

Abstract
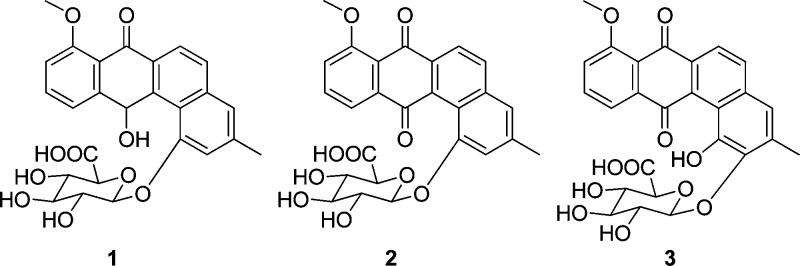
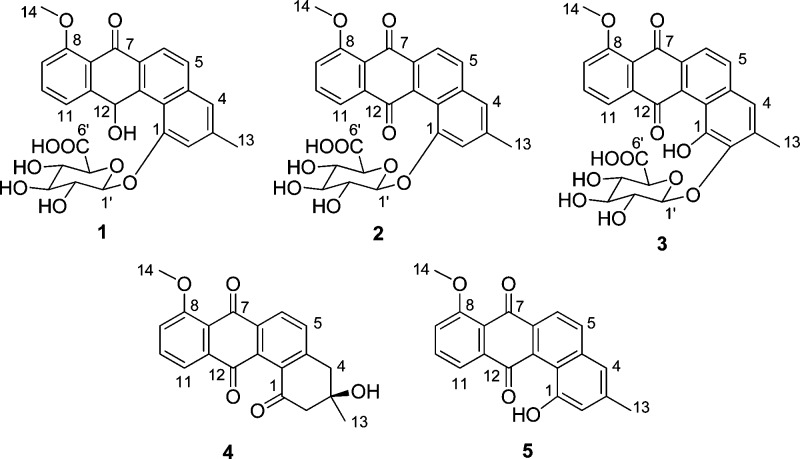
Three new members of the angucycline class of antibiotics, pseudonocardones A–C (1–3), along with the known antibiotics 6-deoxy-8-O-methylrabelomycin (4) and X-14881 E (5) have been isolated from the culture of a Pseudonocardia strain associated with the fungus-growing ant Apterostigma dentigerum. Compounds 4 and 5 showed antibiotic activity against Bacillus subtilis 3610 and liver-stage Plasmodium berghei, while 1–3 were inactive or only weakly active in a variety of biological assays. Compound 5 also showed moderate cytotoxicity against HepG2 cells.
Bacteria of the genus Pseudonocardia associate with fungus-growing ants1 and produce antibiotics that presumptively play a role in suppressing fungal pathogens in ants’ gardens.2,3 While Pseudonocardia belongs to the Actinomycetales, which are well known for their remarkable ability to produce bioactive secondary metabolites, relatively few natural products have been described from Pseudonocardia spp. Antibiotics discovered from Pseudonocardia spp. include dentigerumycin,3 pseudonocardians A and B,4 phenazostatin D,5 and NPP.6 Of these, only dentigerumycin was discovered from a Pseudonocardia strain associated with fungus-growing ants, and these symbionts likely have many more undiscovered natural products. As part of our ongoing effort to explore natural products from bacterial symbionts,3,7−9 we have investigated the natural products from Pseudonocardia sp. EC080529-01, isolated from the cuticle of the fungus-growing ant Apterostigma dentigerum.10 This strain produces three new members of the angucycline family of antibiotics,11 pseudonocardones A (1), B (2), and C (3), along with the known antibiotics 6-deoxy-8-O-methylrabelomycin (4)12 and X-14881 E (5).13
Pseudonocardia sp. EC080529-01 was grown on solid ISP-2 medium, and the agar was extracted with EtOAc followed by MeOH. HPLC-MS analysis of the EtOAc extract revealed two peaks with interesting UV spectra (λmax at 381 and 410 nm, respectively). HPLC-MS analysis of the MeOH extract revealed three additional polar peaks with λmax at 348 nm, 388, and 409 nm, respectively. Production cultures of Pseudonocardia sp. EC080529-01 were grown on solid ISP-2 medium and extracted with EtOAc followed by MeOH. The EtOAc extract was purified (see Experimental Section) to give 6-deoxy-8-O-methylrabelomycin (4, 8.3 mg) and X-14881 E (5, 3.5 mg). The MeOH extract was purified (see Experimental Section) to give pseudonocardones A (1, 2.0 mg), B (2, 0.9 mg), and C (3, 1.8 mg). The known compounds 4 and 5 were identified by 1D and 2D NMR spectroscopy, and their structures were confirmed by comparison of their spectroscopic data with the literature values.12,13
Pseudonocardone A (1) gave a peak in the HRESI(+) MS consistent with a molecular formula of C26H24O10. The NMR data (Figures 1 and 2 and Table 1) obtained for 1 indicated that it was related to the angucycline family of antibiotics and that it was similar in structure to 5. The peak in the UV spectrum of 5 at λmax = 410 nm had shifted to λmax = 348 nm. While compound 5 showed two carbon resonances typical of a quinone (δC 191.3 and 182.0), compound 1 showed only one carbon resonance in this range (δC 186.0). Instead, compound 1 showed a signal for an oxygenated methine resonance (δC 66.5; δH 6.93). This methine resonance showed HMBC correlations to C-6a (δC 132.5), C-7a (δC 120.8), C-11 (δC 123.8), C-11a (δC 147.7), C-12a (δC 139.6), and C-12b (121.6), indicating that C-12 is reduced in 1 to give a hydroquinone tautomer rather than a quinone as found in 5.
Figure 1.

Key COSY and HMBC correlations in the aglycone moiety of pseudonocardone A (1).
Figure 2.

Key COSY and HMBC correlations in the sugar moiety of pseudonocardone A (1).
Table 1. 1H and 13C NMR Data for Pseudonocardones A (1), B (2), and C (3) Recorded in CD3OD at 600 MHz.
| pseudonocardone
A (1) |
pseudonocardone
B (2) |
pseudonocardone
C (3) |
||||
|---|---|---|---|---|---|---|
| position | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) |
| 1 | 156.9 | 154.7 | ||||
| 2 | 114.7 | 7.21, s | 115.8 | 7.25, s | 144.7 | |
| 3 | 140.5 | 141.8 | 134.7 | |||
| 4 | 124.0 | 7.37, s | 122.9 | 7.45, s | 120.3 | 6.95, s |
| 4a | 139.4 | 135.4 | 131.9 | |||
| 5 | 130.0 | 7.78, d (8.8) | 134.4 | 8.06, d (8.2) | 133.0 | 8.97, d (9.4) |
| 6 | 124.5 | 8.10, d (8.2) | 123.0 | 8.16, d (8.8) | 122.8 | 8.17, d (8.8) |
| 6a | 132.5 | 135.0 | 137.1 | |||
| 7 | 186.0 | 182.8 | 183.1 | |||
| 7a | 120.8 | 120.8 | 119.9 | |||
| 8 | 161.1 | 160.3 | 160.5 | |||
| 9 | 112.8 | 7.13, d (8.2) | 118.3 | 7.47, d (8.8) | 119.3 | 7.49, d (8.2) |
| 10 | 136.0 | 7.64, t (7.9) | 136.7 | 7.78, t (7.9) | 136.5 | 7.75, t (7.9) |
| 11 | 123.8 | 7.34, d (8.6) | 119.3 | 7.62, d (7.6) | 121.1 | 7.78, d (7.0) |
| 11a | 147.7 | 139.9 | 138.4 | |||
| 12 | 66.5 | 6.93, s | 189.5 | 190.8 | ||
| 12a | 139.6 | 138.8 | 133.2 | |||
| 12b | 121.6 | 119.5 | 121.0 | |||
| 13 | 21.8 | 2.47, s | 21.6 | 2.52, s | 17.0 | 2.50, s |
| 14 | 49.4 | 3.95, s | 56.4 | 4.01, s | 56.6 | 3.99, s |
| 1′ | 103.8 | 5.23, d (7.6) | 101.8 | 5.26, d (7.0) | 106.6 | 4.76, d (7.6) |
| 2′ | 75.0 | 3.93, t (8.8) | 74.5 | 3.58, m | 75.1 | 3.71, dd (9.1, 7.9) |
| 3′ | 77.1 | 3.63, t (9.4) | 77.1 | 3.59, m | 77.2 | 3.50, t (9.1) |
| 4′ | 73.1 | 3.76, t (9.4) | 72.6 | 3.60, m | 72.9 | 3.63, t (9.4) |
| 5′ | 76.7 | 4.16, d (10.0) | 76.1 | 3.98, d (9.4) | 76.8 | 3.55, d (10.0) |
| 6′ | 173.0 | 172.6 | 173.5 | |||
Additional signals were present in the NMR spectra of 1 that could not be attributed to the angucycline core. Subtracting the atoms accounted for by the hydroquinone substructure showed that the remaining fragment had to account for C6H9O6. A series of oxygenated methine resonances in the HSQC spectrum (δC 73.1–77.1 and δH 3.63–4.16) and a resonance typical of an anomeric carbon (δC 103.8 and δH 5.23) suggested that 1 was glycosylated. An HMBC correlation from the anomeric proton at δH 5.23 to δC 156.9 (C-1) showed that 1 was glycosylated at the oxygen atom attached to C-1. A series of COSY and HMBC correlations (Figure 2) revealed that the sugar was a hexose. Finally, HMBC correlations from δH 3.76 (H-4′) and 4.16 (H-5′) to a carbonyl carbon at δC 173.0 (C-6′) showed that the sugar had been oxidized to the carboxylic acid at C-6′.
The relative configuration of the sugar residue in 1 was determined by coupling constants and a NOESY experiment. Large coupling constants (J values ranged from 7.6 to 10.0 Hz; see Table 1) indicated that all of the protons must be axial, and therefore the sugar corresponds to β-glucuronic acid. This assignment was supported by NOESY correlations between δH 5.23 (H-1′) and δH 3.63 (H-3′), between δH 5.23 (H-1′) and δH 4.16 (H-5′), between δH 3.93 (H-2′) and δH 3.76 (H-4′), and between δH 3.63 (H-3′) and δH 4.16 (H-5′) (Figure 3). The absolute configuration of the sugar moiety was not determined. The absolute configuration of C-12 was also not determined.
Figure 3.

NOESY correlations in the sugar moiety of pseudonocardone A (1).
Pseudonocardone B (2) gave a peak in the HRESI(+) MS consistent with a molecular formula of C26H22O10. The molecular formula of 2 differed from that of 1 by the loss of two hydrogen atoms. The UV spectrum of 2 also differed significantly from that of 1, with the low-energy λmax having shifted from 348 nm in 1 to 388 nm in 2. The NMR data obtained for 2 were very similar to that of 1, suggesting that the compounds were closely related. The NMR signals corresponding to the C-12 oxygenated methine present in 1 were absent from the NMR spectra of 2. Instead, the resonance at δH 7.62 (H-11) showed an HMBC to a carbon at δC 189.5 (C-12), typical of a quinone carbon, revealing that 2 is the quinone analogue of 1.
Pseudonocardone C (3) gave a peak in the HRESI(+) MS consistent with a molecular formula of C26H22O11. The molecular formula of 3 differed from the molecular formula of 2 by the addition of an oxygen atom. The UV and NMR data obtained for 3 were very similar to that of 2, suggesting that they are closely related. The aromatic singlet present in 2 at δH 7.25 (H-2) was absent in the NMR spectrum of 3, and the other aromatic singlet at δH 7.45 (H-4) was shifted upfield to δH 6.95 (H-4) in 3. The methyl resonance at δH 2.50 showed an HMBC correlation to a downfield carbon at δC 144.7 (C-2), suggesting that C-2 was substituted with an oxygen atom. An HMBC correlation from the anomeric proton at δH 4.76 (H-1′) to C-2 showed that the sugar moiety was attached to C-2 instead of to C-1 as found in 1 and 2. The lack of protons within three bonds from C-1 made the assignment of this position impossible to determine from the HMBC data. However, in order to satisfy the molecular formula of 3, C-1 must be oxygenated as it is in 1, 2, and 5.
Compounds 1–5 were tested for antibiotic activity against Escherichia coli K12, Bacillus subtilis 3610, Candida albicans, and Saccharomyces cerevisiae. Compounds 4 and 5 were active against B. subtilis 3610 with MIC values of 25 and 3.13 μg/mL, respectively. None of the compounds showed any activity against E. coli, C. albicans, or S. cerevisiae at concentrations as high as 50 μg/mL. Compounds 1–5 were also tested in a liver-stage malaria assay recently developed in one of our laboratories.14 Compounds 4 and 5 were active against liver-stage Plasmodium berghei with IC50 values of 18.5 and 3.0 μM, respectively. Finally, compounds 1–5 were tested for cytotoxicity against HepG2 cells. Compound 5 was active against HepG2 cells with an IC50 value of 36.1 μM. The glycoside analogues (1–3) were completely inactive against B. subtilis, E. coli, C. albicans, S. cerevisiae, and HepG2 cells at concentrations as high as 50 μg/mL and showed only weak activity against liver-stage P. berghei with IC50 values of 38, 50, and >100 μM, respectively. The lack of activity for the glycosylated analogues provides insight into the structure–activity relationships of this family of compounds. A comparison of the activity of 2 with that of 5 shows that adding the glucuronic acid moiety at C-1 completely abolishes cytotoxic and antibiotic activity. Glycosylation of antibiotics has been proposed as one possible mechanism of self-resistance,15,16 and this might explain the lack of biological activity observed for 1–3.
Table 2. Cytotoxic Activities of 1–5 against HepG2 Cells and Antibiotic Activities of 1–5 against P. berghei, E. coli, B. subtilis, C. albicans, and S. cerevisiaea.
| HepG2 | P. berghei | E. coli | B. subtilis | C. albicans | S. cerevisiae | |
|---|---|---|---|---|---|---|
| compound | IC50 (μM) | IC50 (μM) | MIC (μg/mL) | MIC (μg/mL) | MIC (μg/mL) | MIC (μg/mL) |
| 1 | >100 | 38 | >50 | >50 | >50 | >50 |
| 2 | >100 | 50 | >50 | >50 | >50 | >50 |
| 3 | >100 | >100 | >50 | >50 | >50 | >50 |
| 4 | >100 | 18.5 | >50 | 25 | >50 | >50 |
| 5 | 36.1 | 3.0 | >50 | 3.13 | >50 | >50 |
Data represent the average of two experiments each performed in triplicate. The MIC is defined as the lowest concentration that gave less than 5% of the maximum growth.
Experimental Section
General Experimental Procedures
An Agilent 1200 Series HPLC system equipped with a diode array detector and a Phenomenex C18 column (5 μm, 250 × 21.2 mm) was used for preparative HPLC. For HPLC-MS analysis, an Agilent HPLC system equipped with a diode array detector and a 6130 Series quadrupole mass spectrometer was used with a Phenomenex C18 (5 μm, 100 × 4.6 mm) column. The following gradient was used for HPLC-MS analysis: 0–5 min, isocratic 10% CH3CN + 0.1% formic acid; 5–25 min, linear gradient from 10% CH3CN + 0.1% formic acid to 100% CH3CN + 0.1% formic acid. NMR spectra were recorded in CD3OD (for compounds 1–4) or CD2Cl2 (for compound 5) at 600 MHz and referenced to the internal solvent peak at δH 3.30 and δC 49.0 or δH 5.32 and δC 53.8, respectively. High-resolution mass spectrometry (HR-MS) was performed at the University of Illinois Urbana–Champaign Mass Spectrometry Facility.
Isolation of Pseudonocardia sp. EC080529-01
An ant colony of A. dentigerum was collected from Pipeline Road, Panama, on May 29, 2008, and placed in a sterile Petri dish with moist cotton. After allowing the nest to stabilize for a few days, the Pseudonocardia symbiont from this colony was isolated directly from the mesoternal lobe of a worker by scraping bacteria off the cuticle of the ant using a sterile scalpel and plating on chitin media following the methods of Caldera and Currie, and identified as Pseudonocardia sp. based on multilocus sequencing.10
Cultivation of Pseudonocardia sp. EC080529-01
Production cultures of Pseudonocardia sp. EC080529-01 were grown on solid ISP-2 medium (per liter: yeast extract, 4 g; malt extract, 10 g; glucose, 4 g) in 12 Petri plates (150 × 20 mm, 1.2 L total) for 7 d at 30 °C. The solid agar was cut into small cubes and soaked in EtOAc (1.2 L) overnight. The EtOAc was filtered and dried in vacuo to give the crude EtOAc extract. The solid agar was re-extracted overnight with MeOH (1.2 L), and the MeOH was filtered and dried in vacuo to give the crude MeOH extract. The crude EtOAc extract was dissolved in 90% MeOH–H2O (20 mL) and passed through a C18 column, eluting with additional 90% MeOH–H2O, in order to remove nonpolar components. The eluent from this column was diluted with H2O to give a final MeOH concentration of 60%. This solution was passed through another C18 column and washed with additional 60% MeOH–H2O solution, followed by 100% MeOH. The 60% MeOH–H2O fraction was purified by preparative HPLC using the following gradient: 0–5 min, isocratic 20% CH3CN–H2O; 5–60 min, linear gradient from 20% CH3CN–H2O to 100% CH3CN to give pure 4 (8.3 mg). The 100% MeOH fraction from this C18 column was purified by preparative HPLC using the following gradient: 0–10 min, isocratic 50% CH3CN–H2O; 10–60 min, linear gradient from 50% CH3CN–H2O to 100% CH3CN to give pure 5 (3.5 mg). The crude MeOH extract was dissolved in H2O and passed through an HP-20 column. The HP-20 column was washed with water to remove polar components, and the compounds of interest were then eluted with 100% MeOH. The 100% MeOH fraction was dried in vacuo, redissolved in 60% MeOH–H2O, and passed through a C18 column to remove nonpolar components. The eluent from this column was diluted with H2O to give a final MeOH concentration of 30%. This solution was passed through another C18 column and washed with additional 30% MeOH–H2O, followed by 100% MeOH. The 100% MeOH fraction from this C18 column was purified by reversed-phase HPLC using the following gradient: 0–10 min, isocratic 10% CH3CN–H2O + 0.1% formic acid; 10–60 min linear gradient from 10% CH3CN–H2O + 0.1% formic acid to 100% CH3CN + 0.1% formic acid to give pure 1 (2.0 mg), 2 (0.9 mg), and 3 (1.8 mg).
Pseudonocardone A (1):
colorless solid (2.0 mg); [α]20D −11 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 310 nm (3.11), 279 (sh), 271 nm (3.59); 1H NMR (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD), see Table 1; (+)-HRESI m/z 497.1438 [M + H]+ (calcd for C26H25O10, 497.1448).
Pseudonocardone B (2):
yellow solid (0.9 mg); [α]20D −2 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 377 nm (3.52), 305 nm (3.87); 1H NMR (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD), see Table 1; (+)-HRESI m/z 495.1295 [M + H]+ (calcd for C26H23O10, 495.1291).
Pseudonocardone C (3):
orange solid (1.8 mg); [α]20D −3 (c 0.09, MeOH); UV (MeOH) λmax (log ε) 403 nm (3.53), 314 nm (3.96); 1H NMR (600 MHz, CD3OD) and 13C NMR (150 MHz, CD3OD) see Table 1; (+)-HRESI m/z 511.1229 (calcd for C26H23O11, 511.1240).
Antibiotic Assays
The appropriate test organism was grown in a 5 mL culture overnight in either LB medium (for E. coli and B. subtilis) or YPD medium (for C. albicans and S. cerevisiae) at 30 °C. In each case, the overnight culture was diluted with additional sterile medium (LB or YPD) to an OD600 of 0.01. Compounds 1–5 were dissolved in DMSO to give a concentration of 5 mg/mL and 2-fold serially diluted. These solutions (1 μL) were added to the wells of a 96-well plate, followed by the diluted culture of the test organism (99 μL) to give a final compound concentration ranging from 50 to 0.1 μg/mL. The cultures were allowed to grow for 24 h at 30 °C before the OD600 was measured using a plate reader. The MIC was defined as the lowest concentration that gave less than 5% of the maximum OD600. Each antibiotic assay was performed in duplicate. Dynemicin A was used as a positive control and gave MIC values against E. coli, B. subtilis, C. albicans, and S. cerevisiae of 313, 0.16, 156, and 156 ng/mL, respectively.
Cytotoxic Assay
Compounds were tested for activity against HepG2 human hepatoma cells (ATCC) that were maintained in DMEM (Invitrogen), 10% FBS (Sigma), and 1% antibiotic–antimycotic (Invitrogen) in a standard tissue culture incubator (37 °C, 5% CO2). For assays, compounds 1–5 (in DMSO) were added in triplicate to 15 000 cells in a 384-well plate. The final concentration of DMSO was 1%, and compounds varied from 0 to 50 μg/mL. Cells were incubated with the compounds for 2 days at 37 °C, and then liver cell viability was assessed with CellTiter-Glo (Promega). The relative signal intensity of each sample was evaluated with an EnVision (PerkinElmer) system.
Liver-Stage P. berghei Assay
Liver-stage P. berghei assays were performed using a luciferase-expressing sporozoite strain of P. berghei ANKA. Parasites were obtained from dissection of Plasmodium-infected Anopheles stephensi mosquitoes (New York University Langone Medical Center Insectary). Malaria parasites (4000 sporozoites) were used to infect HepG2 cells (15 000 cells) in a 384-well plate in the presence of compounds 1–5 in triplicate. The final concentration of DMSO was 1%, and compounds varied from 0 to 50 μg/mL. Cells were incubated with the compounds for 2 days at 37 °C, and then relative parasite load was determined after addition of Bright-Glo (Promega). Data analysis for HepG2 toxicity and liver-stage malaria activity was carried out using GraphPad Prism, and curves were fit with a standard inhibition dose–response curve to generate an IC50 value. All statistical results are the mean IC50 value averaged from two independent experiments. Atovaquone was used as a positive control and gave an IC50 in blood stage assays of 0.3 nM.
Acknowledgments
This work was supported by NIH RC4GM096347 (C.R.C. and J.C.), GM086258 (J.C.), and NSF CAREERDEB-747002 (C.R.C.), and a Ruth L. Kirschstein National Research Service Award F32GM093510 (E.R.D.).
Supporting Information Available
HPLC-MS traces for compounds 1–3 and NMR spectra for compounds 1–5. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cafaro M. J.; Currie C. R. Can. J. Microbiol. 2005, 51, 441–446. [DOI] [PubMed] [Google Scholar]
- Currie C. R.; Scott J. A.; Summerbell R. C.; Malloch D. Nature 1999, 398, 701–704. [Google Scholar]
- Oh D.-C.; Poulsen M.; Currie C. R.; Clardy J. Nat. Chem. Biol. 2009, 5, 391–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Tian X.; Niu S.; Zhang W.; Chen Y.; Zhang H.; Yang X.; Zhang W.; Li W.; Zhang S.; Ju J.; Zhang C. Mar. Drugs 2011, 9, 1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey R. P.; Kock I.; Helmke E.; Laatsch H. Z. Naturforsch. B 2003, 58b, 692–694. [Google Scholar]
- Lee M.-J.; Kong D.; Han K.; Sherman D. H.; Bai L.; Deng Z.; Lin S.; Kim E.-S. Appl. Microbiol. Biotechnol. 2012, 95, 157–168. [DOI] [PubMed] [Google Scholar]
- Oh D.-C.; Scott J. J.; Currie C. R.; Clardy J. Org. Lett. 2009, 11, 633–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh D.-C.; Poulsen M.; Currie C. R.; Clardy J. Org. Lett. 2011, 13, 752–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr G.; Poulsen M.; Klassen J. L.; Hou Y.; Wyche T. P.; Bugni T. S.; Currie C. R.; Clardy J. Org. Lett. 2012, 14, 2822–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldera E. J.; Currie C. R.. Am. Nat. 2012, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharel M. K.; Pahari P.; Shepherd M. D.; Tibrewal N.; Nybo S. E.; Shaaban K. A.; Rohr J. Nat. Prod. Rep. 2012, 29, 264–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigihara Y.; Koizumi Y.; Tamamura T.; Homma Y.; Isshiki K.; Dobashi K.; Naganawa H.; Takeuchi T. J. Antibiot. 1988, 41, 1260–1264. [DOI] [PubMed] [Google Scholar]
- Maehr H.; Liu C. M.; Liu M.; Perrotta A.; Smallheer J. M.; Williams T. H.; Blount J. F. J. Antibiot. 1982, 35, 1627–1631. [DOI] [PubMed] [Google Scholar]
- Derbyshire E. R.; Prudêncio M.; Mota M. M.; Clardy J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8450–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Sherman D. H.; Liu H.-W. J. Am. Chem. Soc. 1998, 120, 9374–9375. [Google Scholar]
- Zhao L.; Beyer N. J.; Borisova S. A.; Liu H.-W. Biochemistry 2003, 42, 14794–14804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.