB1 receptor actions are likely to be independent of their regulatory role of neurotransmitter release and neuronal activity. The CB1 mRNA expression pattern in the developing mouse brain is summarized in figure 1. CB1 is present in the telecenphalon from E11.5 and its early expression is also observed in the developing spinal cord. During cortical development, CB1 receptors are present in pioneer neurons that populate the marginal zone of the dorsal cortex (figure 2). In particular, CB1 is present in Cajal–Retzius cells (E12.5) that are characterized by reelin expression [14,15]. Reelin is well known for its role as an instructive signalling cue that, among other actions, promotes radial migration of differentiating neurons. At embryonic day E13.5–E14.5, mouse developing cortex shows higher CB1 receptor expression in the intermediate zone and developing cortical plate (figure 2), where postmitotic neuroblasts and differentiating neurons are located, and expresses early neuronal markers such as class III β-tubulin [13,14]. At these stages, the CB1 receptor is also present in the subpial area of the ganglionic eminences and the primordium of the hippocampus [15]. Later, CB1 receptors are heterogeneously distributed through cortical layers and the hippocampus, in both excitatory glutamatergic projection neurons, as identified by vGlut1 expression, and cholecystokinin (CCK)-expressing GABAergic interneurons colabelled with vGlut3 [16–18]. CB1+CCK+ interneurons derived from the ganglionic eminences follow tangential migratory routes from the ventral telencephalon and reach the developing cortex, hippocampus and amygdala [15,19–21]. The regulatory role of the ECB system in development of excitatory and inhibitory neuronal lineages is also conserved in the adult brain, in which CB1 receptors are functional in cortical excitatory projecting neurons and inhibitory GABAergic interneurons [8,22].
Figure 1.

Expression pattern of the CB1 cannabinoid receptor mRNA at different developmental stages. CB1 mRNA in situ hybridization in the developing mouse nervous system is shown at the indicated stages. BG, basal ganglia; BS, brainstem; Cx, cortex; Hpc, hippocampus; Hth, hypothalamus; Pa, pallium; sPa, subpallium; SC, spinal cord. Published with permission of Allen Developing Mouse Brain Atlas, Seattle (WA), Allen Institute for Brain Science. Copyright ©2009. Available at: http://developingmouse.brain-map.org.
Figure 2.

CB1 cannabinoid receptor expression during cortical development. The CB1 receptor is present in the developing cortex, showing increasing expression levels from undifferentiated to differentiated projection neurons (PNs). The CB1 receptor is present in Cajal–Retzius cells of the marginal zone (MZ) and apical and basal progenitors in the ventricular and subventricular (VZ/SVZ) proliferative area. Representative immunofluorescence images showing the colocalization of the CB1 receptor in radial glial (RG) progenitors and intermediate amplifying progenitor cells (IPCs) as identified with Sox2 and Tbr2 antibodies, respectively [13] (copyright National Academy of Sciences, USA 2009). Higher expression levels of the CB1 receptor are evident in maturing neurons that have reached the CP, that correspond to locally generated PNs. CB1 receptor is present in certain interneuron (IN) populations that reach the pallium upon tangential migration from the ganglionic eminences. Image background corresponds to a representative in situ hybridization of the CNR1 mRNA at E.16.5 (by C. Hoffman and B. Lutz, Johannes Gutenberg University Mainz, Germany).
The expression and functionality of the ECB system has also been characterized in human brain development [23,24]. In human foetal brain, in situ hybridization and binding assays evidence a heterogeneous pattern of CB1 receptor expression with preferential limbic expression and high levels throughout the cerebral cortex, hippocampus, caudate nucleus, putamen and cerebellum. CB1 receptors are present at gestational week 9 in the subventricular zone (SVZ) and Cajal–Retzius cells of the marginal zone [25]. In the second trimester of gestation, intense labelling for CB1 receptors is evident in the hippocampal CA region [24]. High densities of CB1 receptors are detected during prenatal development in fibre-enriched areas that later in the adult brain are practically devoid of these receptors [23]. Overall, the early expression and functionality of the CB1 receptor during nervous system development and its transient and atypical localization in prenatal stages suggest a specific role of the ECB system in human brain development, with potential implications in neuropsychiatric disorders [6,26].
3. The CB1 cannabinoid receptor in neural stem/progenitor cells
Neural stem and progenitor cells of different embryonic brain areas express a functional ECB system. ECBs are actively produced in the neurogenic niche of the developing cortex and engage CB1 receptors on NPs of the ventricular zone (VZ; figure 2), as identified by the expression of the neuroepithelial marker nestin and the transcription factor Sox2 [13,27]. Intermediate progenitor cells of the SVZ, characterized by the expression of the transcription factor Eomes/Tbr2, that contribute to the generation of pyramidal cells in all layers of the cerebral cortex [28], are also targeted by CB1 receptors (Díaz-Alonso et al. 2012, unpublished results). CB1 receptors are present in dividing cells identified by 5-bromo-2′-deoxyuridine labelling, the expression of endogenous cell cycle markers (Ki-67, phosphorylated-histone 3) and the phosphorylation of vimentin (a marker of radial progenitor cell division) [13,29,30]. These observations indicate that the CB1 receptor present in both apical radial progenitors and basal intermediate progenitor cells, albeit at low expression levels when compared to differentiated neurons, exerts a regulatory role in progenitor cell fate. Whereas in the developing chick embryonic CB1 receptor expression follows neuronal differentiation and, at least in the spinal cord, might be restricted to postmitotic neurons [31,32], its expression pattern in the nervous system of the zebrafish is suggestive of its involvement in neurogenesis [33].
Neurospheres (non-adherent in vitro culture of NP cells) from embryonic and postnatal development stages express CB1 receptors and the anandamide (AEA)-degrading enzyme fatty acid amide hydrolase (FAAH), and elevations in their intracellular Ca2+ concentration increase ECB production [29]. In addition, the CB2 receptor and diacylglycerol lipase (DAGL), the enzyme responsible for 2AG generation, are also functional in NP cultures [34,35]. AEA and 2-arachidonoylglycerol (2AG) can act therefore in an autocrine or paracrine manner on NPs or surrounding neighbour cells. DAGL expressed at embryonic stages is preferentially located in axon growth cones and is later redistributed to dendrites where it controls the 2AG retrograde neuromodulatory signalling role [36]. During corticogenesis, as well as in the developing retina, ECB production by N-acyl phosphatidylethanolamine–phospholipase D (NAPE-PLD; one of the enzymes responsible for AEA generation) and DAGL participate in axon guidance [37,38]. In vitro studies in neuroblastoma cells confirmed the positive action of 2AG production in neurite outgrowth and the existence of different mechanisms of action according to the metabolic origin of 2AG [39]. Unfortunately, the precise contribution of the two DAGL enzyme isoforms (α and β) in neural development remains to be clarified (see accompanying paper by Doherty et al.). In the adult, SVZ DAGLα is present in ependymal cells that are intimately related to neural stem cells, and mediates 2AG generation involved in the regulation of neurogenesis [40]. The analysis and characterization of the DAGL locus identified the minimal core promoter sequence and the involvement of the transcriptional regulator specificity protein Sp1 in DAGLα expression. High expression levels of DAGLα in the NSC line Cor-1 rapidly decrease through their differentiation into GABAergic neuronal cells [41], whereas in neuroblastoma cells retinoic acid-induced neuronal-like differentiation increases first DAGLα expression and later DAGLβ [39]. In the developing forebrain, monoacylglycerol lipase (MGL) expression is preferentially observed in the thalamus, thus restricting local 2AG levels and relieving non-permissive axonal growth of corticothalamic projections [42].
Although not discussed here in detail, the regulatory role of the CB1 receptor in neuronal generation and maturation in the embryonic brain is preserved in the neurogenic niches of the adult brain. NPs in adult neurogenic brain areas also express the CB1 receptor and produce ECB ligands [29,30,43]. CB1 receptors are expressed in NP cells of the subgranular zone (SGZ) and SVZ, in which they drive progenitor proliferation and tune neural differentiation. These findings indicate that the role of ECBs as developmental signalling cues is conserved in the mature nervous system [44].
(a). The CB1 cannabinoid receptor drives neural progenitor cell proliferation
CB1 receptor activity in NPs regulates cell proliferation and survival. In vitro, the use of neurosphere cultures of embryonic cortical NPs derived from knockout mice has shown that inactivation of the CB1 receptor, as well as of the CB2 receptor, reduces cell proliferation and impairs self-renewal [29,34]. Accordingly, pharmacological regulation with selective CB1 and CB2 receptor agonists or antagonists exerts a positive or negative action, respectively, on NP cell division [29,30,34,40,45]. In vivo, CB1 receptor loss of function induces alterations of cortical and hippocampal development [20,29] and, whereas CB1-null mice have reduced cortical progenitor proliferation, in FAAH-deficient mice the opposite is observed [13,29]. Abnormal cortical development in CB1-deficient mice is characterized by defective SVZ/VZ pyramidal progenitor proliferation and radial migration, deficits in axonal navigation and aberrant corticofugal projections [13]. The role of the ECB system in the regulation of pyramidal NP cell expansion during cortical development is also recapitulated in brain slices, in which pharmacological regulation of CB1 receptors or genetic manipulation of the ECB tone disrupts proper pyramidal neuron generation [13]. NP proliferation from other brain areas such as the cerebellum is also dependent on CB1 receptor activation [45].
4. Cannabinoid signalling in neural progenitor/stem cells
CB1 receptor signalling in neural cells has been extensively studied, but the existence of selective CB1 receptor-mediated signalling mechanisms in progenitor cells remains to be investigated in detail. CB1 receptor-evoked signal transduction pathways can be divided into two large categories: canonical signalling via the classical repertoire of heterotrimeric Gi protein partners, and crosstalk with other membrane receptor-dependent signalling events (in particular those elicited by neurotrophin/growth factor receptors). Current understanding of the signal transduction mechanisms regulated by CB1 receptors in NPs is summarized in figure 3.
Figure 3.
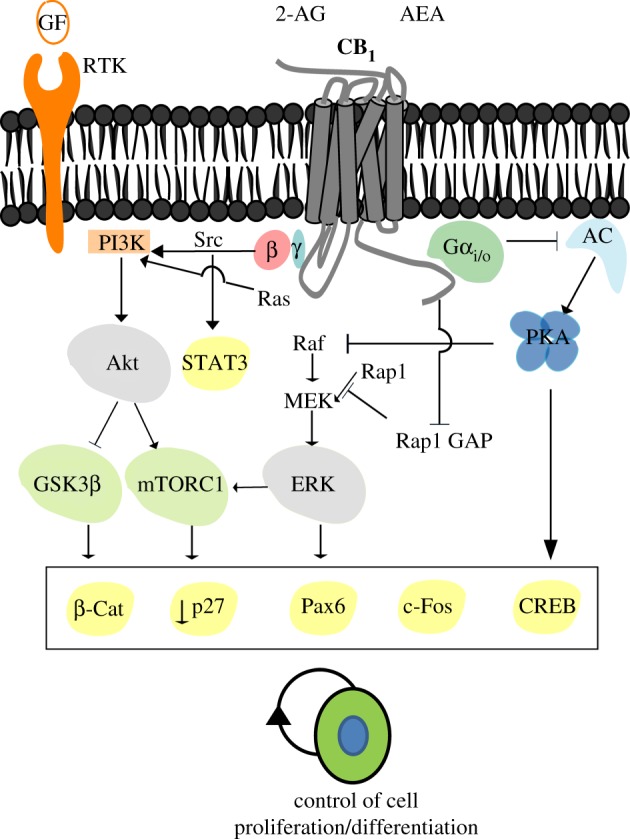
CB1 cannabinoid receptor signalling and regulation of neural stem/progenitor cell proliferation. CB1 receptors are coupled to Gi proteins, thereby mediating the inhibition of adenylyl cyclase (AC) and protein kinase A (PKA). CB1 receptor coupling to Gi signalling is also associated with activation of the extracellular signal-regulated kinase (ERK) pathway via different mechanisms (see text for details). Direct activation of the PI3K/Akt and ERK pathways by CB1 receptors may converge, thus synergizing with their activation by other receptors such as growth factor receptors with tyrosine kinase activity (RTK). CB1 receptor-induced activation of RTKs can occur by promoting the processing of membrane-bound growth factor inactive precursors to yield active growth factors, or by activating intracellular Src family protein kinases. In some circumstances, CB1 activity can antagonize RTK-mediated ERK signalling (see [46,47] for further details). Activation of the CB1 receptor ultimately controls different transcriptional regulators, including CREB, STAT-3, PAX-6 and β-catenin. The CB1 receptor may also regulate mammalian target of rapamycin complex 1 (mTORC1) in NPs as it occurs in differentiated neurons.
(a). CB1 cannabinoid receptor signalling mechanism and cell proliferation
The CB1 receptor-mediated proliferative and pro-survival actions have been attributed, at least in part, to the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt axis and extracellular signal-regulated protein kinase (ERK; figure 3) [48]. The CB1 receptor, via canonical Gi-mediated inhibition of adenylyl cyclase, decreases cAMP concentration, and this in turn plays a prominent role by de-inhibiting the ERK pathway by protein kinase A [49,50]. In addition, G protein βγ subunits liberated upon CB1 receptor activation stimulate the ERK pathway in a PI3K-dependent manner [51]. Therefore, both regulation of cAMP levels and PI3K signalling contribute to CB1-mediated ERK activation. However, the mechanisms of CB1 receptor-mediated ERK activation are multiple and interconnected, thus providing a rather complex scenario. It is likely that, at different time points, upon CB1 receptor activation ERK activation may occur by different mechanisms [52]. According to this model, early ERK activation would be strongly dependent on cAMP levels, activation of members of the cytosolic tyrosine kinase Src family and transactivation of tyrosine kinase receptors. In cerebellar granular progenitor cells, CB1 receptor coupling to the PI3K/Akt pathway is followed by the activation of the glycogen synthase kinase-3β/β-catenin pathway [45]. CB1 receptor activation therefore increases β-catenin nuclear localization and the activation of lymphoid enhancer factor/T-cell factor transcription factors induces proliferation, thereby modulating cell cycle regulatory genes such as cyclin D1.
CB1 signalling in neural cells may also involve the activation of mammalian target of rapamycin complex 1 (mTORC1), a serine/threonine protein kinase that regulates cell growth, proliferation and survival [53]. CB1 receptor stimulation in hippocampal GABAergic neurons activates mTORC1 and downstream p70S6K in pyramidal neurons that, by controlling protein synthesis, is responsible for some amnesic effects of Δ9-tetrahydrocannabinol administration [54,55]. Therefore, CB1 receptor-induced mTORC1 and protein synthesis regulation can explain some long-term cannabinoid actions on neuronal plasticity and cognition. The role of CB1 receptors in mTORC1 signalling during brain development remains unknown, although CB2 receptors have recently been shown to be coupled to mTORC1 activation in NP cells both in the developing cortex and in the SGZ of the adult hippocampus [56]. At postnatal stages, mTORC1 signalling is known to be involved in oligodendrocyte differentiation and myelination [53,57], and the ECB system drives oligodendroglial differentiation and cell survival at least partially via mTORC1 regulation [58]. In contrast to neurons and progenitor cells, in which mTORC1 is activated by cannabinoid receptors [54], in transformed glioma cells cannabinoids, via tribbles homologue 3, inhibit the Akt/mTORC1 axis and can switch on an autophagy programme that results in cell death by apoptosis [59,60].
How this diversity of intracellular CB1 receptor signalling mechanisms in neural cells is regulated remains poorly understood. CB1 receptors may form homo- or heterodimers with other G-protein-coupled receptors [61,62] and this may shift intracellular signalling coupling. Importantly, although CB2 receptors share some of the CB1 receptor signalling effectors (inhibition of cAMP production, ERK and PI3K/Akt activation), their opposite pattern of expression during NP cell differentiation (i.e. NPs are CB1lowCB2+, while differentiated neurons are CB1+CB2neg) may lead to different ratios of homo- and heterodimers that can promote alternative cell fate decisions according to the major signalling pathway engaged.
(b). CB1 cannabinoid receptor crosstalk with other extracellular signalling pathways
CB1 receptors have been shown to crosstalk with growth factor and neurotrophin signalling events at different levels (figure 3). CB1 receptor activation is associated to changes in growth factor expression, and can regulate tyrosine kinase growth factor receptors by direct transactivation mechanisms. In the adult nervous system, CB1 receptor expression is involved in the regulation of the levels of the neurotrophin brain-derived neurotrophic factor (BDNF), and thus CB1-deficient mice have reduced hippocampal BDNF levels under basal circumstances, which could explain some of the neuronal plasticity and emotional alterations shown in those animals [63–65]. Transactivation of growth factor receptors with tyrosine kinase activity (EGFR, Trk B and others) has been shown to be involved in some CB1 receptor-mediated neurodevelopmental actions [52]. CB1 receptor-induced transactivation can be mediated by growth factor or cytokine (e.g. TNFα) expression or their processing and shedding from inactive membrane-bound precursors [66,67]. Moreover, transactivation can occur via cytosolic tyrosine kinases of the Src family and this mechanism may influence interneuron migration [20]. Growth factor levels are also regulated by cannabinoid signalling under different neurodegenerative paradigms, such as hippocampal and striatal excitotoxicity, in which BDNF, fibroblast growth factor 2 (FGF2) and epidermal growth factor (EGF) are tuned by CB1 receptors [68–70]. Reciprocally, FGF receptors promote axonal growth and guidance via DAGL activation and 2AG generation [71].
CB1 receptor activation can also lead to the regulation of small G proteins and subsequent control of cytoskeleton and microtubule dynamics, which may be responsible for cannabinoid actions on neuritogenesis and synaptogenesis. Activation of CB1 receptors can induce either neurite outgrowth or retraction [72–76]. CB1 receptors are enriched in the axonal growth cones of GABAergic interneurons at late gestation and, when activated, they induce a chemorepulsive collapse of axonal growth cones by activating RhoA [37,73]. CB1 receptor-induced neurite outgrowth in neuroblastoma Neuro2A cells occurs via Rap1, Src and the signal transducer and activator of transcription 3 (STAT 3) [74,76]. CB1 receptor activation and IL6 receptor signalling exert a synergistic effect in cAMP–responsive element binding protein (CREB) and STAT3 activation that enforces neurite outgrowth [77]. In the retina, the CB1 receptor induces growth cone collapse in a mechanism involving the intracellular trafficking of the deleted in colorectal cancer receptor [38]. Nerve growth factor-induced neurite outgrowth of PC12 cells is inhibited by CB1 receptor modulation of Trk A/Rap1/B-Raf-mediated sustained ERK activation [72]. The recent demonstration that recruitment of the Gi-interacting protein GRIN (G-protein-regulated inducer of neurite outgrowth) upon CB1 receptor activation can determine the signalling output of FGF stimulation, by allowing Sprouty-mediated inhibition of ERK signalling [46], may reconcile the apparent conflicting results of CB1 receptors mediating a positive or inhibitory action in neurite outgrowth and ERK activation. In summary, further investigation on the role of recently described CB1 receptor interacting proteins (i.e. CRIP1, AP3 and others) will shed light on cannabinoid signalling mechanisms [78] and may clarify the different neurodevelopmental actions of CB1 receptor activity. Importantly, the different kinetics and intensity of signal transduction pathways engaged by the CB1 receptor in a particular cellular context can induce different NP cell fate decisions, for example from proliferation and self-renewal (acute ERK activation) to neural differentiation (sustained ERK activation).
5. The CB1 cannabinoid receptor and neural differentiation
The diversity of neurodevelopmental actions of the CB1 receptor is suggestive of a regulatory role of the ECB system in neural cell differentiation and morphogenesis. CB1 receptor activity has been associated to the regulation of different neural cell types' development, including neurons and glial cells. Genetic elimination of the CB1 receptor at embryonic stages induces alterations of long-range subcortical axonal projections, but the particular mechanisms responsible for this deficit in CB1 knockout cells are as yet unknown and may include: (i) defective VZ/SVZ pyramidal progenitor cell proliferation; (ii) impairment of radial migration; (iii) neuronal differentiation alterations; and (iv) axonal pathfinding disturbance. Inhibition of 2AG synthesis reduced vGlut1 expression and altered the expression of the glutamatergic synapse markers SNAP25 and synaptophysin [13]. However, this finding alone does not prove a regulatory role of CB1 receptors in neuronal differentiation. CB1 receptor expression increases with neuronal cell differentiation and thus increased or reduced CB1 expression are likely to occur in parallel with changes in the expression of other neuronal markers. Although at embryonic stages CB1 receptor ablation results in reduced neurogenesis [29,30], at postnatal stages manipulation of the ECB system interferes with astrocyte and oligodendrocyte development [27,79,80]. In these studies, altered neural cell populations upon CB1 signalling manipulation are observed concomitantly with reduced progenitor cell proliferation. These observations raise the question of whether the CB1 receptor tunes lineage selection of undifferentiated cells or acts by merely expanding specific NP populations.
(a). CB1 cannabinoid receptor-mediated regulation of gene expression
CB1 receptor activation can regulate more than 20 transcription factors that are part of the gene expression signatures involved in NP maintenance, neuronal commitment and maturation [81]. CB1 receptor signalling converges onto the activation of STAT3, a transcription factor responsible for gene expression regulation that is involved in cannabinoid-induced neurite outgrowth and ERK activation [76]. In neuroblastoma cells, CB1 receptor-induced STAT3 activation relies on PI3K-dependent activation of the transcription factor Pax6 [81], a paired box family member essential for the generation of glutamatergic neurons and cortical neurogenesis [82]. In addition, CB1 receptor prevents the inhibitory effect of breast cancer resistance associated on neuritogenesis [81]. During cortical development and pyramidal neurogenesis, CB1 receptors are also able to modulate Pax6 and Tbr2 transcriptional activity in VZ/SVZ progenitors (Díaz-Alonso et al. 2012, unpublished results). Noteworthy, chronic administration of a Δ9-tetrahydrocannabinol analogue severely disrupted chick neural development, and this was associated to gene expression changes of critical neurogenic transcription factors, including Krox20, Otx2, Pax6 and Sox2 [11]. Unfortunately, the involvement of the CB1 receptor in these actions was not investigated. CB1 receptor activity in differentiating cortical neurons is coupled by as yet unknown mechanisms [83] to the modulation of the neurogenic transcription factor code Ctip2-Satb2 (figure 4) [84]. CB1 receptors are positively coupled to COUP-TF II interacting protein 2 (Ctip2) and negatively to Satb2-mediated repression of Ctip2. Thus, CB1 receptor activity tunes the transcriptional neurogenic programme responsible for upper and lower cortical neuron differentiation, and CB1 receptor inactivation results in reduced Ctip2+ corticospinal projection neuron development that affects in turn motor function in adulthood [83].
Figure 4.

CB1 cannabinoid receptor signalling and neuronal differentiation. CB1 receptor activity in differentiating cortical neurons is coupled by as yet unknown mechanisms to the modulation of the neurogenic transcription factor code Ctip2-Satb2. CB1 receptors are positively coupled to COUP-TF II interacting protein 2 (Ctip2) and negatively to Satb2-mediated repression of Ctip2. Thus, CB1 receptor activity tunes the transcriptional neurogenic programme responsible for upper and lower cortical neuron differentiation. Transcription factors involved in cortical laminar specification regulated by CB1 receptor are indicated in bold letters.
The involvement of the CB1 receptor in embryonic neuronal development [85], but also in postnatal astrogliogenesis [27] and oligodendrocyte survival and myelination [80,86], suggests that CB1 receptor signalling could also target still unknown pro-gliogenic transcription factors [3]. ECB signalling may be involved in tumour-initiating stem cell decisions of proliferation versus cell cycle exit and differentiation [87], and CB1 receptor regulation of STAT3 is a likely candidate to mediate CBI regulation of astrogliogenesis [88]. In summary, the CB1 receptor exerts a dual role, pro-neurogenic in some cases and pro-gliogenic in others, thus indicating that differences in the intrinsic progenitor features and/or in the surrounding niche may be responsible for alternative CB1 receptor-driven neurogenic outcomes.
6. Pathophysiological implications of the neurodevelopmental role of CB1 cannabinoid receptors
The neurodevelopmental role of the ECB system reveals that altered cannabinoid signalling, due to either hyper- or hypo-function of the CB1 receptor, can exert long-lasting consequences in adult brain neuronal function by modifying the actively developing brain. Neurodevelopmental disorders can originate by subtle or severe alterations of various neurogenic processes, including neuronal generation, migration, maturation and connectivity that are responsible for adult brain dysfunction [89]. Among developmental disorders, cortical alterations constitute an important example of how embryonic deficits affect adult neurological function. As previously discussed, CB1 receptor signalling plays a regulatory role in different neural cell fate processes involved in these pathologies. Genetic polymorphisms of cannabinoid receptors can induce subtle changes during development by influencing signalling strength or duration and later, when synaptic transmission ensues, by influencing the appropriate balance of neuronal activity. Likewise, mutations of ECB-metabolizing enzymes, including degrading (FAAH, ABHD6/12, MGL) or synthesizing enzymes (NAPE-PLD, DAGL), may result in less active enzymes that would increase or reduce ECB tone and signalling. In this regard, FAAH polymorphisms have been associated with drug abuse behaviours [90,91]. A recent proof of concept of this notion is the involvement of ABDH12 mutations that associate with the neurodegenerative disease polyneuropathy, hearing loss, ataxia, retinitis pigmentosa and cataract (PHARC) that occurs with concomitant demyelination and cerebellar ataxia [92].
CB1 receptor signalling can be influenced as well by prenatal exposure to marijuana-derived cannabinoids or by contact with drugs targeting either directly or indirectly the ECB system. The neurobiological consequences of plant-derived cannabinoid intake on pre- and postnatal stages have been recently reviewed from the perspective of animal models and humans [6,93], and indicate that the brain burst period is of especial susceptibility. According to the developmental stage in which CB1 receptor signalling is functional, its interference may affect different neural cell populations, including neuronal generation and specification (embryonic stages) [13,37], glial development (postnatal stages) [27,80] and neuronal maturation and connectivity [13,32,94]. Blockade of the CB1 receptor when the neurogenic wave responsible for deep cortical neuronal generation is active affects corticospinal neuronal specification, thereby tuning subcerebral- versus callosal neuron-projections and thus skilled motor function in adulthood [83]. In addition, CB1 receptor expression, first in white matter and later in postnatal grey matter, participates in whisker barrel map development of the somatosensory cortex, supporting the contribution of the CB1 receptor for the appropriate integration of sensory information input [95]. In summary, the regulatory role of the CB1 receptor in cortical development processes has the potential to exert significant impact on adult brain function [96,97].
Developmental interference of cannabinoid signalling can influence human emotion-, threat- and reward-related brain function at different levels [6,26]. Polymorphisms of the CNR1 gene, which encodes the CB1 receptor, may reduce or enhance G-protein-mediated signalling and have been associated to major depression, psychoses and schizophrenia [98,99]. Unexpectedly, polymorphisms of the CB2 receptor-encoding gene, CNR2, may associate with depressive syndromes and schizophrenia [100]. Changes in the appropriate number, specification or migration of projection neurons and interneurons will result in modifications of neuronal activity that in turn will be followed by a more generalized neurochemical unbalance. The glutamatergic neuronal dysfunction hypothesis of schizophrenia [101] suggests that malfunction of the CB1 receptors in pyramidal neurogenesis may contribute to the pathogenesis of psychoses or schizophrenia symptoms. Malfunction of the ECB system may be one of the causes underlying neuronal dysfunction, but alternatively the CB1 receptor and ECB-metabolizing enzymes are also likely to adapt to aberrant neuronal homeostasis as an attempt to counteract the changes of neuronal transmission [102]. Thus, cortical glutamic acid decarboxylase 67 deficiency, a typical neurochemical marker of schizophrenia, results in lower CB1 receptor expression. It remains unknown whether these kind of ECB system adaptations exert positive effects to cope with those alterations, or worsen the pathological processes.
(a). Neurodevelopmental disorders: epileptogenesis
One of the most common consequences of cortical development alterations is the appearance of epileptic foci due to alterations in neuronal excitability [89,103]. Considering the dual role of the CB1 receptor in the generation and maturation of excitatory and inhibitory neurons it can be predicted that CB1 receptor-dependent signalling alterations during development would impact the appropriate excitation/inhibition balance of the mature brain. Ablation of the CB1 receptor interferes with cortical progenitor proliferation [29], the correct specification of upper/lower cortical neurons [83] and axonal growth and fasciculation [13,32]. Thus, unbalanced CB1 receptor activity and its consequences in cortical pyramidal neurogenesis may elicit epileptic syndromes similar to those associated with cortical dysplasia, tuberous sclerosis or heterotopias [89]. Deletion of doublecortin, a microtubule-associated protein characteristic of migrating neuroblasts that is responsible for lissencephaly, interferes with excitatory neuron radial migration [104], induces lamination alterations and has a profound impact on neuronal excitability [105]. These findings suggest that exacerbated excitotoxicity in CB1-deficient mice [68] and the involvement of the ECB system in seizure threshold and epilepsy [106,107] may, at least in part, be due to developmental cortical alterations that result in unbalanced excitation/inhibition activity.
In addition to excitatory neuronal alterations, unbalanced generation of interneuron populations contribute to developmental epilepsies [103]. As the ECB system is involved in the development and morphogenesis of inhibitory neurons [15,20], it is likely that these developmental alterations may be responsible for changes in the susceptibility to epileptogenesis. Disruption of cortical interneuron development is known to exert GABAergic cell type-specific deficits, epilepsy and behavioural dysfunction [108,109]. Thus, the decrease in the number of interneurons and disruption of appropriate inhibitory synapse development observed in Dlx1-deficient mice, a homeodomain transcription factor essential during embryonic development for the production of forebrain GABAergic interneurons, is associated with a reduction of GABA-mediated inhibitory postsynaptic currents, electrographic seizures and cortical dysrhythmia in vivo [109]. Ablation of neurogenic transcription factors during development interferes with cortical excitation/inhibition balance and, for example, COUP-TFI knockout mice display altered balance of the development of medial versus caudal ganglionic eminence interneurons [110]. Whether the CB1 receptor plays a role in the differentiation and development of the different interneuron populations is still unknown. However, defective CB1 receptor function in CCK+vGlut3+ basket neuron development would conceivably affect the excitation/inhibition balance by interfering with interneuron-mediated inhibition. In agreement with this notion, experimental models of epilepsy result in predominant loss of CCK+CB1+ basket interneurons [111], and indiscriminate loss of local-circuit hippocampal interneurons triggers network hyperexcitability, loss of CA1 pyramidal cells and hippocampal epileptiform seizures [112]. Chronic cannabinoid administration induces alterations of CCK+ interneuron density in the hippocampus and cortex [15,20] that are likely to interfere with the balance of inhibition/excitation and thus may result in the development of epileptogenic foci.
Once neuronal activity is established, the absence or interference with CB1-mediated neuromodulation would constitute a major mechanism for unbalanced neuronal activity through the disruption of excitatory and inhibitory activity [8,22]. CB1 receptor engagement by retrograde ECB messengers is a key regulator of synaptic plasticity, both of inhibitory synapses (depolarization-induced suppression of inhibition and long-term depression of inhibitory transmission) and excitatory synapses (depolarization-induced suppression of excitation and long-term depression of excitatory transmission) [8,22,113]. Thus, CB1 receptor blockade induces epileptic discharges that have been attributed to the absence of depolarization-induced suppression of GABA postsynaptic currents [114]. CB1 receptors are involved in limbic hyperexcitability and fever-induced seizures through the potentiation of depolarization-induced suppression of inhibition in CCK+ interneurons [105,115]. In addition, CB1 receptors expressed solely in excitatory hippocampal vGlut1 neurons can allow protection from kainic acid-induced seizures [18,68]. It is important to note that, as within the early stages of brain development GABA is excitatory instead of inhibitory, CB1 receptor activation and subsequent inhibition of GABA release would result in different outcomes depending on the developmental stage in which the ECB system function is altered.
7. Conclusions
Developmental neurobiology studies have started to elucidate the contribution of CB1 receptor signalling in appropriate nervous system formation. These studies have underscored the active role of ECBs as local cues of neurogenic niches that, via the CB1 receptor, drive progenitor cell proliferation/cell cycle progression, control neuronal migration and tune neuronal differentiation/specification. At early developmental stages, the CB1 receptor and a precisely regulated ECB tone act as signalling cues in neurogenic niches [44,84]. CB1 receptor activity exerts a critical regulatory role in different neural cell fate decisions, i.e. (i) cell cycle progression and proliferation; (ii) neural cell specification; and (iii) migration and morphogenesis. Dysfunction of the ECB system may be a determinant of seizure onset and epileptogenesis as a consequence of unbalanced excitatory and inhibitory neurotransmission [116,117]. At postnatal stages, acute or long-lasting CB1 receptor-mediated neuromodulation upon cannabinoid exposure or altered ECB signalling interferes with neuronal maturation and tunes neuronal connectivity and developing circuits, which may in turn exert relevant consequences on adult neuronal function [6]. In summary, the CB1 receptor exerts a key regulatory role in cortical developmental and this may have significant consequences in adult brain function, including the tuning of an appropriate balance of neuronal excitation/inhibition activity and the susceptibility to suffer neuropsychiatric disorders.
Acknowledgements
Research in our laboratory is financially supported by Ministerio de Ciencia e Innovación (PLE2009-0117 to I.G.-R. and SAF2009-08403 to M.G.), Comunidad de Madrid-Universidad Complutense de Madrid (S2011/BMD-2308, S2011/BMD-2336 and 950344 to M.G. and I.G.-R.). J.D.-A. is supported by Fondo de Investigaciones Sanitarias. We are grateful to our collaborators and previous members of our lab that significantly contributed to part of the work summarized here.
References
- 1.Sur M., Rubenstein J. L. 2005. Patterning and plasticity of the cerebral cortex. Science 310, 805–810 10.1126/science.1112070 (doi:10.1126/science.1112070) [DOI] [PubMed] [Google Scholar]
- 2.O'Leary D. D., Chou S. J., Sahara S. 2007. Area patterning of the mammalian cortex. Neuron 56, 252–269 10.1016/j.neuron.2007.10.010 (doi:10.1016/j.neuron.2007.10.010) [DOI] [PubMed] [Google Scholar]
- 3.Guillemot F., Molnar Z., Tarabykin V., Stoykova A. 2006. Molecular mechanisms of cortical differentiation. Eur. J. Neurosci. 23, 857–868 10.1111/j.1460-9568.2006.04626.x (doi:10.1111/j.1460-9568.2006.04626.x) [DOI] [PubMed] [Google Scholar]
- 4.Miller F. D., Gauthier A. S. 2007. Timing is everything: making neurons versus glia in the developing cortex. Neuron 54, 357–369 10.1016/j.neuron.2007.04.019 (doi:10.1016/j.neuron.2007.04.019) [DOI] [PubMed] [Google Scholar]
- 5.Eiraku M., Sasai Y. In press Self-formation of layered neural structures in three-dimensional culture of ES cells. Curr. Opin. Neurobiol. [DOI] [PubMed] [Google Scholar]
- 6.Jutras-Aswad D., DiNieri J. A., Harkany T., Hurd Y. L. 2009. Neurobiological consequences of maternal cannabis on human fetal development and its neuropsychiatric outcome. Eur. Arch. Psychiat. Clin. Neurosci. 259, 395–412 10.1007/s00406-009-0027-z (doi:10.1007/s00406-009-0027-z) [DOI] [PubMed] [Google Scholar]
- 7.D'Souza D. C., Sewell R. A., Ranganathan M. 2009. Cannabis and psychosis/schizophrenia: human studies. Eur. Arch. Psychiat. Clin. Neurosci. 259, 413–431 10.1007/s00406-009-0024-2 (doi:10.1007/s00406-009-0024-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heifets B. D., Castillo P. E. 2009. Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306 10.1146/annurev.physiol.010908.163149 (doi:10.1146/annurev.physiol.010908.163149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun X., Dey S. K. 2008. Aspects of endocannabinoid signaling in periimplantation biology. Mol. Cell Endocrinol. 286(Suppl. 1), S3–S11 10.1016/j.mce.2008.01.002 (doi:10.1016/j.mce.2008.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang S., et al. 2007. Expression and function of cannabinoid receptors CB1 and CB2 and their cognate cannabinoid ligands in murine embryonic stem cells. PLoS ONE 2, e641. 10.1371/journal.pone.0000641 (doi:10.1371/journal.pone.0000641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Psychoyos D., Hungund B., Cooper T., Finnell R. H. 2008. A cannabinoid analogue of Δ9-tetrahydrocannabinol disrupts neural development in chick. Birth Defects Res. B Dev. Reprod. Toxicol. 83, 477–488 10.1002/bdrb.20166 (doi:10.1002/bdrb.20166) [DOI] [PubMed] [Google Scholar]
- 12.Berrendero F., GarciaGil L., Hernandez M. L., Romero J., Cebeira M., de Miguel R., Ramos J. A., Fernández-Ruiz J. J. 1998. Localization of mRNA expression and activation of signal transduction mechanisms for cannabinoid receptor in rat brain during fetal development. Development 125, 3179–3188 [DOI] [PubMed] [Google Scholar]
- 13.Mulder J., et al. 2008. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl Acad. Sci. USA 105, 8760–8765 10.1073/pnas.0803545105 (doi:10.1073/pnas.0803545105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitalis T., Laine J., Simon A., Roland A., Leterrier C., Lenkei Z. 2008. The type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. Eur. J. Neurosci. 28, 1705–1718 10.1111/j.1460-9568.2008.06484.x (doi:10.1111/j.1460-9568.2008.06484.x) [DOI] [PubMed] [Google Scholar]
- 15.Morozov Y. M., Torii M., Rakic P. 2009. Origin, early commitment, migratory routes, and destination of cannabinoid type 1 receptor-containing interneurons. Cereb. Cortex 19(Suppl. 1), i78–i89 10.1093/cercor/bhp028 (doi:10.1093/cercor/bhp028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katona I., Urban G. M., Wallace M., Ledent C., Jung K. M., Piomelli D., Mackie K., Freund T. F. 2006. Molecular composition of the endocannabinoid system at glutamatergic synapses. J. Neurosci. 26, 5628–5637 10.1523/JNEUROSCI.0309-06.2006 (doi:10.1523/JNEUROSCI.0309-06.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lafourcade M., Elezgarai I., Mato S., Bakiri Y., Grandes P., Manzoni O. J. 2007. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE 2, e709. 10.1371/journal.pone.0000709 (doi:10.1371/journal.pone.0000709) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monory K., et al. 2006. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 51, 455–466 10.1016/j.neuron.2006.07.006 (doi:10.1016/j.neuron.2006.07.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morozov Y. M., Freund T. F. 2003. Post-natal development of type 1 cannabinoid receptor immunoreactivity in the rat hippocampus. Eur. J. Neurosci. 18, 1213–1222 10.1046/j.1460-9568.2003.02852.x (doi:10.1046/j.1460-9568.2003.02852.x) [DOI] [PubMed] [Google Scholar]
- 20.Berghuis P., et al. 2005. Endocannabinoids regulate interneuron migration and morphogenesis by transactivating the TrkB receptor. Proc. Natl Acad. Sci. USA 102, 19 115–19 120 10.1073/pnas.0509494102 (doi:10.1073/pnas.0509494102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bodor A. L., Katona I., Nyiri G., Mackie K., Ledent C., Hajos N., Freund T. F. 2005. Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J. Neurosci. 25, 6845–6856 10.1523/JNEUROSCI.0442-05.2005 (doi:10.1523/JNEUROSCI.0442-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hashimotodani Y., Ohno-Shosaku T., Kano M. 2007. Endocannabinoids and synaptic function in the CNS. Neuroscientist 13, 127–137 10.1177/1073858406296716 (doi:10.1177/1073858406296716) [DOI] [PubMed] [Google Scholar]
- 23.Mato S., Del Olmo E., Pazos A. 2003. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain. Eur. J. Neurosci. 17, 1747–1754 10.1046/j.1460-9568.2003.02599.x (doi:10.1046/j.1460-9568.2003.02599.x) [DOI] [PubMed] [Google Scholar]
- 24.Wang X., Dow-Edwards D., Keller E., Hurd Y. L. 2003. Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience 118, 681–694 10.1016/S0306-4522(03)00020-4 (doi:10.1016/S0306-4522(03)00020-4) [DOI] [PubMed] [Google Scholar]
- 25.Zurolo E., Iyer A. M., Spliet W. G., Van Rijen P. C., Troost D., Gorter J. A., Aronica E. 2010. CB1 and CB2 cannabinoid receptor expression during development and in epileptogenic developmental pathologies. Neuroscience 170, 28–41 10.1016/j.neuroscience.2010.07.004 (doi:10.1016/j.neuroscience.2010.07.004) [DOI] [PubMed] [Google Scholar]
- 26.Galve-Roperh I., Palazuelos J., Aguado T., Guzman M. 2009. The endocannabinoid system and the regulation of neural development: potential implications in psychiatric disorders. Eur. Arch. Psychiat. Clin. Neurosci. 259, 371–382 10.1007/s00406-009-0028-y (doi:10.1007/s00406-009-0028-y) [DOI] [PubMed] [Google Scholar]
- 27.Aguado T., et al. 2006. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 26, 1551–1561 10.1523/JNEUROSCI.3101-05.2006 (doi:10.1523/JNEUROSCI.3101-05.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowalczyk T., et al. 2009. Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb. Cortex 19, 2439–2450 10.1093/cercor/bhn260 (doi:10.1093/cercor/bhn260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguado T., et al. 2005. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 19, 1704–1706 [DOI] [PubMed] [Google Scholar]
- 30.Jiang W., Zhang Y., Xiao L., Van Cleemput J., Ji S. P., Bai G., Zhang X. 2005. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J. Clin. Invest. 115, 3104–3116 10.1172/JCI25509 (doi:10.1172/JCI25509) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Begbie J., Doherty P., Graham A. 2004. Cannabinoid receptor, CB1, expression follows neuronal differentiation in the early chick embryo. J. Anat. 205, 213–218 10.1111/j.0021-8782.2004.00325.x (doi:10.1111/j.0021-8782.2004.00325.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson S., Chambers D., Hobbs C., Doherty P., Graham A. 2008. The endocannabinoid receptor, CB1, is required for normal axonal growth and fasciculation. Mol. Cell Neurosci. 38, 89–97 10.1016/j.mcn.2008.02.001 (doi:10.1016/j.mcn.2008.02.001) [DOI] [PubMed] [Google Scholar]
- 33.Lam C. S., Rastegar S., Strahle U. 2006. Distribution of cannabinoid receptor 1 in the CNS of zebrafish. Neuroscience 138, 83–95 10.1016/j.neuroscience.2005.10.069 (doi:10.1016/j.neuroscience.2005.10.069) [DOI] [PubMed] [Google Scholar]
- 34.Palazuelos J., Aguado T., Egia A., Mechoulam R., Guzman M., Galve-Roperh I. 2006. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 20, 2405–2407 10.1096/fj.06-6164fje (doi:10.1096/fj.06-6164fje) [DOI] [PubMed] [Google Scholar]
- 35.Molina-Holgado F., Rubio-Araiz A., Garcia-Ovejero D., Williams R. J., Moore J. D., Arevalo-Martin A., Gómez-Torres O., Molina-Holgado E. 2007. CB2 cannabinoid receptors promote mouse neural stem cell proliferation. Eur. J. Neurosci. 25, 629–634 10.1111/j.1460-9568.2007.05322.x (doi:10.1111/j.1460-9568.2007.05322.x) [DOI] [PubMed] [Google Scholar]
- 36.Gao Y., et al. 2010. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 30, 2017–2024 10.1523/JNEUROSCI.5693-09.2010 (doi:10.1523/JNEUROSCI.5693-09.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berghuis P., et al. 2007. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 316, 1212–1216 10.1126/science.1137406 (doi:10.1126/science.1137406) [DOI] [PubMed] [Google Scholar]
- 38.Argaw A., et al. 2011. Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J. Neurosci. 31, 1489–1499 10.1523/JNEUROSCI.4134-09.2011 (doi:10.1523/JNEUROSCI.4134-09.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung K. M., Astarita G., Thongkham D., Piomelli D. 2011. Diacylglycerol lipase-α and -β control neurite outgrowth in neuro-2a cells through distinct molecular mechanisms. Mol. Pharmacol. 80, 60–67 10.1124/mol.110.070458 (doi:10.1124/mol.110.070458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goncalves M. B., et al. 2008. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol. Cell Neurosci. 38, 526–536 10.1016/j.mcn.2008.05.001 (doi:10.1016/j.mcn.2008.05.001) [DOI] [PubMed] [Google Scholar]
- 41.Walker D. J., Suetterlin P., Reisenberg M., Williams G., Doherty P. 2010. Down-regulation of diacylglycerol lipase-alpha during neural stem cell differentiation: identification of elements that regulate transcription. J. Neurosci. Res. 88, 735–745 [DOI] [PubMed] [Google Scholar]
- 42.Keimpema E., et al. 2010. Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. J. Neurosci. 30, 13 992–14 007 10.1523/JNEUROSCI.2126-10.2010 (doi:10.1523/JNEUROSCI.2126-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin K., Xie L., Kim S. H., Parmentier-Batteur S., Sun Y., Mao X. O., Childs J., Greenberg D. A. 2004. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol. Pharmacol. 66, 204–208 10.1124/mol.66.2.204 (doi:10.1124/mol.66.2.204) [DOI] [PubMed] [Google Scholar]
- 44.Galve-Roperh I., Aguado T., Palazuelos J., Guzman M. 2007. The endocannabinoid system and neurogenesis in health and disease. Neuroscientist 13, 109–114 10.1177/1073858406296407 (doi:10.1177/1073858406296407) [DOI] [PubMed] [Google Scholar]
- 45.Trazzi S., Steger M., Mitrugno V. M., Bartesaghi R., Ciani E. 2010. CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3beta/beta-catenin signaling. J. Biol. Chem. 285, 10 098–10 109 10.1074/jbc.M109.043711 (doi:10.1074/jbc.M109.043711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hwangpo T. A., Jordan J. D., Premsrirut P. K., Jayamaran G., Licht J. D., Iyengar R., Neves S. R. 2012. G-protein-regulated inducer of neurite outgrowth (GRIN) modulates Sprouty protein repression of mitogen-activated protein kinase (MAPK) activation by growth factor stimulation. J. Biol. Chem. 287, 13 674–13 685 10.1074/jbc.M111.320705 (doi:10.1074/jbc.M111.320705) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rueda D., Navarro B., Martinez-Serrano A., Guzman M., Galve-Roperh I. 2002. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the RaP1/B-Raf/ERK pathway. J. Biol. Chem. 277, 46 645–46 650 [DOI] [PubMed] [Google Scholar]
- 48.Galve-Roperh I., Aguado T., Palazuelos J., Guzman M. 2008. Mechanisms of control of neuron survival by the endocannabinoid system. Curr. Pharm. Des. 14, 2279–2288 10.2174/138161208785740117 (doi:10.2174/138161208785740117) [DOI] [PubMed] [Google Scholar]
- 49.Davis M. I., Ronesi J., Lovinger D. M. 2003. A predominant role for inhibition of the adenylate cyclase/protein kinase A pathway in ERK activation by cannabinoid receptor 1 in N1E-115 neuroblastoma cells. J. Biol. Chem. 278, 48 973–48 980 10.1074/jbc.M305697200 (doi:10.1074/jbc.M305697200) [DOI] [PubMed] [Google Scholar]
- 50.Derkinderen P., Valjent E., Toutant M., Corvol J. C., Enslen H., Ledent C., Trzaskos J., Caboche J., Girault J. A. 2003. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J. Neurosci. 23, 2371–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galve-Roperh I., Rueda D., Gomez Del Pulgar T., Velasco G., Guzman M. 2002. Mechanism of extracellular signal-regulated kinase activation by the CB(1) cannabinoid receptor. Mol. Pharmacol. 62, 1385–1392 10.1124/mol.62.6.1385 (doi:10.1124/mol.62.6.1385) [DOI] [PubMed] [Google Scholar]
- 52.Dalton G. D., Howlett A. C. 2012. Cannabinoid CB1 receptors transactivate multiple receptor tyrosine kinases and regulate serine/threonine kinases to activate ERK in neuronal cells. Br. J. Pharmacol. 165, 2497–2511 10.1111/j.1476-5381.2011.01455.x (doi:10.1111/j.1476-5381.2011.01455.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoeffer C. A., Klann E. 2010. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75 10.1016/j.tins.2009.11.003 (doi:10.1016/j.tins.2009.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puighermanal E., Marsicano G., Busquets-Garcia A., Lutz B., Maldonado R., Ozaita A. 2009. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat. Neurosci. 12, 1152–1158 10.1038/nn.2369 (doi:10.1038/nn.2369) [DOI] [PubMed] [Google Scholar]
- 55.Puighermanal E., Busquets-Garcia A., Maldonado R., Uzaita A. 2012. Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Phil. Trans. R. Soc. B 367, 3254–3263 10.1098/rstb.2011.0384 (doi:10.1098/rstb.2011.0384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palazuelos J., Ortega Z., Diaz-Alonso J., Guzman M., Galve-Roperh I. 2012. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 287, 1198–1209 10.1074/jbc.M111.291294 (doi:10.1074/jbc.M111.291294) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou J., et al. 2011. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell. 20, 97–108 10.1016/j.devcel.2010.11.020 (doi:10.1016/j.devcel.2010.11.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gomez O., Sanchez-Rodriguez A., Le M., Sanchez-Caro C., Molina-Holgado F., Molina-Holgado E. 2011. Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br. J. Pharmacol. 163, 1520–1532 10.1111/j.1476-5381.2011.01414.x (doi:10.1111/j.1476-5381.2011.01414.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carracedo A., et al. 2006. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 9, 301–312 10.1016/j.ccr.2006.03.005 (doi:10.1016/j.ccr.2006.03.005) [DOI] [PubMed] [Google Scholar]
- 60.Salazar M., et al. 2009. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest. 119, 1359–1372 10.1172/JCI37948 (doi:10.1172/JCI37948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Callen L., et al. 2012. Cannabinoid receptors CB1 and CB2 form functional heteromers in the brain. J. Biol. Chem. 287, 20 851–20 865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rozenfeld R., et al. 2012. Receptor heteromerization expands the repertoire of cannabinoid signaling in rodent neurons. PLoS ONE 7, e29239. 10.1371/journal.pone.0029239 (doi:10.1371/journal.pone.0029239) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsicano G., et al. 2002. The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534 10.1038/nature00839 (doi:10.1038/nature00839) [DOI] [PubMed] [Google Scholar]
- 64.Aso E., Ozaita A., Valdizan E. M., Ledent C., Pazos A., Maldonado R., Valverde O. 2008. BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J. Neurochem. 105, 565–572 10.1111/j.1471-4159.2007.05149.x (doi:10.1111/j.1471-4159.2007.05149.x) [DOI] [PubMed] [Google Scholar]
- 65.Bergami M., Rimondini R., Santi S., Blum R., Gotz M., Canossa M. 2008. Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc. Natl Acad. Sci. USA 105, 15 570–15 575 10.1073/pnas.0803702105 (doi:10.1073/pnas.0803702105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hart S., Fischer O. M., Ullrich A. 2004. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 64, 1943–1950 10.1158/0008-5472.CAN-03-3720 (doi:10.1158/0008-5472.CAN-03-3720) [DOI] [PubMed] [Google Scholar]
- 67.Rubio-Araiz A., et al. 2008. The endocannabinoid system modulates a transient TNF pathway that induces neural stem cell proliferation. Mol. Cell Neurosci. 38, 374–380 10.1016/j.mcn.2008.03.010 (doi:10.1016/j.mcn.2008.03.010) [DOI] [PubMed] [Google Scholar]
- 68.Marsicano G., et al. 2003. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88 10.1126/science.1088208 (doi:10.1126/science.1088208) [DOI] [PubMed] [Google Scholar]
- 69.Aguado T., Romero E., Monory K., Palazuelos J., Sendtner M., Marsicano G., Lutz B., Guzman M., Galve-Roperh I. 2007. The CB1 cannabinoid receptor mediates excitotoxicity-induced neural progenitor proliferation and neurogenesis. J. Biol. Chem. 282, 23 892–23 898 10.1074/jbc.M700678200 (doi:10.1074/jbc.M700678200) [DOI] [PubMed] [Google Scholar]
- 70.De March Z., et al. 2008. Cortical expression of brain derived neurotrophic factor and type-1 cannabinoid receptor after striatal excitotoxic lesions. Neuroscience 152, 734–740 10.1016/j.neuroscience.2007.11.044 (doi:10.1016/j.neuroscience.2007.11.044) [DOI] [PubMed] [Google Scholar]
- 71.Williams E. J., Walsh F. S., Doherty P. 2003. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J. Cell Biol. 160, 481–486 10.1083/jcb.200210164 (doi:10.1083/jcb.200210164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rueda D., Navarro B., Martinez-Serrano A., Guzman M., Galve-Roperh I. 2002. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J. Biol. Chem. 277, 46 645–46 650 10.1074/jbc.M206590200 (doi:10.1074/jbc.M206590200) [DOI] [PubMed] [Google Scholar]
- 73.Ishii I., Chun J. 2002. Anandamide-induced neuroblastoma cell rounding via the CB1 cannabinoid receptors. Neuroreport 13, 593–596 10.1097/00001756-200204160-00011 (doi:10.1097/00001756-200204160-00011) [DOI] [PubMed] [Google Scholar]
- 74.Jordan J. D., et al. 2005. Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through G(alpha)o/i-triggered proteasomal degradation of Rap1GAPII. J. Biol. Chem. 280, 11 413–11 421 10.1074/jbc.M411521200 (doi:10.1074/jbc.M411521200) [DOI] [PubMed] [Google Scholar]
- 75.Zhou D., Song Z. H. 2001. CB1 cannabinoid receptor-mediated neurite remodeling in mouse neuroblastoma N1E-115 cells. J. Neurosci. Res. 65, 346–353 10.1002/jnr.1160 (doi:10.1002/jnr.1160) [DOI] [PubMed] [Google Scholar]
- 76.He J. C., Gomes I., Nguyen T., Jayaram G., Ram P. T., Devi L. A., Iyengar R. 2005. The G alpha(o/i)-coupled cannabinoid receptor-mediated neurite outgrowth involves Rap regulation of Src and Stat3. J. Biol. Chem. 280, 33 426–33 434 10.1074/jbc.M502812200 (doi:10.1074/jbc.M502812200) [DOI] [PubMed] [Google Scholar]
- 77.Zorina Y., Iyengar R., Bromberg K. D. 2010. Cannabinoid 1 receptor and interleukin-6 receptor together induce integration of protein kinase and transcription factor signaling to trigger neurite outgrowth. J. Biol. Chem. 285, 1358–1370 10.1074/jbc.M109.049841 (doi:10.1074/jbc.M109.049841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith T. H., Sim-Selley L. J., Selley D. E. 2010. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br. J. Pharmacol. 160, 454–466 10.1111/j.1476-5381.2010.00777.x (doi:10.1111/j.1476-5381.2010.00777.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gomez O., et al. 2010. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 58, 1913–1927 10.1002/glia.21061 (doi:10.1002/glia.21061) [DOI] [PubMed] [Google Scholar]
- 80.Arevalo-Martin A., Garcia-Ovejero D., Rubio-Araiz A., Gomez O., Molina-Holgado F., Molina-Holgado E. 2007. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur. J. Neurosci. 26, 1548–1559 10.1111/j.1460-9568.2007.05782.x (doi:10.1111/j.1460-9568.2007.05782.x) [DOI] [PubMed] [Google Scholar]
- 81.Bromberg K. D., Ma'ayan A., Neves S. R., Iyengar R. 2008. Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science 320, 903–909 10.1126/science.1152662 (doi:10.1126/science.1152662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Osumi N., Shinohara H., Numayama-Tsuruta K., Maekawa M. 2008. Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cells 26, 1663–1672 10.1634/stemcells.2007-0884 (doi:10.1634/stemcells.2007-0884) [DOI] [PubMed] [Google Scholar]
- 83.Diaz-Alonso J., et al. In press The CBI cannabinoid receptor drives corticospinal motor neuron differentiation through the Ctip2/Satb2 transcriptional regulation axis. J. Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Molyneaux B. J., Arlotta P., Menezes J. R., Macklis J. D. 2007. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437 10.1038/nrn2151 (doi:10.1038/nrn2151) [DOI] [PubMed] [Google Scholar]
- 85.Harkany T., Guzman M., Galve-Roperh I., Berghuis P., Devi L. A., Mackie K. 2007. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci. 28, 83–92 10.1016/j.tips.2006.12.004 (doi:10.1016/j.tips.2006.12.004) [DOI] [PubMed] [Google Scholar]
- 86.Molina-Holgado E., Vela J. M., Arevalo-Martin A., Almazan G., Molina-Holgado F., Borrell J., Guaza C. 2002. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 22, 9742–9753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aguado T., Carracedo A., Julien B., Velasco G., Milman G., Mechoulam R., Alvarez L., Guzmán M., Galve-Roperh I. 2007. Cannabinoids induce glioma stem-like cell differentiation and inhibit gliomagenesis. J. Biol. Chem. 282, 6854–6862 10.1074/jbc.M608900200 (doi:10.1074/jbc.M608900200) [DOI] [PubMed] [Google Scholar]
- 88.Fukuda S., Abematsu M., Mori H., Yanagisawa M., Kagawa T., Nakashima K., Yoshimura A., Taga T. 2007. Potentiation of astrogliogenesis by STAT3-mediated activation of bone morphogenetic protein-Smad signaling in neural stem cells. Mol. Cell Biol. 27, 4931–4937 10.1128/MCB.02435-06 (doi:10.1128/MCB.02435-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pang T., Atefy R., Sheen V. 2008. Malformations of cortical development. Neurologist 14, 181–191 10.1097/NRL.0b013e31816606b9 (doi:10.1097/NRL.0b013e31816606b9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sipe J. C., Chiang K., Gerber A. L., Beutler E., Cravatt B. F. 2002. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc. Natl Acad. Sci. USA 99, 8394–8399 10.1073/pnas.082235799 (doi:10.1073/pnas.082235799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hariri A. R., Gorka A., Hyde L. W., Kimak M., Halder I., Ducci F., Ferrell R. E., Goldman D., Manuck S. B. 2009. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol. Psychiat. 66, 9–16 10.1016/j.biopsych.2008.10.047 (doi:10.1016/j.biopsych.2008.10.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fiskerstrand T., et al. 2010. Mutations in ABHD12 cause the neurodegenerative disease PHARC: an inborn error of endocannabinoid metabolism. Am. J. Hum. Genet. 87, 410–417 10.1016/j.ajhg.2010.08.002 (doi:10.1016/j.ajhg.2010.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schneider M. 2009. Cannabis use in pregnancy and early life and its consequences: animal models. Eur. Arch. Psychiat. Clin. Neurosci. 259, 383–393 10.1007/s00406-009-0026-0 (doi:10.1007/s00406-009-0026-0) [DOI] [PubMed] [Google Scholar]
- 94.Wu C. S., Zhu J., Wager-Miller J., Wang S., O'Leary D., Monory K., Lutz B., Mackie K., Lu H.-C. 2010. Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur. J. Neurosci. 32, 693–706 10.1111/j.1460-9568.2010.07337.x (doi:10.1111/j.1460-9568.2010.07337.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li L., Bender K. J., Drew P. J., Jadhav S. P., Sylwestrak E., Feldman D. E. 2009. Endocannabinoid signaling is required for development and critical period plasticity of the whisker map in somatosensory cortex. Neuron 64, 537–549 10.1016/j.neuron.2009.10.005 (doi:10.1016/j.neuron.2009.10.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ramocki M. B., Zoghbi H. Y. 2008. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455, 912–918 10.1038/nature07457 (doi:10.1038/nature07457) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heng J. I., Moonen G., Nguyen L. 2007. Neurotransmitters regulate cell migration in the telencephalon. Eur. J. Neurosci. 26, 537–546 10.1111/j.1460-9568.2007.05694.x (doi:10.1111/j.1460-9568.2007.05694.x) [DOI] [PubMed] [Google Scholar]
- 98.Martinez-Gras I., et al. 2006. (AAT)n repeat in the cannabinoid receptor gene, CNR1: association with schizophrenia in a Spanish population. Eur. Arch. Psychiat. Clin. Neurosci. 256, 437–441 10.1007/s00406-006-0665-3 (doi:10.1007/s00406-006-0665-3) [DOI] [PubMed] [Google Scholar]
- 99.Ponce G., Hoenicka J., Rubio G., Ampuero I., Jimenez-Arriero M. A., Rodriguez-Jimenez R., Palomo T, Ramos J. A., 2003. Association between cannabinoid receptor gene (CNR1) and childhood attention deficit/hyperactivity disorder in Spanish male alcoholic patients. Mol. Psychiat. 8, 466–467 10.1038/sj.mp.4001278 (doi:10.1038/sj.mp.4001278) [DOI] [PubMed] [Google Scholar]
- 100.Onaivi E. S., et al. 2008. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS ONE 3, e1640. 10.1371/journal.pone.0001640 (doi:10.1371/journal.pone.0001640) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Paz R. D., Tardito S., Atzori M., Tseng K. Y. 2008. Glutamatergic dysfunction in schizophrenia: from basic neuroscience to clinical psychopharmacology. Eur. Neuropsychopharmacol. 18, 773–786 10.1016/j.euroneuro.2008.06.005 (doi:10.1016/j.euroneuro.2008.06.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eggan S. M., Hashimoto T., Lewis D. A. 2008. Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch. Gen. Psychiat. 65, 772–784 10.1001/archpsyc.65.7.772 (doi:10.1001/archpsyc.65.7.772) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rakhade S. N., Jensen F. E. 2009. Epileptogenesis in the immature brain: emerging mechanisms. Nat. Rev. Neurol. 5, 380–391 10.1038/nrneurol.2009.80 (doi:10.1038/nrneurol.2009.80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Manent J. B., Wang Y., Chang Y., Paramasivam M., LoTurco J. J. 2009. Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nat. Med. 15, 84–90 10.1038/nm.1897 (doi:10.1038/nm.1897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nosten-Bertrand M., et al. 2008. Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PLoS ONE 3, e2473. 10.1371/journal.pone.0002473 (doi:10.1371/journal.pone.0002473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ludanyi A., et al. 2008. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J. Neurosci. 28, 2976–2990 10.1523/JNEUROSCI.4465-07.2008 (doi:10.1523/JNEUROSCI.4465-07.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen K., et al. 2003. Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron 39, 599–611 10.1016/S0896-6273(03)00499-9 (doi:10.1016/S0896-6273(03)00499-9) [DOI] [PubMed] [Google Scholar]
- 108.Powell E. M., Campbell D. B., Stanwood G. D., Davis C., Noebels J. L., Levitt P. 2003. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J. Neurosci. 23, 622–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cobos I., Calcagnotto M. E., Vilaythong A. J., Thwin M. T., Noebels J. L., Baraban S. C., Rubenstein J. L. R. 2005. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068 10.1038/nn1499 (doi:10.1038/nn1499) [DOI] [PubMed] [Google Scholar]
- 110.Lodato S., et al. 2011. Loss of COUP-TFI alters the balance between caudal ganglionic eminence- and medial ganglionic eminence-derived cortical interneurons and results in resistance to epilepsy. J. Neurosci. 31, 4650–4662 10.1523/JNEUROSCI.6580-10.2011 (doi:10.1523/JNEUROSCI.6580-10.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wyeth M. S., Zhang N., Mody I., Houser C. R. 2010. Selective reduction of cholecystokinin-positive basket cell innervation in a model of temporal lobe epilepsy. J. Neurosci. 30, 8993–9006 10.1523/JNEUROSCI.1183-10.2010 (doi:10.1523/JNEUROSCI.1183-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Antonucci F., et al. 2012. Cracking down on inhibition: selective removal of GABAergic interneurons from hippocampal networks. J. Neurosci. 32, 1989–2001 10.1523/JNEUROSCI.2720-11.2012 (doi:10.1523/JNEUROSCI.2720-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cachope R. 2012. Functional diversity of synaptic plasticity mediated by endocannabinoids. Phil. Trans. R. Soc. B 367, 3242–3253 10.1098/rstb.2011.0386 (doi:10.1098/rstb.2011.0386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bernard C., Milh M., Morozov Y. M., Ben-Ari Y., Freund T. F., Gozlan H. 2005. Altering cannabinoid signaling during development disrupts neuronal activity. Proc. Natl Acad. Sci. USA 102, 9388–9393 10.1073/pnas.0409641102 (doi:10.1073/pnas.0409641102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen K., Neu A., Howard A. L., Foldy C., Echegoyen J., Hilgenberg L., Smith M., Mackie K., Soltesz I. 2007. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J. Neurosci. 27, 46–58 10.1523/JNEUROSCI.3966-06.2007 (doi:10.1523/JNEUROSCI.3966-06.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Katona I., Freund T. F. 2008. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 14, 923–930 10.1038/nm.f.1869 (doi:10.1038/nm.f.1869) [DOI] [PubMed] [Google Scholar]
- 117.Lutz B., Monory K. 2008. Soothing the seizures of children. Nat. Med. 14, 721–722 10.1038/nm0708-721 (doi:10.1038/nm0708-721) [DOI] [PubMed] [Google Scholar]