

Abstract
Keratitis-Icthyosis-Deafness syndrome is a rare congenital disorder characterized by keratitis, ichthyosis, and deafness. We report a 13 year old female child who presented with diffuse alopecia of the scalp and body. There was erythrokeratoderma of face and discrete hyperkeratotic hyperpigmented papulo plaque lesions on the body. Patient also had reticulate hyperkeratosis of palms and soles. There was history of recurrent episodes of folliculitis over the scalp and body. There was no evidence of any malignancy. Eye involvement in the form of bilateral vascularising keratitis was present. There was bilateral mixed hearing loss.
Keywords: Keratitis, deafness, ichthyosis, Keratitis-ichthyosis-deafness
INTRODUCTION
Keratitis-Ichthyosis-Deafness (KID) syndrome is a rare ectodermal disorder of which hardly a hundred cases have been published in literature. The clinical triad of progressive vascularizing keratitis, sensorineural hearing impairment, and skin manifestations (generalized or localized) are better classified as erythrokeratoderma than as ichthyosis.[1] Heterozygous missense mutations in the GJB2 gene localized on chromosome 13q11-q12 encoding a gap junction protein called connexin-26 were found to be associated with the KID syndrome.[2]
CASE REPORT
We present a13-year old female child, born of non-consanguineous parents as a full-term normal vaginal delivery. Her parents noticed that the child had decreased hair over the scalp. She had thick dark skin over the face, upper limbs, buttocks, and lower limbs since birth. There was no apparent history suggestive of a collodion membrane. Pregnancy and delivery were uncomplicated. The thickness of lesions gradually increased as the child grew older. She also complained of recurrent episodes of pustular lesions on the scalp and body. There was history of photophobia, decreased visual acuity, and decreased hearing since childhood and also of poor performance at school. There was no history of loss of teeth or hyperhidrosis/hypohidrosis. Gait was normal. No other congenital abnormality or systemic disease was detected. There was no family history of similar problems. She had two normal siblings.
Examination of the child showed sparse hair over the scalp with evidence of follicular atrophy at places and multiple folliculitis lesions [Figure 1]. Few remaining hair were thin, rough, and hypopigmented. The morphology of the hair shaft was normal on clinical and microscopic examination. Sparse hair were found over axilla and pubis. There was presence of ciliary and supraciliary madarosis. There were multiple hyperpigmented and hyperkeratotic papulo plaque lesions over the scalp, neck, distal forearm, trunk, buttocks, medial aspect of thighs, and lower one third of the extensor aspect of the leg. Bilaterally symmetrical well-defined erythematous non-blanchable,non-scaly plaques were present on both the cheeks [Figure 2]. Both the elbows, knees, and dorsa of hands and feet showed presence of diffuse hyperpigmentation and hyperkeratosis [Figure 3a and b]. There was bilateral palmoplantar keratoderma with a yellowish hue, velvety texture, and prominent dermatoglyphics [Figure 4a and b]. Examination of the oral mucosa was unremarkable. No abnormality was detected in the nails. Her intelligence quotient and stature was estimated to be in normal range.
Figure 1.
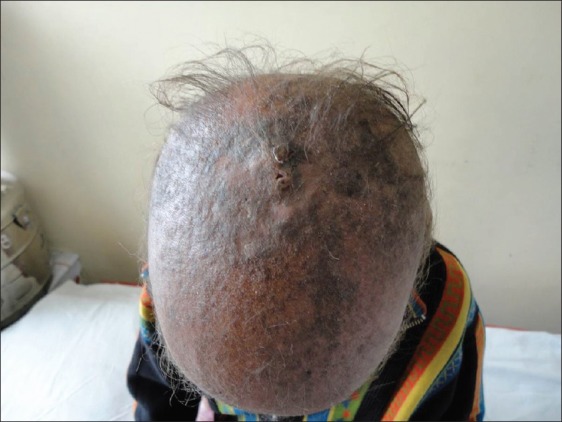
Diffuse alopecia of scalp (a)-cicatricial at places; (b) -Multiple hyperkeratotic hyperpigmented papulo-plaque lesions; (c) few lesions of folliculitis in the healing phase
Figure 2.
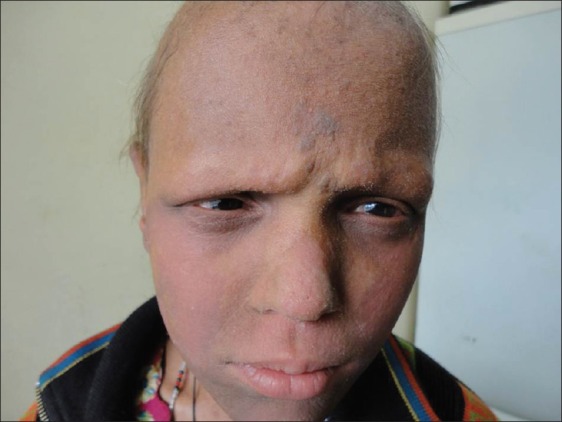
Photophobia and erythrokeratoderma of both cheeks with hypotrichosis of the eyelashes and eyebrows
Figure 3.
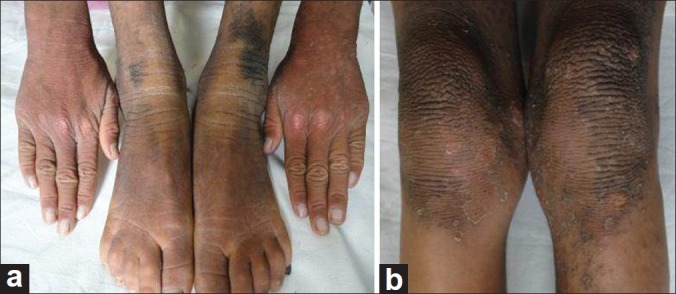
Diffuse hyperpigmentation and hyperkeratosis of the (a) dorsa of hands and feet; (b) knees
Figure 4.
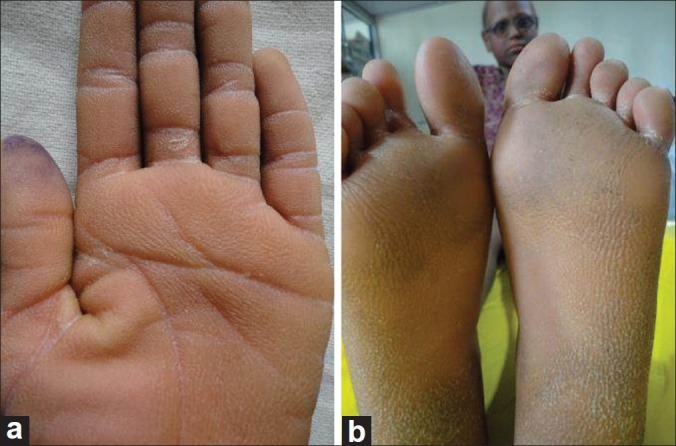
Keratoderma of (a) palm; (b) soles
Ophthalmological examination showed bilateral corneal neovascularization [Figure 5] with evidence of dry eye. Fluorescein angiography showed no abnormality.
Figure 5.
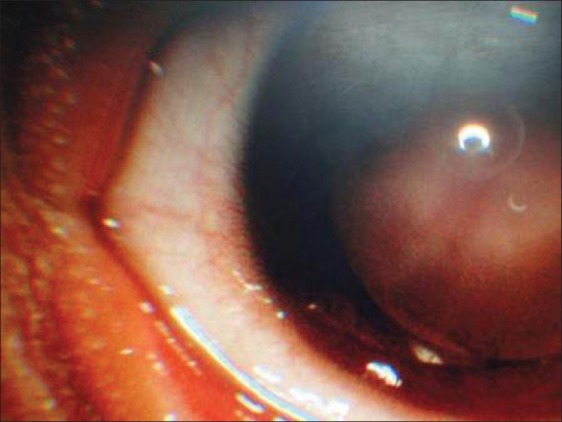
Slit lamp examination showing vascularising keratitis
Otololaryngologic assessment showed bilateral severe mixed hearing loss for higher frequencies.
Routine investigations in the form of complete hemogram, liver function test, renal function test, and chest X-ray were normal. A biopsy specimen from the lower leg showed hyperkeratosis, follicular plugging, and thickening of the granular layer.
Diagnosis of KID syndrome was entertained. Hearing aids, photochromatic lenses, and emollients were given to the patient.
DISCUSSION
KID syndrome was first reported in1915 by Burns as a generalized congenital keratoderma with ocular and mucosal involvement, but the acronym KID syndrome was coined in1981 by Skinner et al. to highlight the main features of the syndrome.[3] Caceres-Rios et al., analysed approximately 70 cases, most of them were sporadic, had been reported and found all of them showing cutaneous and auditory abnormalities, 90% sensorineural deafness, 89% erythrokeratoderma, 79% alopecia, 41% reticulated hyperkeratosis of the palms and soles, and 95% had ophthalmologic defects, most of them (79%) had vascularizing keratitis. They proposed the name keratodermatous ectodermal dysplasia, as the KID acronym does not accurately define this entity and the skin condition does not always show ichthyosis, but rather keratodermatous skin.[1]
Both an autosomal dominant form and an autosomal recessive form have been described, but numerous sporadic cases have also been reported. Heterozygous missense mutations in the GJB2 gene localized on chromosome 13q11-q12 encoding a gap junction protein called connexin-26 were found to be associated with the KID syndrome.[2] Patients are more susceptible to chronic cutaneous bacterial and mycotic infections. These contribute to alopecia, nail dystrophy, and body odor. Patients with KID syndrome are susceptible to severe sepsis and deaths are reported with increased frequency in these patients.[4] An increased oncogenic potential with invasive squamous cell carcinoma (SCC) arising within the hyperkeratotic lesions has been reported in several KID patients.[5] Other disorders as hypohidrosis, dermoskeleton dystrophies, cerebellar hypoplasia, Hutchinson's triad symptoms, cryptorchidism, and peripheral neuropathy may be associated. Cases of carotenemia, generalized cytomegalia infection, malignant histiocytoma, and hair follicle tumours have been described.[6] Hearing loss, mainly sensorineural, is always present, with variable degrees of compromise. It is congenital but is usually detected during infancy or early childhood and has almost always developed by age 7 years. Because of sensorineural deafness, speech development is usually delayed.[7] In previously published research, the main eye symptoms of the KID syndrome are photophobia, irritation, and visual disturbance due to vascularizing keratitis. The eye lesions of the KID syndrome express later than the other alterations, and they may not evolve with symptoms until puberty. Corneal vascularization, which expresses bilaterally but asymmetrically, is very frequent (in more than 80% of cases). Recurring corneal erosions, corneal leucomae, meibomitis, and severe dry eye syndrome are other eye manifestations of KIDS.[8] Intellect is unaffected but the combined disabilities of deafness, blindness, and disfigurement impose severe limitations and hardship on the individual.[6] The closest differential diagnosis of KID syndrome is Ichthyosis Follicularis with Aleopecia and Photophobia (IFAP). There is no palmoplantar keratoderma in IFAP and hearing is normal.[9] Erythrokeratoderma is a manifestation of KID syndrome while photosensitivity is seen in IFAP.
Early diagnosis of KID syndrome is important, and the timely use of hearing aids and speech therapy can prevent damages in the development of speech. Even though the general prognosis seems good, the life-long follow-up of patients is necessary because the KID syndrome can be associated to malignant tumors, particularly SCC.[8] Recently, the follicular occlusion triad (dissecting cellulitis of the scalp, cystic acne, and hidradenitis suppurativa), follicular tumors, and SCCs have been increasingly recognized as manifestations of KID syndrome.[4,5]
Management of KID syndrome requires multidisciplinary care by dermatology, ophthalmology, and otolaryngology services. Topical retinoids and keratolytics have limited benefits. There are several reports of substantial improvement in hyperkeratosis with acitretin therapy, and isotretinoin has variable efficacy in treating the follicular occlusion triad in KID patients.[5,7] Oral antibiotics and antifungal medications are necessary when infections develop and prolonged courses are occasionally required. Close monitoring of the skin and oral mucosa for the development of malignancy is essential. Lubricating and antiinflammatory agents have variable success in managing ocular disease, and cochlear implants have restored hearing in several affected individuals.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Caceres-Rios H, Tamayo-Sanchez L, Duran-Mckinster C, de la Luz Orozco M. Keratitis, ichthyosis, and deafness (KID syndrome): review of the literature and proposal of a new terminology. Pediatr Dermatol. 1996;13:105–13. doi: 10.1111/j.1525-1470.1996.tb01414.x. [DOI] [PubMed] [Google Scholar]
- 2.Maintz L, Betz RC, Allam JP, Wenzel J, Jaksche A, Friedrichs N, et al. Keratitis-ichthyosis-deafness syndrome in association with follicular occlusion triad. Eur J Dermatol. 2005;15:347–52. [PubMed] [Google Scholar]
- 3.Skinner BA, Greist MC, Norins AL. The keratitis, ichthyosis, and deafness (KID) syndrome. Arch Dermatol. 1981;117:285–9. [PubMed] [Google Scholar]
- 4.Gilliam A, Williams ML. Fatal septicemia in an infant with keratitis, ichthyosis, and deafness (KID) syndrome. Pediatr Dermatol. 2002;19:232–6. doi: 10.1046/j.1525-1470.2002.00075.x. [DOI] [PubMed] [Google Scholar]
- 5.Van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis-ichthyosis-deafness syndrome. J Invest Dermatol. 2002;118:724–7. doi: 10.1046/j.1523-1747.2002.01735.x. [DOI] [PubMed] [Google Scholar]
- 6.Maestrini E, Korge BP, Ocaña-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, et al. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237–43. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- 7.Langer K, Konrad K, Wolff K. Keratitis, ichthyosis and deafness (KID) syndrome: Report of three cases and a review of the literature. Br J Dermatol. 1990;122:689–97. doi: 10.1111/j.1365-2133.1990.tb07292.x. [DOI] [PubMed] [Google Scholar]
- 8.Gómez-Faiña P, Ruiz-Viñals AT, Buil-Calvo JA, España-Albelda A, Pazos-López M, Castilla-Céspedes M. Patient with severe corneal disease in KID syndrome. Arch Soc Esp Oftalmol. 2006;81:225–7. doi: 10.4321/s0365-66912006000400010. [DOI] [PubMed] [Google Scholar]
- 9.Traboulsi E, Aaked N, Megarbane H, Megarbane A. Ocular findings in Ichthyosis follicularis, alopecia, photophobia (IFAP) syndrome. Ophthalmic Genet. 2004;25:153–6. doi: 10.1080/13816810490514405. [DOI] [PubMed] [Google Scholar]