

Abstract
Pancreatic cancer is a lethal malignancy with a 5-year survival rate of only 6%. Surgical resection remains the only cure, yet even after resection the 5-year survival is only 20% due to a high recurrence rate. Thus, a high proportion of patients with this disease will ultimately require systemic chemotherapy for advanced pancreatic cancer (APC). While the advent of personalized medicine has resulted in significant advances in the management of many cancer types, the standard of care for pancreatic cancer remains gemcitabine based, with very few exceptions. This article first aims to provide an overview of the benefits and limitations of gemcitabine alone, gemcitabine combinations, and different modes of administration of gemcitabine in APC. It then discusses research, suggesting that pharmacogenomic differences in enzymes that affect gemcitabine transport and metabolism can predict benefit from this drug in pancreatic cancer. Finally, the article outlines novel therapies and combinations that exploit these interindividual variations in gemcitabine metabolism to improve the efficacy of this drug in the management of APC.
Keywords: gemcitabine, metabolism, pancreatic cancer, pharmacogenomics
Gemcitabine in pancreatic cancer
Gemcitabine alone
Pancreatic cancer is the fourth leading cause of cancer-related death in the USA with an estimated incidence of over 43,000 cases in 2010 [Jemal et al. 2010]. Disease stage is highly correlated to prognosis, with median overall survival (OS) for patients with locally advanced or metastatic disease being 9 or 6 months, respectively. The current standard of care for metastatic or unresectable pancreatic cancer is either gemcitabine monotherapy or a gemcitabine combination. The benefit of gemcitabine monotherapy was first demonstrated in 1997 in the Radiation Therapy Oncology Group (RTOG) 9704 trial that randomized patients with untreated advanced pancreatic cancer (APC) to gemcitabine or 5-fluorouracil (5-FU) [Burris et al. 1997]. The primary outcome measure was the clinical benefit response, a composite score that included assessments of pain and analgesic requirements, Karnofsky performance status, and weight. A significant benefit of 23.8% in the gemcitabine group and 4.8% in the 5-FU group (p = 0.0022) was demonstrated, however the improvement in OS seen was small, only 4.2–5.7 months (p = 0.0025) in the gemcitabine arm (Figure 1).
Figure 1.
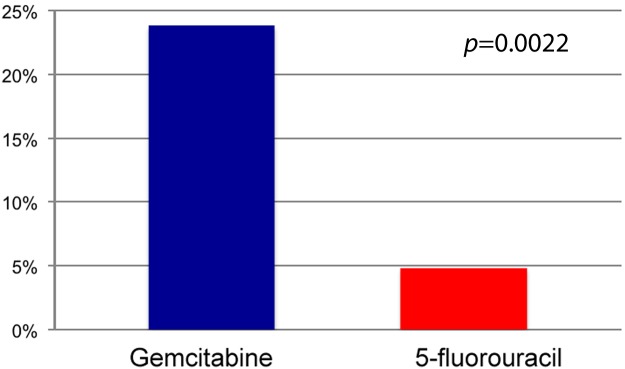
Clinical benefit response with gemcitabine compared with 5-fluoroucracil.
Since then, gemcitabine has been studied in combination with other potentially synergistic agents. However, these studies have resulted in either modest or nonsignificant improvements in OS. A phase III study has also demonstrated improved OS with FOLFIRINOX (5-FU/leucovorin, irinotecan, oxaliplatin) over gemcitabine in the first-line setting. However, only patients with an Eastern Cooperative Oncology Group (ECOG) performance status of 1 and few patients with biliary obstruction were included in this study, which limits the generalizability of these results and will limit its widespread use [Conroy et al. 2011].
Gemcitabine combinations
Among the gemcitabine combinations tested, gemcitabine and capecitabine showed promising results in response rate and progression-free survival but the trend toward improved OS did not reach statistical significance [Cunningham et al. 2009]. Gemcitabine and erlotinib incrementally increased OS from 5.9 to 6.2 months, but the clinical relevance of this is unclear given the increase in side effects (rash and diarrhea) with the combination and the increase in cost [Moore et al. 2007]. Finally, a recent open-label phase I–II trial in 67 patients receiving first-line chemotherapy for APC showed that adding nab-paclitaxel to gemcitabine resulted in a median OS of 12.2 months, longer than previously reported in patients at this stage [Von Hoff et al. 2011]. However, full evaluation of this combination awaits confirmation in a larger phase III clinical trial. Despite the clinical benefit response seen with gemcitabine in APC, the objective response rate is only 9.4% [Conroy et al. 2011]. What factors contribute to a low response rate in gemcitabine-based chemotherapy, and can an understanding of these factors help us stratify patients into gemcitabine-responsive and nonresponsive groups? Additionally, for patients who fall into the gemcitabine-nonresponsive groups, can we exploit the known pathways of gemcitabine resistance to improve the responsiveness of their cancer to gemcitabine?
Fixed-dose-rate gemcitabine
Gemcitabine is a nucleoside analogue that inhibits DNA synthesis and undergoes intracellular activation by deoxycytidine kinase (DCK), yielding gemcitabine monophosphate (dFdCMP), which is then further phosphorylated to its diphosphate (dFdCDP) and triphosphate (dFdCTP) (Figure 2). These inhibit ribonucleotide reductase (RNR) and DNA polymerase, respectively. The initial development of gemcitabine included a detailed determination of its cellular pharmacology in preclinical systems and in humans. Subsequent pharmacokinetic studies showed that gemcitabine given at a fixed dose rate (FDR) intravenous infusion of 10 mg/m2/min produced the highest accumulation of active dFdCTP in the peripheral blood mononuclear cells [Grunewald et al. 1991; Touroutoglou et al. 1998]. The proof of principle was tested in a randomized phase II trial of 92 patients with APC. Treatment was gemcitabine 1500 mg/m2 by FDR or 2200mg/m2 of gemcitabine over 30 min given on days 1, 8, and 15 of a 4-week cycle. The results suggested a better OS with FDR (8 months versus 5 months) and an improvement in the 1-year survival (9% to 29%) but at the expense of increased toxicity [Tempero et al. 2003]. However, a recently reported phase III trial failed to confirm the survival advantage of gemcitabine by FDR over its conventional administration schedule [Poplin et al. 2009].
Figure 2.

Transport and metabolism of gemcitabine.
dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate; dFdC, gemcitabine; dFdCDP, gemcitabine diphosphate; dFdCMP, gemcitabine monophosphate; dFdCTP, gemcitabine triphosphate; dFdU, 2′,2′-difluorodeoxyuridine; dFdUMP, 2′,2′-difluorodeoxyuridine monophosphate; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; hENT1, human equilibrative nucleoside transporter 1.
Mechanisms of resistance to gemcitabine and new drugs
Gemcitabine transport and human equilibrative nucleoside transporter 1
Research into the cellular uptake of gemcitabine has yielded clues to improving its efficacy. Gemcitabine is a polar nucleoside analog that does not readily diffuse across the plasma membrane, and therefore requires the activity of human equilibrative nucleoside transporter 1 (hENT1) to enter cells and exert its cytotoxic effects [Mackey et al. 1998] (Figure 2). Preclinical data in pancreatic cancer cell lines showed that gemcitabine resistance is negatively correlated with hENT1 expression and can be induced by specific inhibitors of hENT1 [Mori et al. 2007].
Clinical data also support the concept that lack of hENT1 may therefore be predictive for resistance to gemcitabine. When patients with uniformly detectable hENT1 on immunohistochemistry of their tumors were compared with patients with regions of absent hENT1, the former group had a longer OS after gemcitabine treatment: 13 months compared with 4 months [Spratlin et al. 2004]. When detecting hENT1 by reverse transcriptase polymerase chain reaction, similar results were seen: levels of hENT1 expression correlated with OS, disease-free survival and time to progression in a dose-dependent fashion [Giovanetti et al. 2006; R. Kim et al. 2011]. hENT1 expression could be either a good prognostic factor or a predictive factor for response to gemcitabine in APC. Evidence supporting the latter came from an analysis of patient tumor tissues from the RTOG 9704 study. In the gemcitabine group, patients with high hENT1 expression had an improved OS compared with those with low hENT1 expression [hazard ratio (HR) 0.40, 95% confidence interval (CI) 0.22–0.75], while in the 5-FU group there was no association between OS and hENT1 expression [Farrell et al. 2008]. These results support the hypothesis that hENT1 is a biomarker for response to gemcitabine, and raise the possibility that hENT1 can be used to stratify patients into groups that derive benefit from gemcitabine and groups that do not. An ongoing study to test this treatment strategy will determine hENT1 levels in patients with resected pancreatic cancer, then treat hENT1-high patients with gemcitabine, and hENT1-low patients with 5-FU [Spratlin, 2011].
One means of enhancing gemcitabine’s efficacy in hENT1 expressing tumors is to bypass the transporter. CO-1.01 is a lipid-conjugated form of gemcitabine that can diffuse across the plasma membrane without the need for hENT1. Intracellular esterases cleave the lipid tail, and gemcitabine then undergoes phosphorylation to its active form. In preclinical studies, CO-1.01 had cytotoxic effects on cancer cell lines in vitro and in human xenografts in nude mice. Dipyridamole, an inhibitor of the hENT1 transporter, protects cell lines against killing by gemcitabine but not by CO-1.01, indicating that CO-1.01 activity was independent of hENT1 [Bergman et al. 2011].
In a phase I study of CO-1.01, seven patients, including two with gemcitabine-refractory pancreatic cancer, had stable disease or tumor shrinkage [Nilsson et al. 2009]. hENT-1 expression was not checked in this study, but in future studies could be used to stratify patients for subgroup analysis. CO-1.01 is well tolerated: the most frequent grade 1–2 toxicities were nausea, vomiting, fatigue and anorexia. Grade 3–4 events included fatigue and neutropenia in 16%, but no episodes of febrile neutropenia occurred. Ongoing studies are a phase IIA international study of CO-1.01 in the first-line setting for APC, and a phase II open-label US study in gemcitabine-refractory APC with low hENT1. The US study will determine whether clinical gemcitabine resistance due to low hENT1 can be overcome with CO-1.01.
Gemcitabine metabolism and deoxycytidine kinase
The intracellular metabolism of gemcitabine can also be exploited to enhance its antitumor effects (Figure 2). Gemcitabine phosphorylation by DCK is the rate-limiting step for gemcitabine activity. dFdCDP and dFdCTP inhibit RNR and DNA polymerase, respectively. dFdCTP also causes single-strand breaks when incorporated into DNA. Inhibiting RNR also depletes deoxycytidine triphosphate, a feedback inhibitor of DCK. This results in increased DCK activity and increased phosphorylation of gemcitabine to its active forms.
Preclinical, in vitro studies have associated lower DCK levels with gemcitabine resistance. Clinical studies include a retrospective study in resected pancreatic cancer, which showed that high DCK and hENT1 correlated with improved OS in gemcitabine-treated but not untreated patients [Marechal et al. 2011]. A novel agent that bypasses both hENT1 and DCK is NUC-1031, a gemcitabine analog to which a phosphoramidate ProTide moiety has been added, allowing for passive diffusion across the plasma membrane. Once in the cell, dFdCMP is generated directly from the drug without a requirement for DCK activity. NUC-1031 is also less sensitive to cytidine aminase, the enzyme by which gemcitabine is metabolized to inactive intermediates. In xenograft mouse models NUC-1031 was active against partially and fully gemcitabine-resistant pancreatic cancer cell lines [McGuignan et al. 2011]. A phase I/II study of this drug is planned, and prospective studies will measure hENT1, deoxycytidine kinase and cytidine aminase activity in tumor biopsies to identify possible biomarkers for response to this new agent.
Gemcitabine and potentially synergistic agents
Though no agents have demonstrated synergy with gemcitabine in clinical trials, preclinical data suggest that there may be subgroups of patients who benefit from combination therapies, and testing combinations in these subgroups may yield further improvements in outcomes.
Preclinical data show that gemcitabine and capecitabine are synergistic in xenograft models because of their complementary mechanisms of action. Capecitabine is an oral prodrug that is converted to 5-FU selectively in tumor tissues by thymidine phosphorylase. 5-FU is then converted to 5-fluoro-deoxyuridine monophosphate (5-F-dUMP), which acts as an inhibitor of thymidylate synthase (TS) and DNA synthesis. Gemcitabine depletes the normal substrate for TS by inhibiting RNR and depleting intracellular pools of deoxyuridine monophosphate (dUMP). 5-F-dUMP outcompetes dUMP for binding to TS, resulting in synergy between gemcitabine and capecitabine. Clinical trials that support this hypothesis include a meta-analysis of three phase III clinical trials that demonstrated a benefit in OS with the gemcitabine–capecitabine combination compared with gemcitabine alone (HR 0.86, 95% CI 0.75–0.98) [Von Hoff et al. 2011]. Retrospective studies have suggested a relationship between the ratio of thymidine phosphorylase to dihydropyrimidine dehydrogenase and the sensitivity of a tumor to capecitabine, and this may be a promising biomarker to identify patients who could benefit from this combination.
Erlotinib was added in combination with gemcitabine because pancreatic tumors often overexpress human epidermal growth factor receptor type 1 (EGFR/HER1). KRAS mutation is one mechanism of primary resistance to EGFR tyrosine kinase inhibitors in non-small cell lung cancers, and KRAS is frequently mutated in pancreatic cancer. A small retrospective study investigated whether KRAS mutation also predicts erlotinib resistance in pancreatic cancer, and found that the survival benefit conferred by gemcitabine–erlotinib could be accounted for by the response in KRAS wild-type tumors alone, while patients with KRAS mutation derived no survival benefit from the combination [S. Kim et al. 2011]. Though the mechanism of synergy between erlotinib and gemcitabine is not well defined, it may involve the restoration of apoptotic pathways by erlotinib which then improves the induction of cell death by gemcitabine [Saif, 2011].
Summary
Personalized medicine has had a profound impact on the treatment of many cancer types. There is growing evidence that interindividual variations in drug metabolism and drug delivery to target tissues alters the efficacy of different chemotherapies. There are now at least two drugs, CO-1.01 and NUC-1031, which show promise as the first personalized therapies for pancreatic cancer. They are both analogs of gemcitabine, a drug with proven activity in pancreatic cancer, but differ from gemcitabine in that they can bypass several mechanisms by which pancreatic tumors may exhibit resistance to this drug. Phase II studies in patients with gemcitabine-refractory disease and in the first-line setting, with appropriate analysis of biomarkers of response, will give us important information on how to best use these rationally designed therapies.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: Dr. Saif received honorarium for a Speakers Bureau from Eli Lilly and Genentech.
Contributor Information
Muhammad Wasif Saif, Director, GI Oncology Program, Tufts University School of Medicine, 800 Washington Street, Box 295, Boston, MA 02111, USA.
Yoomi Lee, Division of Hematology and Oncology, Department of Medicine, Columbia University Medical Center, New York, NY USA.
Richard Kim, Department of Gastrointestinal Oncology, H. Lee Moffitt Cancer Center, Tampa, FL, USA.
References
- Bergman A., Adema A., Balzarini J., Bruheim S., Fichtner I., Noordhuis P., et al. (2011) Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest New Drugs 29: 456–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris H., Moore M., Andersen J., Green M., Rothenberg M., Modiano M., et al. (1997) Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 15: 2403–2413 [DOI] [PubMed] [Google Scholar]
- Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., et al. (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825 [DOI] [PubMed] [Google Scholar]
- Cunningham D., Chau I., Stocken D., Valle J., Smith D., Steward W., et al. (2009) Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 27: 5513–5518 [DOI] [PubMed] [Google Scholar]
- Farrell J., Elsaleh H., Garcia M., Lai R., Ammar A., Regine W., et al. (2008) Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 136: 187–195 [DOI] [PubMed] [Google Scholar]
- Giovanetti E., Del Tacca M., Mey V., Funel N., Nannizzi S., Ricci S., et al. (2006) Transcription analysis of human equilibrative transporter 1 predicts survival in pancreas cancer patients treated with gemcitabine. Cancer Res 66: 3928–3935 [DOI] [PubMed] [Google Scholar]
- Grunewald R., Abbruzzese J., Tarassoff P., Plunkett W. (1991) Saturation of 2′,2′- difluorodeoxycytidine 5′-triphosphate accumulation by mononuclear cells during a phase I trial of gemcitabine. Cancer Chemother Pharmacol 27: 258–262 [DOI] [PubMed] [Google Scholar]
- Jemal A., Siegel R., Xu J., Ward E. (2010) Cancer statistics, 2010. CA Cancer J Clin 60: 277–300 [DOI] [PubMed] [Google Scholar]
- Kim R., Tan A., Lai K., Jiang J., Wang Y., Rybicki L., et al. (2011) Prognostic roles of human equilibrative transporter 1 (hENT-1) and ribonucleoside reductase subunit M1 (RRM1) in resected pancreatic cancer. Cancer 117: 3126–3134 [DOI] [PubMed] [Google Scholar]
- Kim S., Lim D., Jang K., Lim T., Lee J., Choi Y., et al. (2011) Impact of KRAS mutations on clinical outcomes in pancreatic cancer patients treated with first-line gemcitabine-based chemotherapy. Mol Cancer Ther 10: 1993–1999 [DOI] [PubMed] [Google Scholar]
- Mackey J., Mani R., Selner M., Mowles D., Young J., Belt J., et al. (1998) Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 58: 4349–4357 [PubMed] [Google Scholar]
- Marechal R., Bachet J., Mackey J., Demetter P., Graham K., Couvelard A., et al. (2011) Prediction of gemcitabine benefit after curative-intent resection of pancreatic adenocarcinoma using HENT1 and dCK protein expression. J Clin Oncol 29(15 Suppl.): abstract 4024. [Google Scholar]
- McGuigan C., Habib N., Wasan H., Gabra H., Jiao L., Slusarczyk M., et al. (2011) A phosphoramidate ProTide (NUC-1031) and acquired and intrinsic resistance to gemcitabine. J Clin Oncol 29(15 Suppl.): abstract 13540. [Google Scholar]
- Moore M., Goldstein D., Hamm J., Figer A., Hecht J., Gallinger S., et al. (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25: 1960–1966 [DOI] [PubMed] [Google Scholar]
- Mori R., Ishikawa T., Ichikawa Y., Taniguchi K., Matsuyama R., Ueda M., et al. (2007) Human equilibrative nucleoside transporter 1 is associated with the chemosensitivity of gemcitabine in human pancreatic adenocarcinoma and biliary tract carcinoma cells. Oncol Rep 17: 1201–1205 [PubMed] [Google Scholar]
- Nilsson B., Hendlisz A., Castella M., Aamdal S., Dueland S., Nyakas M., et al. (2009) First-in-human study of a novel nucleoside analogue, CP-4126, in patients with advanced solid tumors. J Clin Oncol 27(15 Suppl.): abstract 2577. [Google Scholar]
- Poplin E., Feng Y., Berlin J., Rothenberg M., Hochster H., Mitchell E., et al. (2009) Phase III randomized study of gemcitabine and oxaliplatin versus gemcitabine (fixed dose rate infusion) compared with gemcitabine (30 minute infusion) in patients with pancreatic carcinoma E6201: a trial of the Eastern Cooperative Oncology Group . J Clin Oncol 27: 3778–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saif M. (2011) Does erlotinib restore chemosensitivity to chemotherapy in pancreatic cancer? A case series. Anticancer Res 31: 1039–1042 [PubMed] [Google Scholar]
- Spratlin J. (2011) Biomarker directed adjuvant chemotherapy for resected pancreas cancer. Clinicaltrials.gov http://clinicaltrials.gov/ct2/show/record/NCT01411072 (accessed 19 June 2012).
- Spratlin J., Sangha R., Glubrecht D., Dabbagh L., Young J., Dumontet C., et al. (2004) The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res 10: 6956–6961 [DOI] [PubMed] [Google Scholar]
- Tempero M., Plunkett W., Ruiz Van Haperen V., Hainsworth J., Hochster H., Lenzi R., et al. (2003) Randomized phase II comparison of dose intense gemcitabine: thirty-minute infusion and fixed dose rate infusion in patients with pancreatic adenocarcinoma. J Clin Oncol 21: 3402–3408 [DOI] [PubMed] [Google Scholar]
- Touroutoglou N., Gravel D., Raber M., Plunkett W., Abbruzzese J. (1998) Clinical results of a pharmacodynamically-based strategy for higher dosing of gemcitabine in patients with solid tumors. Ann Oncol 9: 1003–1008 [DOI] [PubMed] [Google Scholar]
- Von Hoff D., Ramanathan R., Borad M., Laheru D., Smith L., Wood T., et al. (2011) Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 29: 4548–4554 [DOI] [PMC free article] [PubMed] [Google Scholar]