

Abstract
Genetically encoded calcium indicators (GECIs) are powerful tools for systems neuroscience. Recent efforts in protein engineering have significantly increased the performance of GECIs. The state-of-the art single-wavelength GECI, GCaMP3, has been deployed in a number of model organisms and can reliably detect three or more action potentials in short bursts in several systems in vivo. Through protein structure determination, targeted mutagenesis, high-throughput screening, and a battery of in vitro assays, we have increased the dynamic range of GCaMP3 by severalfold, creating a family of “GCaMP5” sensors. We tested GCaMP5s in several systems: cultured neurons and astrocytes, mouse retina, and in vivo in Caenorhabditis chemosensory neurons, Drosophila larval neuromuscular junction and adult antennal lobe, zebrafish retina and tectum, and mouse visual cortex. Signal-to-noise ratio was improved by at least 2- to 3-fold. In the visual cortex, two GCaMP5 variants detected twice as many visual stimulus-responsive cells as GCaMP3. By combining in vivo imaging with electrophysiology we show that GCaMP5 fluorescence provides a more reliable measure of neuronal activity than its predecessor GCaMP3. GCaMP5 allows more sensitive detection of neural activity in vivo and may find widespread applications for cellular imaging in general.
Introduction
Calcium is a ubiquitous second messenger, playing an essential role in excitable cells and signal transduction. Calcium ions (Ca2+) enter neurons during action potential (AP) firing and synaptic input. AP firing and synaptic inputs can therefore be assessed, sometimes quantitatively, by measuring changes in intracellular [Ca2+] (Yasuda et al., 2004). Genetically encoded calcium indicators (GECIs) (Mank and Griesbeck, 2008) and small molecule calcium-sensitive dyes (Cobbold and Rink, 1987) are both used to report [Ca2+] changes; but GECIs have the advantage that they enable chronic, noninvasive imaging of defined cells and compartments (Mao et al., 2008). State-of-the-art GECIs include the Förster resonance energy transfer (FRET) indicators D3cpVenus (D3cpV) (Palmer et al., 2006), TN-XXL (Mank et al., 2008), and YC3.60 (Nagai et al., 2004), and the single-wavelength sensor GCaMP3 (Tian et al., 2009). GCaMP3 is based on circularly permuted green fluorescent protein (cpGFP), calmodulin (CaM), and the Ca2+/CaM-binding “M13” peptide (M13pep). Several versions of the original GCaMP sensor (Nakai et al., 2001) have been published (Ohkura et al., 2005; Tallini et al., 2006; Akerboom et al., 2009). Recent versions include “GCaMP4.1” (Shindo et al., 2010), which was used to image Xenopus gastrulation, but no sequence information or comparison with other GECIs is published. “GCaMP-HS” consists of GCaMP2 with a subset of the “superfolder GFP” mutations (Pédelacq et al., 2006) and was used for imaging zebrafish motor neurons (Muto et al., 2011), but was also not compared with other sensors. The “G-GECO” sensors (Zhao et al., 2011) were created from GCaMP3 by random mutagenesis; they show ∼2× greater fluorescence increase in purified protein (Ca2+-saturated vs Ca2+-free) but were not tested in neurons. However, the sensors are dimmer than GCaMP3 in both the Ca2+-free and Ca2+-bound states, which can complicate imaging.
GCaMP3 has been used to detect activity in large neuronal populations in the motor cortex (Tian et al., 2009), barrel cortex (O'Connor et al., 2010), and hippocampus (Dombeck et al., 2010) of behaving mice. Long-term imaging of GCaMP3 has revealed learning-related circuit changes in vivo (Huber et al., 2012). GCaMP3 imaging has also been used to probe dendritic excitation in layer 5 dendrites in vivo (Xu et al., 2010; Mittmann et al., 2011), light-evoked responses in populations of neurons in mouse retina (Borghuis et al., 2011), zebrafish tectum (Del Bene et al., 2010), and walking Drosophila (Chiappe et al., 2010; Seelig et al., 2010), among others. However, GCaMP3 remains an imperfect GECI. Single APs are not reliably detected in vivo, and detection of active cells lags synthetic indicators (Tian et al., 2009). GCaMP5 was engineered from GCaMP3 using a combination of structure-guided design (Akerboom et al., 2009) and semirational library screening. We have enhanced sensitivity by increasing the dynamic range of the fluorescence response (ΔF/F = (F − F0)/F0)), the Ca2+-saturated brightness, and the Ca2+ affinity. We characterized 12 new GCaMP5s in vitro and in vivo under a wide variety of conditions. Each sensor has improved properties relative to the parent sensor GCaMP3. The ideal GCaMP5 indicator for a given study can be selected from this set according to particular experimental requirements.
Materials and Methods
Mutagenesis.
Site-directed mutagenesis of GCaMP3 was carried out using the method of Kunkel (1991) or the QuikChange methodology (Agilent Technologies). Single-stranded uracil-containing DNA template of pRSET-GCaMP3 for Kunkel mutagenesis was produced according to established protocols. Mutants were confirmed by DNA sequencing.
Animal use: All experiments were conducted according to protocols approved by the Institutional Animal Care & Use and Institutional Biosafety Committees of the Howard Hughes Medical Institute, Janelia Farm Research Campus, and of the corresponding committees at the other institutions.
Escherichia coli lysate screen.
Libraries were generated using primers containing degenerate codons (NNS) following the site-directed mutagenesis protocols described above and transformed into E. coli XL1-Blue (Stratagene/Agilent). The following day, colonies were scraped off plates, combined, and the plasmid library was isolated with Qiagen Miniprep kits, following the procedures provided, eluting in 100 μl water. One microliter of the library was subsequently transformed into E. coli BL21 (DE3) (EMD4 Biosciences), plated on 244 × 244 mm square LB-agar plates containing 100 μg/ml ampicillin, and grown for 20 h at 30°C. Colonies were selected using a colony picker (QPix2XT; Genetix) and grown in 800 μl ZYM-5052 medium (Studier, 2005) containing 100 μg/ml ampicillin in 96 deep-well blocks for 48 h at 30°C, shaking vigorously at 700 rpm. Two microliters was taken from each well and mixed into a fresh deep-well block containing 800 μl LB medium + 100 μg/ml ampicillin, grown overnight at 37°C, pelleted, and stored at 4°C for sequence analysis. The E. coli BL21 (DE3) cultures in the deep-well blocks containing the overexpressed mutants were pelleted by centrifugation (4000 × g, 20 min, 4°C), frozen, thawed, resuspended in lysis buffer (20 mm TRIS, pH 8.0, 100 mm NaCl, 1 mg/ml lysozyme, 1.5 Kunitz units/ml DNAseI from Qiagen) and subsequently incubated at 30°C for 2–4 h, shaking. Lysates were clarified by centrifugation (4000 × g, 30 min, 4°C), and 100 μl was taken from each well into Greiner Bio-One black 96-well fluorescence plates (Greiner) in duplicate. To one plate 1 μl 100 mm CaCl2 was added (final Ca2+ concentration ∼1 mm), to the other 1 μl 100 mm EGTA, pH 7.4. Fluorescence was measured in a Tecan Sapphire2 Spectrophotometer (Tecan), at 485 nm excitation and 510 nm emission, 5 nm slits, gain = 90 V.
Protein expression and purification.
Cloning, expression in E. coli, and purification was performed essentially as before (Akerboom et al., 2009). Briefly, for expression in BL21 (DE3), pRSETa-GCaMP variants were transformed to BL21 (DE3), and single colonies were grown for 3 d in ZYM-5052 media (Studier, 2005) at 25°C, after which cells were pelleted by centrifugation. Cells were lysed by resuspending them in 4 × w/v lysis buffer 2 (20 mm TRIS.HCl, pH 8.0, 100 mm NaCl), followed by a freeze–thaw cycle and subsequent cell rupture by passing the cell suspension three times through a precooled (4°C) Avestin Emulsiflex-C5 (Avestin) and finally a 15 s sonication step at 30 mW amplitude on ice (Fisher Dismembrator Model 100 equipped with 3 mm tip). Lysate was clarified by centrifugation (30,000 × g, 4°C, 45 min.), and cell-free extract was incubated on a rotary incubator with 5% (v/v) Profinity IMAC Ni-NTA resin (Bio-Rad) at 4°C for 4–16 h. Resin was allowed to settle in 20 ml disposable columns (Bio-Rad), and was washed with 20 ml lysis buffer 2, followed by 10 ml wash buffer (20 mm TRIS.HCl, pH 8.0, 100 mm NaCl, 10 mm imidazole). Proteins were eluted into elution buffer (20 mm TRIS.HCl, pH 8.0, 100 mm NaCl, 300 mm imidazole) and subsequently dialyzed extensively into lysis buffer 2 using Spectra/Por membrane tubing (Spectrum Laboratories), with a molecular weight cutoff (MWCO) of 10,000 Da, at 4°C. Concentration and purity was determined using Agilent Protein 230 chips on an Agilent 2100 Bioanalyzer (Agilent Technologies) as well as NuPAGE Novex BIS-TRIS SDS-PAGE gels (Invitrogen). Proteins were concentrated when necessary using Millipore Centrifugal Filter Units of 10,000 Da MWCO (Millipore).
pH titrations.
Five microliters purified GCaMP solution (∼100 μm) was added to 100 μl premixed, pH titrated, triple-buffer (10 mm glycine, 10 mm sodium citrate, 10 mm TRIS.HCl) containing either 3 mm BAPTA or 1 mm CaCl2, in duplicate. Fluorescence was measured in 96-well Greiner Bio-One transparent fluorescence plates in a Tecan Sapphire2 Spectrophotometer (Tecan), at 485 nm excitation and 510 nm emission, 5 nm slits, gain = 90 V.
Excitation/emission spectroscopy.
Five microliters purified GCaMP solution (∼100 μm) was added to 100 μl of either zero-free calcium buffer containing the following (in mm): 10 EGTA, 100 KCl, and 30 MOPS, pH 7.2 or 39 μm free calcium buffer containing the following (in mm): 10 CaEGTA, 100 KCl, and 30 MOPS, pH 7.2 (Invitrogen). Excitation spectra were taken from 240 to 550 nm, emission was 600 nm. Emission spectra were taken from 470 to 700 nm, excitation wavelength was 430 nm (gain = 80 V, 5 nm slit widths). Spectra were normalized to the calcium-saturated excitation and emission maxima, respectively. For absorbance measurements, 200 μl protein was dialyzed into 20 mm TRIS.HCl, pH 8.0, 100 mm NaCl, using Slide-a-Lyzer mini dialysis units with a 2000 Da MWCO (Pierce). The absorbance of 100 μl protein with 2 μl of either 100 mm CaCl2 or 100 mm EGTA added was measured from 240 to 700 nm, blanked against the chemically identical dialysis buffer containing 2 μl of 100 mm CaCl2 or 2 μl 100 mm EGTA. Spectra were normalized using the absorbance at 280 nm.
Calcium titrations.
Calcium affinity assays were performed by mixing different volumes of the zero-free calcium buffer containing the following (in mm) 10 EGTA, 100 KCl, and 30 MOPS, pH 7.2 and 39 μm free-calcium buffer containing the following (in mm): 10 CaEGTA in 100 KCl, and 30 MOPS, pH 7.2 from the calcium calibration buffer kit (Invitrogen) according to the manufacturer's instructions. Measurements were performed by mixing 3 μl purified GCaMP (∼100 μm) with 100 μl of different ratios of zero-free calcium buffer and 39 μm free-calcium buffer (Invitrogen) in 96-well transparent Greiner Bio-One plates and measuring the fluorescence at 485 nm excitation and 510 nm emission, 5 nm slits, gain = 90 V in duplicate as described earlier.
GCaMP photophysics.
Photophysical properties for GCaMP2, GCaMP3, and the GCaMP5 variants A, D, and G were investigated in buffer solutions in the presence or absence of free Ca2+. Absorption and emission properties, including quantum yield, were determined using a UV/VIS spectrometer (Lambda 35; PerkinElmer) and a fluorescence spectrometer (LS-55; PerkinElmer). Two-photon-excited properties, including fluorescence excitation spectra, two-photon cross section, fluorescence decay time, and emission spectroscopy, were conducted with laser pulses from an 80 MHz Ti:Sapphire laser (Chameleon Ultra II; Coherent). In all cases, near-IR laser pulses were focused into a solution containing the GCaMP proteins via a 60×, 1.2 NA water-immersion objective of an inverted epifluorescence microscope (IX81; Olympus), slightly overfilling the objective back aperture. Fluorescence generated by two-photon excitation was collected by the same 60× objective used to excite the proteins, and after passing through a shortpass filter (FF01–720SP; Semrock) and a bandpass filter (FF01–550/88; Semrock), was directed to the input face of a fiber-coupled avalanche photodiode (APD). Two different APDs were used, one optimized for speed for lifetime measurements (model PDF CCTB; Micro Photon Devices), and one optimized for low noise for fluorescence correlation spectroscopy (FCS) and spectral measurements (SPCM-AQRH-14-FC; PerkinElmer) coupled to a 100 μm core multimode fiber (AFS105/125Y; Thorlabs). Output pulses generated by the fast-timing APD were fed to a TCSPC board (TimeHarp200; Picoquant). Output pulses from the low-noise APD were fed to an external autocorrelator (Flex03LQ-01; www.correlator.com) for spectra and FCS measurements. Emission spectra were recorded from an additional microscope port using a fiber-coupled 0.3 m spectrograph/CCD (model SP2358 with Pixis 256 CCD camera; Princeton Instruments).
Control of the amount of laser power delivered to the sample in the focal plane of the microscope was accomplished by rotating the linear polarization of the laser output using an achromatic ½-wave plate (AHWP05M-980; Thorlabs), mounted in a computer-controlled rotation stage, followed by passing this beam through an angle-fixed Glan-Laser polarizer (GL10-B; Thorlabs). We calibrated the system before measurements, where for each laser wavelength (in 10 nm increments), the laser power at the focus of the microscope objective was measured while the ½-wave plate was angle adjusted, calibrating at each wavelength a specific power at the focus with a specific orientation of the wave plate. Laser wavelength and power adjustments, as well as data acquisition, were run under computer control. Data analysis was performed using Origin 8.0 software (OriginLab), and FCS fits were performed using a custom software package (V. Iyer, unpublished).
Samples were prepared by dilution from stock solutions of purified proteins into buffers, at either pH 7.25 or pH 9.5, to characterize the fluorescence properties under physiological pH and at elevated pH where most of the protein chromophores are in the deprotonated form in the presence of Ca2+. The pH 7.25 buffers (30 mm MOPS, 100 mm KCl) were either purchased as part of a Ca2+-calibration kit (C-3008MP; Invitrogen), that contains either 10 mm K2EGTA or 10 mm CaEGTA, or prepared from MOPS and KCl stock solutions (Mediatech) and contain either 1 mm CaCl2 or 0.5 mm EGTA or 1 mm BAPTA. The pH 9.5 buffer (30 mm CHES, 100 mm KCl) was prepared from stock chemicals and was supplemented with either 1 mm CaCl2 or 0.5 mm EGTA. For measurements using very low amounts of protein (below 200 nm) such as FCS measurements and one-photon fluorescence measurements, we added 0.1 mg/ml bovine serum albumin (BSA) as a blocking agent to the buffer solutions to prevent loss of proteins from solution to the nearby glass or coverslip surfaces via adsorption over the course of the measurements; this eliminated any decrease in fluorescence signal over the course of the measurements.
Protein concentration determination for biophysical analysis.
The concentration of chromophore-forming proteins was determined by two methods: alkali-denaturation (Ward, 2005), and two-photon-excited FCS. For both methods, absorption spectra of either enhanced GFP (EGFP) or GCaMP protein solutions were taken in the UV/VIS spectrometer at either pH 7.25 or pH 9.5. In the second method using FCS, GCaMP stock solutions were diluted several thousand fold into pH 9.5 buffer (30 mm CHES, 100 mm KCl) containing 1 mm CaCl2, and excited at 960 nm in a fluorescence microscope over a range of laser powers. At each laser power, the mean fluorescence rate <F> and its fluctuations were recorded for 50–200 s, and the autocorrelation G(τ) of the fluorescence signal was computed. A fit to the autocorrelation function G(τ), based on a diffusion model, determines the diffusion coefficient of the proteins, and the mean number of fluorophores in the excitation volume, given by <N> = 1/G(τ →0) (Schwille et al., 1999). This measurement is repeated for a sample containing EGFP at known concentration (determined by alkali denaturation) and dilution in pH 9.5 buffer, providing a reference between a known concentration and a number of fluorophores in the excitation volume.
Quantum yield.
Quantum yield (QY) was determined for the fluorescent proteins in both pH 7.25 and 9.5 buffer in the presence of 1 mM CaCl2 using standard methods that measure the optical absorption and total fluorescence yield of samples at a fixed wavelength for both sample and a standard fluorescein with a QY of 0.93 in aqueous 0.1 N NaOH (Magde et al., 2002) with approximately the same emission spectrum and emission peak.
Fluorescence decay and lifetime.
The fluorescence lifetime was measured by time-correlated single-photon counting (TCSPC) using two-photon excitation at 960 nm in a fluorescence microscope setup, where detector pulses from the fast-timing APD and trigger signals from a PIN diode monitoring the laser pulse train were fed to the TCSPC board. To achieve improved performance, a pulse-picker (Model 350–160; Conoptics) was inserted in the beam to reduce the laser pulse frequency from 80 to 20 MHz. The fluorescence lifetime of GCaMP samples was determined in either pH 9.5 buffer supplemented with CaCl2 or EGTA as described earlier, or pH 7.25 buffer (Invitrogen buffer with either 10 mm CaEGTA or 10 mm EGTA). The lifetime reference for the system was fluorescein (e−1 lifetime τ = 4.1 ± 0.1 ns); in our setup the fluorescein decay was well fit to a single-exponential decay (4.0 ns, χ2 = 1.08). Measured fluorescent decays were fit to a single-exponential decay curve, or to a two-exponential decay, which improved the goodness-of-fit.
Two-photon excitation spectra.
Two-photon excitation spectra and ΔF/F were measured for the GCaMPs in the presence and absence of free calcium at pH 7.25 and 9.5, respectively, as described above, at 1 μm protein concentration. Two-photon spectra are taken with constant laser power delivered to the sample, although due to a wavelength-dependent pulse width of the femtosecond pulses, and changes in focal spot size (focused beam diameter scales as the excitation wavelength), the laser intensity varies gradually across the spectrum. We do not correct for this variation in intensity. Together with each run of GCaMP samples, a reference two-photon excitation spectrum of fluorescein was recorded, allowing us to determine the absolute two-photon cross section of the GCaMPs using published cross sections measured for these fluorophores (Xu and Webb, 1996; Drobizhev et al., 2011).
Peak brightness per molecule.
Fluorophores can be characterized by their specific brightness under two-photon excitation, measured in counts per second per molecule, at a specific laser intensity and wavelength. This is the average fluorescence rate detected per fluorescent molecule, and is measured using two-photon-excited FCS. This quantity reaches a maximum or peak value as the laser intensity is increased, beyond which the fluorescence rate per molecule decreases with higher intensity, due to photobleaching of the fluorophores in the volume of the focused laser beam. While the peak brightness will strongly depend on the molecular environment (in vitro vs intracellular/in vivo), this value can be used as a quantitative measure in comparing the photostability of different fluorophores.
To determine the peak brightness, GCaMPs were diluted to nominally 50 nm in pH 9.5 buffer containing either 1 mm CaCl2 or 0.5 mm EGTA. Solutions also contained 0.1 mg/ml BSA to prevent adsorption of fluorophores to the nearby coverslip surface. As a control, EGFP at 50 nm was prepared and measured in the same buffer, without CaCl2 or EGTA. Measurements were taken for a series of laser powers (with power measured in the focal plane) at 940 nm, where fluorescence time course data was acquired for 50–200 s at each laser intensity. For FCS, we used the low-noise APD. The output of the APD was fed to an autocorrelator and associated software to generate two quantities: the time-average fluorescence rate <F> and the measured autocorrelation G(τ) of the fluorescence data. FCS theory equates the quantity 1/G(0) to <N>, the average number of emitting molecules in the excitation volume. By acquiring <F> and G(t) simultaneously for each protein over a range of laser intensities, and fitting G(0) to determine <N>, we can define the two-photon brightness at each intensity as <F>/<N>, the effective detected fluorescence rate per emitting molecule at each intensity. This quantity has a maximum value or peak brightness, since as the intensity is raised, saturation and photobleaching begin to diminish the fluorescence rate.
Intensity dependence of ΔF/F under two-photon excitation.
Without using FCS, GCaMPs can be characterized by the power dependence of their (ΔF/F)max. For these measurements, GCaMPs were diluted to 0.5 μm in pH 7.25 buffer ± Ca2+ and fluorescence recorded for laser excitation at 940 nm under increasing power over the range of 0.5 mW–70 mW.
Human embryonic kidney cell assay.
For expression in human embryonic kidney (HEK) 293 cells, DNA was PCR amplified from the pRSETa-GCaMP construct, purified, and digested with BglII and NotI (NEB), and ligated into digested pEGFP-N1 (Takara-Bio), which had been digested with BglII and NotI to remove EGFP, resulting in pCMV-GCaMPs. Successful clones, with the EGFP gene replaced with the gene coding for GCaMP, were confirmed by sequence analysis, and plasmids were prepared using the endo-free plasmid Maxi kit (Qiagen). Equal amounts of plasmid for each GCaMP5 variant were transfected into HEK293 cells using the 96-well Nucleofector protocol in Amaxa plates (Lonzo), with each variant in 16 wells for eight duplicate measurements. Cells were grown and incubated at 37°C for 2 d, after which growth medium was aspirated and replaced with 100 μl prewarmed (37°C) cell buffer (1× TBS, 2 mm CaCl2). Whole-well fluorescence response traces evoked by addition of 100 μl acetylcholine in cell buffer (dilution series 10−2−10−10 m) were recorded by the liquid handling 96-well Hamamatsu FDSS.
Adeno-associated virus production.
For cloning in pAAV, DNA was PCR amplified from pCMV-GCaMP clones, purified and digested with BamHI and HindIII (NEB), gel-purified, and ligated into predigested pAAV-humanSynapsin-1 vector. Ligations were transformed into E. coli Stbl2 (Invitrogen) and plates were incubated for 2 d at 30°C. Insertions were verified by DNA sequencing. Virus was produced by standard protocols.
Construct availability: constructs and AAV viruses for GCaMP5G are available from AddGene (http://www.addgene.org) or the University of Pennsylvania Vector Core (http://www.med.upenn.edu/gtp/vector_core.shtml), respectively. The Janelia Farm GECI Project website (http://www.janelia.org/team-project/geci-project) has additional information about construct characterization and availability.
Crystal structure determination.
GCaMP proteins were expressed in E. coli BL21 (DE3) using the pRSETa plasmid and purified by immobilized metal ion affinity and size exclusion chromatography as described (Rodríguez Guilbe et al., 2008; Akerboom et al., 2009). Proteins were concentrated to 5–10 mg/ml for crystallization as described earlier. Crystallization was carried out at room temperature by sitting-drop vapor diffusion using commercially available sparse-matrix screens (Hampton Research) by mixing 1.2 μl of protein solution with 1.2 μl of precipitant in 96-well plates. Crystals were cryoprotected before x-ray data collection. X-ray data were reduced using Mosflm (Leslie, 1992)/Scala (Collaborative Computational Project, Number 4, 1994) or D*Trek (Pflugrath, 1999) (see Table 2). Each structure was solved by molecular replacement using Phaser (McCoy et al., 2007), or simple rigid body refinement starting from previously published GCaMP structures using the CCP4 package (Collaborative Computational Project, Number 4, 1994). Iterative cycles of model building in Coot (Emsley and Cowtan, 2004) and refinement in Refmac/CCP4 (Collaborative Computational Project, Number 4, 1994) led to the final models described in Table 2. Protein structures have been deposited in the Protein Data Bank (http://www.rcsb.org); accession codes are shown in Table 2.
Table 2.
Crystallization and structure determination of GCaMP variants; x-ray data collection and refinement statistics
| GCaMP2-T116V, D381Y | GCaMP5A | GCaMP5G | GCaMP5H | GCaMP2-LIA | GCaMP3-KF | |
|---|---|---|---|---|---|---|
| PDB ID | 3SG2 | 3SG3 | 3SG4 | 3SG5 | 3SG6 | 3SG7 |
| Oligomeric state | Monomer | Monomer | Monomer | Dimer | Dimer | Monomer |
| Data collection | ||||||
| Radiation source | APS 31-ID | APS 31-ID | APS 31-ID | APS 31-ID | Copper anode | ALS 8.20.2 |
| Wavelength (Å) | 0.9793 | 0.9793 | 0.9793 | 0.9793 | 1.5418 | 1.0000 |
| Space group | P41212 | P41212 | P41212 | C2 | C2 | P212121 |
| Unit cell dimensions | ||||||
| a, b, c (Å) | 119.6, 119.6, 96.9 | 120.4, 120.4, 98.0 | 120.2, 120.2, 97.4 | 128.6, 46.0, 67.4 | 129.0, 47.5, 68.7 | 57.4, 61.5, 124.0 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 100.2, 90 | 90, 99.7, 90 | 90, 90, 90 |
| Resolution (Å) | 23.8–2.0 (2.11–2.0) | 20.0–2.1 (2.21–2.1) | 19.7–2.4 (2.53–2.4) | 20.0–1.9 (2.0–1.9) | 28.0–1.7 (1.76–1.7) | 50.0–1.9 (1.97–1.9) |
| Rsym | 0.088 (0.574) | 0.132 (0.612) | 0.138 (0.670) | 0.088 (0.472) | 0.070 (0.468) | 0.068 (0.825) |
| I/σI | 20.8 (4.3) | 15.3 (4.9) | 14.7 (3.7) | 10.9 (3.5) | 15.9 (1.6) | 10.2 (2.0) |
| Completeness (%) | 99.9 (100) | 99.9 (100) | 99.8 (100) | 99.6 (100) | 93.9 (73.9) | 95.5 (95.6) |
| Redundancy | 13 (11.4) | 13.9 (13.4) | 13.7 (9.3) | 5.5 (5.5) | 6.5 (2.1) | 7.2 (7.1) |
| Refinement | ||||||
| Resolution (Å) | 2.00 | 2.10 | 2.40 | 1.90 | 1.70 | 1.90 |
| Unique reflections | 45514 | 40352 | 26965 | 29184 | 40438 | 32056 |
| Rwork/Rfree | 0.173/0.207 | 0.165/0.199 | 0.172/0.213 | 0.180/0.225 | 0.203/0.249 | 0.184/0.230 |
| No. atoms (B-factors (Å2)) | 3465 (33.0) | 3470 (33.6) | 3396 (36.5) | 3228 (19.8) | 3358 (19.6) | 3396 (21.2) |
| Protein | 3182 (32.6) | 3187 (33.4) | 3167 (36.5) | 3089 (18.9) | 3082 (18.3) | 3206 (20.1) |
| Ligand/ion | 4 (27.5) | 4 (31.1) | 4 (33.5) | 30 (49.6) | 4 (30.4) | 4 (34.7) |
| Water | 279 (36.7) | 279 (35.9) | 225 (37.0) | 109 (34.5) | 272 (35.2) | 186 (39.6) |
| RMSD values | ||||||
| Bond lengths (Å) | 0.027 | 0.028 | 0.024 | 0.022 | 0.025 | 0.024 |
| Bong angles (°) | 2.02 | 2.09 | 1.91 | 1.90 | 2.02 | 1.98 |
| Ramachandran plot | ||||||
| Favored/outliers (%) | 97.7/0.3 | 95.9/0.3 | 95.6/0.5 | 99.2/0 | 98.7/0 | 97.7/0.3 |
Structures of GCaMP variants have been deposited in the Protein Data Bank (http://www.pdb.org/).
Size exclusion chromatography.
To check the extent of in vitro dimerization of GCaMP5A, GCaMP5B, and GCaMP5G compared with GCaMP2 and GCaMP3, an aliquot of each purified mutant was adjusted to 150 μm in lysis buffer 2, after which EGTA was added to 1 mm final concentration. This mixture was allowed to equilibrate at room temperature for 30 min, after which CaCl2 was added to 2.5 mm final concentration. Aliquots (100 μl) of each GCaMP were then injected onto a Superdex 200 10/300 GL column (GE Healthcare) with 20 mm Tris, pH 8.0, 100 mm NaCl, and 2 mm CaCl2 as the running buffer, and protein was eluted at a flow rate of 0.5 ml/min.
Hippocampal neuronal culture imaging.
Primary cultures of hippocampal neurons were obtained from P0 rat pups by dissection, papain-based dissociation, and plating onto Matrigel-coated (BD Biosciences), 24-well glass-bottom plates (MatTek) and then cultured in DMEM/B27 medium (Invitrogen). SIV-based lentiviral vectors containing hsyn1-GCaMP variant-IRES-nls-mCherry-WPRE-SV40 polyadenylation signal (hsyn1: human synapsin-1 promoter) constructs were produced through quadruple transfection of HEK293T cells in 10 cm plates. Harvested lentiviral particles were used to infect hippocampal neuronal cultures on the third day in vitro for 16 h, and medium was replaced with DMEM/B27/4 μM AraC (Invitrogen and Sigma). On day 16–18 in vitro, infected neurons were stimulated using a custom-built, 24-well multiplexed field stimulator with platinum wires and imaged using an Olympus IX81 motorized, inverted microscope (10× objective, 0.4 NA, Chroma ET-GFP or ET-TxRed filter sets) Prior Scientific H117 ProScanII motorized stage; Cairn Research optical feedback OptoLED illumination system; and an EMCCD camera (Andor iXon+ 897, 34.8 frames per second). Field stimuli were delivered at 40 V, 83 Hz, 1 ms pulses for the following trains: 1, 2, 3, 5, 10, 20, 40, 80, and 160 field stimuli. The whole system was automated using MetaMorph (MM; Molecular Devices) and MATLAB (MathWorks) software. Imaging buffer included the following (in mm): 145 NaCl, 2.5 KCl, 10 glucose, 10 HEPES, pH 7.4, 2 CaCl2, 1 MgCl2, 0.01 3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (Tocris Bioscience), 0.01 6-cyano-7-nitroquinoxaline-2,3-dione (Tocris Bioscience), 0.01 gabazine (Tocris Bioscience), and 1 α-methyl-4-carboxyphenylglycine (Tocris Bioscience). Images were processed and analyzed using custom software.
Astrocyte imaging.
Methods were identical to those described previously (Shigetomi et al., 2010b). Briefly, we used an Olympus IX71 microscope equipped with an IXON DV887DCS EMCCD camera (Andor), epifluorescence condenser, control unit, and Polychrome V monochromator (TILL Photonics). The control of excitation and image acquisition was achieved using TILLVision software. We used an Olympus 60× 1.45 NA objective lens. Images were typically taken every 1 s. Exposure time and pixel binning were optimized to visualize fluorescence signals for each experiment (maximum binning was 4 × 4). Cultures were perfused with recording buffer containing the following (in mm): 110 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 10 d-glucose, and 10 HEPES at pH 7.4 (adjusted with NaOH).
For imaging astrocyte activity in neuron/astrocyte cocultures, Lck-GCaMP3 and Lck-GCaMP5G were subcloned into an AAV vector driven by the astrocyte-specific GFAP promoter(Brenner et al., 1994). Neuron-astrocyte coculture was infected at day 3 in vitro and cells were imaged 10 d postinfection.
Mouse retina in vitro imaging.
Methods were identical to Borghuis et al. (2011).
Caenorhabditis elegans imaging.
The same experimental protocol was used as in previous GCaMP experiments (Tian et al., 2009). All recordings are from the AWCon sensory neuron. After 10 s of observation, odor ([IAA] was 10−4 v/v) was added and animals imaged for 50 s. Odor was delivered for 5 min; during the last 10 s of odor addition, and for 50 s after odor removal, animals were imaged to observe the neuron's response to odor removal (which activates the AWCon neuron).
Expression levels of C. elegans transgenes from extrachromosomal arrays can show considerable animal-to-animal variation, complicating analysis of imaging results. To linearize the imaging measurements and improve comparisons across different expression levels, we first selected AWC pixels whose intensity exceeded nonfluorescent background, then calculated average pixel intensity.
Drosophila larval neuromuscular junction preparation.
To allow imaging access to transgenic presynaptic neuromuscular junction (NMJ) boutons that expressed variants of GCaMP3, third instar Drosophila larvae were dissected using methods similar to those described previously (Jan and Jan, 1976). Genetic constructs were prepared in the pJFRC7–20XUAS-IVS vector (Pfeiffer et al., 2010) and inserted in the VK00005 site (Venken et al., 2006) to allow expression in motor neurons using the OK6-Gal4 driver (Aberle et al., 2002). The combination of OK6-Gal4, VK00005, and pJFRC7–20XUAS-IVS provided good labeling of Type 1b and 1s, but not type II boutons in heterozygous animals (+/yellow1 white1118; +/OK6-Gal4; +/UAS-GCaMPxx). Actively wandering larvae were dissected in ice-cold Schneider's insect medium (Sigma), pH shifted to 7.2 using NaOH. A 35 mm Petri dish previously one-third filled with Sylgard (Dow Corning), was used to pin down the cuticle and body wall muscles. Utmost care was taken during dissection to reduce the potential for muscle movement during imaging (ensuring central longitudinal muscles were not contacted with instruments at any time, cuticle stretching was sufficient to keep the preparation in place but not so much to cause spontaneous calcium release, choosing pin insertion locations that minimized potential damage to imaging region). Imaging commenced within 15 min of the segmental nerves being cut close to the ventral nerve cord. Before imaging, the Schneider's insect medium was replaced with HL6 (Macleod et al., 2002) supplemented with 2 mm Ca2+, an osmolarity of ∼340 ± 10 mOsm, pH of ∼7.2 ± 0.05, and 7 mm l-glutamic acid added to reduce muscle movement (Macleod et al., 2004). During imaging, pH and temperature were monitored, with pH values ranging from ∼7.4–7.6 (start to end) while temperature stayed relatively constant (∼22 ± 0.5°C).
Drosophila larval NMJ stimulation parameters.
Cut segmental nerves were drawn by suction into a heat-polished glass pipette, ∼12 μm internal diameter (Macleod et al., 2002), which was connected to an ISO-Flex stimulator (A.M.P.I.) to deliver suprathreshold electrical pulses (∼2.9 V, ∼5 mA). After 3 s of baseline, each stimulus was 2 s in duration with individual 300 μs impulses delivered at a frequency of 1, 5, 10, 20, 40, 80, and 160 Hz. The interstimulus interval was ∼20 s and each stimulus frequency was repeated five times, with a pause of ∼35 s, before moving to a higher stimulus frequency. The stimulus order was kept constant as higher frequency stimuli were potentially deleterious and outside the normal range of firing frequencies (Chouhan et al., 2010).
Drosophila larval NMJ acquisition hardware and software.
Wide-field Ca2+ imaging was performed on an upright Olympus BX61-WI microscope using a 40× water-immersion objective (0.8 NA), a 2× lens extender (EX2C; Computar), and an Andor EMCCD camera (Model DU897 BV, 512 × 512 pixels, 30 FPS, 100 EMgain, 5.2× pregain, −70°C, 32.9 ms exposure; Andor Technology) with 7% illumination from a DC powered X-CITE exacte light source (Lumen Dynamics). The fluorescence filter set used was BrightLine 3035B (472/30, 495, 520/35; Semrock Rochester). The imaging system was controlled by custom-written journals in MM (version 7.7.5; Molecular Devices) while the stimulation and image timing was controlled (as a slave) by MATLAB (MathWorks) using a USB-6343, X Series DAQ (National Instruments) and custom-written software, Ephus (Suter et al., 2010).
Fields of view (FOVs) were exclusively from Muscle 13 due to its high native firing rate (Chouhan et al., 2010), using predominantly segment A4; however, segments A3 and A5 were used when four boutons (not including the tip bouton) were not simultaneously in focus or present. The left and right sides of the animal were taken as separate samples to increase throughput of data collection, with the second imaging session ∼20 min after the first. Once each FOV was selected and the corresponding nerve drawn into the glass pipette and connected to the stimulator, the MM controlled data acquisition.
An image-based auto-focusing routine (Adjust Focus) was used to find the optimal focus position using the Brenner algorithm based on nearest-neighbor pixel values (Brenner et al., 1976). MM was configured to start the camera trigger and simultaneously start the stimulus timer in Ephus via a USB X Series DAQ. Using a Uniblitz shutter (Model VS25S1ZM1R3–24; Vincent Associates) set into the light path and controlled by either MM or Ephus, 11 dark frames were collected (shutter closed) first, followed by 394 frames with an open shutter. The image-based auto-focusing routine was repeated between each of the five replicate trials for each stimulus frequency acquired, to account for any small movements in the z-axis.
Along with the image data, we collected analog signals for the temperature and pH of the bath, the current, and the voltage of the stimulus and the frame signals from the camera. Continuously monitoring temperature and pH enabled constant conditions across experiments for different calcium indicators. Recording the current, voltage and frame signals at 10 kHz enabled precise calculation of the timing of the calcium response.
Drosophila larval NMJ image analysis.
MATLAB was used for all analysis. We performed background subtraction before calculating fractional changes in fluorescence from baseline values (ΔF/F0). During the 15 min FOV acquisitions, we infrequently observed either slow XY drifts in the muscle position or actual muscle contractions, normally in neighboring muscles. This was despite 7 mm l-glutamic acid being present, which normally prevented muscle movement in acquisitions of <3 min (Macleod et al., 2004). In the rare cases that we observed noticeable changes in Z-position (<1:1000 trials), which were detected by changes in baseline fluorescence (F0), we discarded the data from the entire FOV. In order not to discard FOVs due to XY movement, we developed a custom MATLAB analysis routine that accounted for image movement between images in a stack (one stimulus, using cross-correlation; Guizar-Sicairos et al., 2008) and also between trials, by independently tracking each segmented region of interest (ROI) centroid, and moving the ROIs based on the centroid movement between trials. We only included data from each FOV if the ΔF/F0 continually increased with increasing stimulation and peaked at 80 Hz, as previously observed from intracellular recordings (Chouhan et al., 2010). When this was not observed, presumably due to muscle damage or overstretching during the dissection, the data from the entire FOV was discarded.
For each GCaMP construct, 10 FOV that met the quality control parameters described above were used. These FOV were collected from seven animals (i.e., more than one FOV collected from one animal). To describe the performance of an indicator, boutons within an FOV were averaged, replicate trials of a stimulus frequency were averaged, and the 10 FOV were averaged.
Preparation and odor delivery for adult Drosophila experiments.
Flies were reared on standard cornmeal agar medium. We used the Gal4/UAS system (Brand et al., 1994) to direct the expression of the calcium sensors to projection neurons (PNs). GH146-Gal4 flies were a gift from L. Luo (Stanford University, Stanford, CA). All animals were adult females, 3–5 d after eclosion. Adult flies were dissected using previously described methods (Jayaraman and Laurent, 2007). Flies were anesthetized in a vial on ice until movement stopped (<15 s) and then gently inserted into a hole in a piece of aluminum foil. Small drops of wax (55°C) were used to suspend the fly in the hole, with the edge of foil defining a horizontal plane around the head and thorax, from the first antennal segment anteriorly to the scutellum posteriorly. The dorsal side of the foil was bathed in saline, while the ventral side (including antennae and maxillary palps) remained dry and accessible to odors. A window was cut in the dorsal head cuticle between the eyes, extending from the ocelli to the first antennal segment. Fat and air sacs dorsal and anterior to the brain were removed, but the perineural sheath was left intact. The proboscis was affixed with a small drop of wax to a strand of human hair to limit brain movement. Spontaneous leg movements were typically observed in this preparation for the duration of the recording (2–3 h). The saline composition used in all olfactory experiments contained the following (in mm): 103 NaCl, 3 KCl, 5 N-tris(hydroxymethyl) methyl-2-aminoethane-sulfonic acid, 10 trehalose, 10 glucose, 26 NaHCO3, 1 NaH2PO4, 2.0 CaCl2, and 4 MgCl2, adjusted to 275 mOsm, pH 7.4.
Odors (different concentrations of octanol) were delivered using a custom-made odor-delivery system designed by Dmitry Rinberg, and a Teflon nozzle (entry diameter 1/8”) directed toward the antennae. Odors were delivered at different concentrations diluted in paraffin oil (paraffin oil alone, 0.001, 0.01, 0.1, 1.0, and 10%) in a constant stream of air (1 L/min) with an additional 10% dilution in air. For each concentration, five replicate deliveries were performed and the data averaged. Odor delivery times were measured using a mini-PID (Aurora Scientific). Odors were presented for 1 s. All comparisons of sensor performance were made using experiments with identical odor presentation times. The results reported are based on data obtained from five GCaMP3-expressing flies (six antennal lobes; ALs) and five GCaMP5-expressing flies (six ALs).
Calcium imaging in adult Drosophila.
We imaged on a two-photon microscope using PrairieView software and an Olympus 40×, 0.8 NA LUMPlanFL/IR objective. A mode-locked Ti:Sapphire Chameleon Ultra II laser (Coherent) tuned to 920 nm was used for excitation. Fluorescence was collected using photomultiplier tubes (Hamamatsu) after bandpass filtering. Images were acquired in frame scan mode (20 Hz) for a single plane of one AL. Fluorescence time series were then obtained by averaging across the spatial extent of the glomerulus in the frame. In all cases fluorescence changes were calculated relative to baseline fluorescence levels as determined by averaging >2 s just before odor presentation.
In vivo imaging of visually evoked calcium transients in larval zebrafish tectal neuropil.
mitfa−/− (nacre) zebrafish larvae (Lister et al., 1999) expressing Gal4 under the control of the pan-neuronal elavl3/HuC promoter (elavl3:Gal4) were injected at 1–8 cell embryonic stage with a solution containing 0.04% phenol red, 50 ng/μl transposase mRNA, and 25 ng/μl plasmid DNA with the coding sequence of GCaMP5A or GCaMP5G downstream of 14 upstream activation sequences (UASs) and flanked by two Tol2 sequences for stable genomic integration. An elavl3:Gal4, UAS:GCaMP3 stable line (Del Bene et al., 2010) was used for experiments with GCaMP3.
Six days postfertilization (dpf) larvae were embedded in 2% low melting point agarose and imaged at 7 dpf at 28°C. A custom-built two-photon microscope equipped with a mode-locked Ti:Sapphire Chameleon UltraII laser (Coherent) tuned to 920 nm and controlled by ScanImage v3.6 software (Pologruto et al., 2003) was used for acquiring image time series at 17 Hz. Visual stimuli (moving vertical gray bars on black background) were generated with Vision Egg software (Straw, 2008) and presented with an 800 × 600 pixel organic light-emitting diode (OLED; eMagin) with a green-light filter to the left eye of the larvae. Imaging was performed in the right (contralateral) tectal neuropil. Image time series were x-y motion-corrected with a program written in MATLAB (Dombeck et al., 2007) and analyzed with ImageJ.
In vivo two-photon imaging of visually evoked calcium transients in larval zebrafish tectal somata.
Five and 6 dpf AB/nacre larval zebrafish expressing GCaMP2, GCaMP3, or GCaMP5G under the elavl3 promoter were paralyzed by immersing them in 1 mg/ml solution of bungarotoxin dissolved in E3 fish embryo water and were subsequently embedded in 2% low melting point agarose in a 35 mm Petri dish. They were placed in a custom two-photon microscope and imaged using a Mai Tai HP Ti-Sapphire laser tuned to 950 nm. The visual stimulus used for the experiment consisted of a light dot (0.5 mm × 0.5 mm) projected, using an amber (590 nm) LED mounted into a miniature LCOS projector, onto an opal glass screen directly underneath the larvae. Stimulus light was filtered with a narrow bandpass filter. Each fish was run through one stimulus set with the laser off to detect stimulus bleed-through, which was always negligible. The dot appeared to the left or right of the larva and moved in a straight line at a speed of 3 mm/s until it disappeared on the opposite side. The larva was located in the middle of the dot's trajectory and positioned perpendicular to this trajectory, with the point of closest approach of the dot being 0.5 mm rostral to the larva.
The experimental protocol consisted of 1 min darkness, followed by a presentation every 30 s of the moving dot, alternating between left to right and right to left. There were 10 such presentations (5 in each direction). The experiment concluded with 1 min of darkness, and therefore lasted 7 min in total. Individual frames were captured at 138.32 ms per frame (7.23 Hz), using a quad-interlaced scan pattern that ensured that each cell was sampled evenly at four times this frame rate.
Movies were assessed for x-y drift during the experiment (usually <1 pixel), and a subpixel translation correction was applied using MATLAB software (David Heeger, New York University). Neuronal somata were detected based on their dark nuclei. Mean images were smoothed with a Gaussian, and local minima were detected. These were classified as cell nuclei if the ratio of the brightness 3 pixels from the center was >3.5 × the brightness 1 pixel from the center, i.e., they look like a bright ring around a dark center, and they were sufficiently bright (>17,500 photons detected per experiment). Fluorescence was then averaged over a 7 × 7 pixel square. Baseline fluorescence (F) was defined as the average fluorescence in the 50 frames immediately preceding each left-right stimulus.
In vivo imaging of visually evoked calcium transients in larval zebrafish retinal bipolar cell terminals.
All procedures were carried out according to the UK Animals (Scientific Procedures) Act 1986 and approved by the UK Home Office. Fish were maintained on a 14:10 h light/dark cycle at a temperature of 28°C and bred naturally. Transient expression of GCaMP2 or GCaMP5s was driven by constructs containing the Ribeye-A promoter (Dreosti et al., 2009), which targets ribbon synapses of sensory neurons. Constructs at a concentration of 0.084 μg/μl were injected into embryos at the 1–4 cell stage. To inhibit melanin formation, larvae were kept in 200 μm 1-phenyl-2-thiourea (Sigma) from 28 h postfertilization. Imaging was carried out as described previously (Dreosti et al., 2009). Briefly, whole zebrafish larvae (9–11 dpf) were immobilized in 2.5% low melting point agarose on a glass coverslip. The temperature of the room was 23−24°C. Bipolar cell terminals were imaged in vivo using a custom-built two-photon microscope equipped with a mode-locked Chameleon titanium–sapphire laser tuned to 915 nm (Coherent) with an Olympus LUMPlanFI 40× water-immersion objective (NA 0.8). Emitted fluorescence was captured through both the objective and a substage oil condenser, filtered through an HQ 520/60 nm 2P GFP emission filter (Chroma Technology), and detected by a set of GaAsP photodiodes (Hamamatsu). Scanning and image acquisition were controlled under ScanImage v.3.6 software (Pologruto et al., 2003). Movies were processed using the SARFIA suite of analysis routines (Dorostkar et al., 2010) running in Igor Pro 7 (Wavemetrics). These routines begin with the automated extraction of the fluorescence change in each terminal by defining ROIs using a filtering algorithm based on a Laplacian transform.
Light stimuli were delivered using an amber LED (590 nm, Phillips Luxeon, 350 mA, 3 V) filtered through a 600/10 nm BP filter (Thorlabs) and projected through a light guide onto the surface of the bath, very close to eye of the fish. The stimulus time course was controlled in Igor Pro v. 4.01 (WaveMetrics) and time locked to image acquisition through ScanImage. The mean intensity of light stimuli was ∼2 × 105 photons/μm2/s, which corresponds to a low photoptic intensity.
Mouse preparation for in vivo imaging.
Mice were anesthetized using isoflurane (3% for induction, 1.5–2% during surgery). A circular craniotomy (2–3 mm diameter) was placed above V1 (centered 2.7 mm lateral from lambda suture). Oregon Green BAPTA-1 (OGB-1)-AM (Invitrogen) was injected as previously described (Stosiek et al., 2003; Komiyama et al., 2010; Zariwala et al., 2011). A custom titanium head post was fixed to the skull using black dental cement (Contemporary Ortho-Jet). The craniotomy was covered with agarose (1.2–1.5%) and a round glass coverslip (Warner Instrument, 5 mm diameter, #1 thickness) was cemented to the skull to reduce motion of the exposed brain. For cell-attached recording, the exposed brain was covered with ∼1 mm thick agarose without a coverslip. The animal was then transferred to the imaging setup, where it was placed on a warm blanket (37°C) and kept anesthetized using 0.5% isoflurane and sedated with chlorprothixene (20–40 μl at 0.33 mg/ml, i.m.) (Niell and Stryker, 2008).
In vivo mouse imaging and electrophysiology.
Imaging was performed with a custom-built two-photon microscope (designs available at http://research.janelia.org/Svoboda/). The light source was a Mai Tai 80 femtosecond pulsed laser (Spectra-Physics) running at 900 nm for GCaMPs and 800 nm for OGB-1. The objective was a 40× dipping lens (Olympus, 40×, 0.8 NA). Image acquisition was performed using ScanImage 3.7 (http://www.scanimage.org) (Pologruto et al., 2003). Images (512 × 250 pixels, 250 × 250 μm) were collected at 4 Hz.
In vivo cell-attached recording was performed using glass pipettes (∼5–7 MΩ) filled with solution containing the following (in mm): 125 NaCl, 5 KCl, 10 glucose, 10 HEPES, 2 CaCl2, 2 MgSO4, and 0.1 Alexa Fluor 594). Signals were amplified using an AxoPatch 200B amplifier (Molecular Devices), filtered at 5 kHz, and digitized at 10 kHz. Images (32 × 20 pixels, 20 × 20 μm) were acquired at 50 Hz.
Mouse visual stimuli.
The moving grating stimuli were generated using the Psychophysics Toolbox (Brainard, 1997; Pelli, 1997) in MATLAB. Each stimulus trial consisted of a 4 s blank period (uniform gray at mean luminance) followed by a 4 s drifting sinusoidal grating (0.05 cycles per degree, 1 Hz temporal frequency). The visual stimuli were synchronized to individual image frames using frame-start pulses provided by ScanImage 3.7. The gratings were presented through an LCD monitor (30 × 40cm), placed 25 cm in front of the center of the right eye of the mouse. The monitor subtended an angle of ±38° horizontally and −20° to +38° vertically around the eye of the mouse. For cell-attached recording, we used a smaller LCD monitor (12 × 16 cm) placed 10 cm in front of the right eye. Each stimulus trial consisted of a 4 s blank period followed by a 2 s drifting square wave grating (0.05 cycles per degree, 2 Hz temporal frequency).
Mouse data analysis.
Slow drifts in brain position in the X and Y directions were corrected using the TurboReg plug-in in ImageJ (Thévenaz et al., 1998). All remaining analyses were performed in MATLAB. ROIs corresponding to visually identifiable cell bodies were selected using a semi-automated algorithm. For GCaMP3, GCaMP5G, and GCaMP5K, ring-shaped ROIs were placed at the cytosolic regions of the cells (excluding the nucleus; GCaMP expression is typically restricted to the cytoplasm; Tian et al., 2009). For OGB-1, circular ROIs covering the whole soma were used. The fluorescence time course of each cell was measured by averaging all pixels within the ROI. The neuropil contamination was corrected using a published method (Kerlin et al., 2010); in short, the neuropil signal Fneuropil (t) surrounding each cell was measured by averaging the signal of all pixels within a 20 μm circular region from the cell center (excluding all selected cells). The true fluorescence signal of a cell body was estimated as follows:
with r = 0.7 throughout the study. After neuropil correction, the ΔF/F0 of each trial was calculated as (F − F0)/F0, where F0 is the baseline fluorescence signal averaged over a 2 s period immediately before the start of grating stimulation. Visually responsive neurons were defined using ANOVA across blank and eight direction periods (p < 0.01) (Ohki et al., 2005). Of the responsive cells, orientation-selective cells were defined by ANOVA across eight direction periods (p < 0.01).
The orientation selectivity index (OSI), tuning width, and direction selectivity index (DSI) were calculated for visually responsive cells. First, the preferred orientation (θpref) of the cell was determined as the angle of the grating that produced the strongest response. The orientation-tuning curve was constructed by measuring the mean ΔF/F0 over the 4 s stimulus period for each orientation. We then fitted the tuning curve as the sum of two Gaussian functions centered on θpref and θpref + π with equal width σ, different amplitudes A1 and A2, and a constant baseline B (Niell and Stryker, 2008). The value of σ was required to be larger than 15° to reflect the limit of our stimulus set (45° separation) in resolving sharper tuning. The OSI was defined as follows:
 |
where Rpref and Rortho are the response amplitude at the preferred (θpref) and the orthogonal orientation . Tuning width was defined as the half-width at half-maximum of the fitted Gaussian (). Finally, DSI was calculated as follows:
 |
where Ropposite is the response in the opposite direction (θpref + π).
For simultaneous imaging and cell-attached recording, ring-shaped ROIs were placed at the cytosolic regions of the cells. Neuropil signals were measured from the image region with the cell excluded. Neuropil compensation was performed as above (r = 0.7) before calculating ΔF/F. For visual-evoked response (see Fig. 9A–E), the baseline fluorescence (F0) was measured over a 1 s period immediately before the start of grating stimulation. For responses to a few isolated AP (Fig. 9E,F), we searched the spike trace for events with 1, 2, and 3 APs within a 200 ms window, and no other APs during a 1 s period before and a 0.5 s period after the first AP. The baseline fluorescence (F0) was measured over a 0.2 s period before the first AP. AP detection was quantified using template matching with the average trace of 1, 2, and 3 AP events as templates for detecting 1, 2, and 3 APs, respectively. The 0 AP traces (478 traces) were taken from the same fluorescence recordings during periods where no AP was detected for at least 2 s. The decision variable was the projection of the fluorescence traces along the direction of the template vector. Detection efficiency was defined as the fraction of correctly detected events given a 5% false positive rate.
Figure 9.
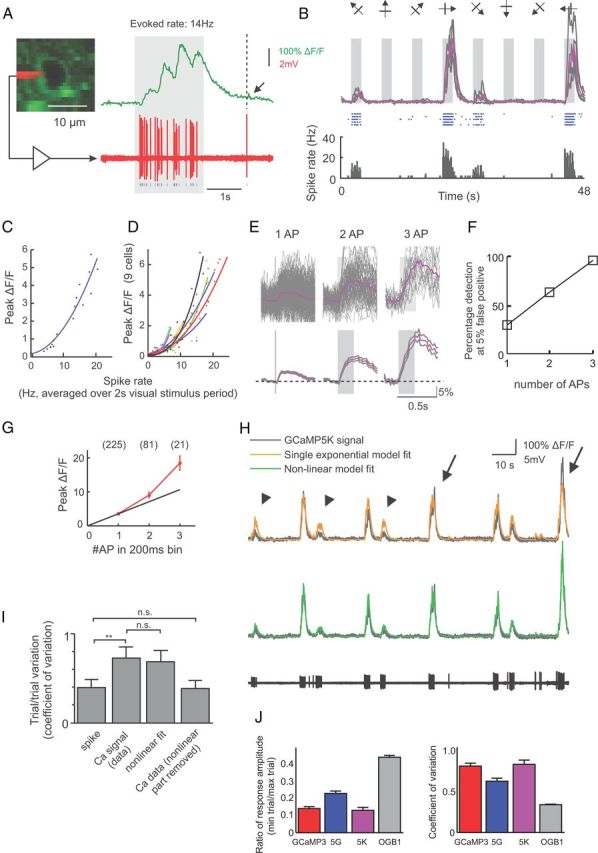
Relationship between spiking and 5K signal in vivo. A, Visually evoked 5K response (top) and simultaneously recorded spikes (bottom) in a layer 2/3 pyramidal cell in V1. Arrow, Putative single spike-induced signal. B, 5K responses (top: gray, individual trials; purple, average of 5 trials) and corresponding spike raster (middle) and peristimulus time histogram (bottom) during the presentation of eight oriented grating stimuli. C, Peak GCaMP5K response during 2 s visual stimulation as a function of spike rate. D, Peak GCaMP5K response plotted against spike rate for nine cells. E, Average GCaMP5K response to 1, 2, and 3 APs within 200 ms search windows. Gray traces are mean ± SEM (n = 225, 81, and 21 for 1, 2, and 3 APs, respectively). F, Spike detection efficiency. G, Peak ΔF/F response to 1, 2, and 3 APs. H, Single exponential model (yellow trace) and nonlinear model (green trace) fit of the GCaMP5K signal (gray) from the simultaneously recorded spike response (black trace, bottom). Arrows, Underestimation of large events. Arrowheads, Overestimation of small events by the single exponential model. I, Trial-to-trial variability of GCaMP5K (coefficient of variation) during repeated presentation of preferred stimuli calculated using different measures. (**p = 0.0012; n.s., nonsignificant; n = 7 cells). J, Trial-to-trial variability of fluorescent responses at preferred orientation quantified for all visually responsive neurons, for all four calcium indicators.
The single exponential model fit of the GCaMP5K signal (Fig. 9H) was calculated by linear convolution of the detected spike point process with a single exponential kernel: . The amplitude A and the decay time constant τ were adjusted to minimize the mean square error of the fit. For the nonlinear model, the output of the linear mode x(t) = s(t)·h(t) was passed to a nonlinear stage to generate the final output as follows:
The four parameters A, τ, b, and c were adjusted independently to minimize the mean square error.
Statistical methods.
Unless specified otherwise, all statistical methods were implemented in MATLAB or Microsoft Excel, using standard packages.
Results
Structure-guided engineering of GCaMP5s and biophysical characterization
A small family of GCaMP5 variants was produced from the GCaMP3 scaffold by combining improvements generated from site-directed mutagenesis at the cpGFP/CaM proto-interface and targeted library screening at the M13pep-cpGFP and cpGFP-CaM linkers. We also tested the effect of mutations to the M13 peptide and near the third Ca2+-binding site of CaM (Fig. 1A, Table 1). To facilitate engineering, we solved the crystal structure of several GCaMP2 and GCaMP3 variants (Table 2).
Figure 1.
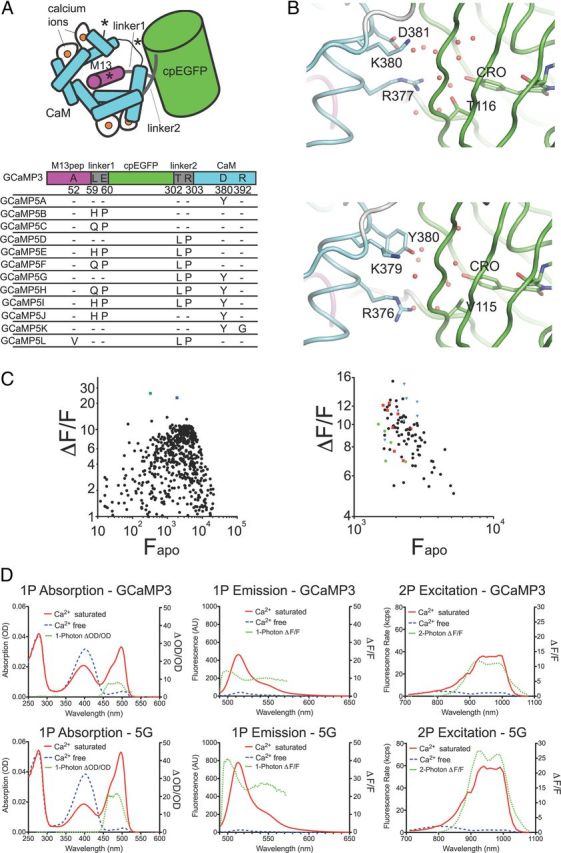
Design of GCaMP5s. A, Schematic of the GCaMP3 structure with sites of engineering shown. B, Structural effects of the D381Y mutation (D380Y in GCaMP3 numbering). Chromophore environment at the cpGFP/CaM interface in GCaMP2 (top, PDB 3EVR)(Akerboom et al., 2009) and GCaMP5G (bottom, PDB 3SG4) structure reported here. Structures are shown as a diagram and sticks colored by domain (cpGFP, green; linker, white; CaM, cyan). Selected portions of the model around the GFP chromophore (CRO) are represented as sticks with ordered water molecules represented as red spheres. C, (ΔF/F)max versus Fapo for both linker 1 variants of GCaMP3 (left) and linker 2 variants of GCaMP3 (right) in bacterial lysate. Left, The green square denotes L1-Gln-Pro, the blue square denotes L1-His-Pro. Right, Linker variants L2-Pro-X are depicted as red squares, L2-X-Pro as blue triangles, and original GCaMP3 linker variants (L2-Thr-Arg) as green dots. D, One-photon absorption (left), one-photon emission (middle), and two-photon excitation (right) spectra of both GCaMP3 (top) and GCaMP5G (bottom). Calcium-free spectra are depicted by dashed blue lines and calcium-saturated spectra by solid red lines. Dashed green lines depict (ΔF/F)max, plotted on the right axis.
Table 1.
In vitro/in vivo characteristics of GCaMP5 variants
| GCaMP variant | Mutationsa | Max. in vitro ΔF/F | Fbase, Fmax HEKb | Ratio (Fmax/Fbase) HEKb | Baseline brightness cultured neurons (%)c | Ca2+ affinity (Kd) | pKa (sat/apo) | Hill coefficient | kond |
|---|---|---|---|---|---|---|---|---|---|
| GCaMP2 | — | 5.1 ± 0.1 | ND | ND | ND | 545 ± 32 nm | 7.02 ± 0.01/8.72 ± 0.02 | 1.8 ± 0.1 | ND |
| GCaMP3 | Deletion R2, M65K, T115V, N362D | 12.3 ± 0.4 | 1 ± 0.03 | 2.9 ± 0.1 | 100 ± 1.6 | 405 ± 9 nm | 6.97 ± 0.01/8.40 ± 0.02 | 2.1 ± 0.1 | 8.3 ± 1.5 s−1 |
| 2.9 ± 0.11 | 33 ± 3 s−1 | ||||||||
| GCaMP5A | D380Y | 17.4 ± 1.2 | 0.95 ± 0.07 | 3.8 ± 0 0.02 | 57.3 ± 1.0 | 307 ± 12 nm | 6.77 ± 0.02/8.70 ± 0.04 | 2.7 ± 0.1 | ND |
| 3.7 ± 0.15 | |||||||||
| GCaMP5B | L59H, E60P | 23.8 ± 3.4 | 0.48 ± 0.02 | 3.9 ± 0.2 | 35.2 ± 0.8 | ND | ND | ND | ND |
| 1.87 ± 0.09 | |||||||||
| GCaMP5C | L59Q, E60P | 35.1 ± 0.9 | 0.49 ± 0.02 | 4.5 ± 0.1 | ND | ND | ND | ND | ND |
| 2.2 ± 0.08 | |||||||||
| GCaMP5D | T302L, R303P | 22.0 ± 1.8 | 0.34 ± 0.02 | 3.3 ± 0.1 | 37.5 ± 0.5 | 730 ± 18 nm | 7.43 ± 0.02/8.91 ± 0.05 | 2.5 ± 0.1 | 7.8 ± 1.5 s−1 |
| 1.1 ± 0.3 | 75.3 ± 2.6 s−1 | ||||||||
| GCaMP5E | L59H, E60P, T302L, R303P | 40.9 ± 2.2 | 0.19 ± 0.01 | 10.8 ± 0.3 | 30.1 ± 0.4 | ND | ND | ND | ND |
| 2.0 ± 0.14 | |||||||||
| GCaMP5F | L59Q, E60P, T302L, R303P | 162 ± 3.5 | 0.17 ± 0.03 | 7.1 ± 0.3 | ND | ND | ND | ND | ND |
| 1.2 ± 0.13 | |||||||||
| GCaMP5G | T302L, R303P, D380Y | 32.7 ± 1.5 | 0.40 ± 0.05 | 5.5 ± 0.4 | 61.6 ± 1.3 | 460 ± 11 nm | 6.96 ± 0.02/9.14 ± 0.05 | 2.5 ± 0.2 | 6.5 ± 1.0 s−1 |
| 2.1 ± 0.11 | 58.1 ± 1.1 s−1 | ||||||||
| GCaMP5H | L59Q, E60P, T302L, R303P, D380Y | 158 ± 12 | 0.01 ± 0.01 | 48.6 ± 35.3 | ND | ND | ND | ND | ND |
| 0.44 ± 0.02 | |||||||||
| GCaMP5I | L59H, E60P, T302L, R303P, D380Y | 42.0 ± 3.1 | 0.03 ± 0.02 | 28.9 ± 19.2 | 39.7 ± 6.5 | ND | ND | ND | ND |
| 0.71 ± 0.02 | |||||||||
| GCaMP5J | L59H, E60P, D380Y | ND | ND | ND | 42.2 ± 1.4 | ND | ND | ND | |
| GCaMP5K | D380Y, R392G | 9.4 ± 0.14 | ND | ND | 197.2 ± 7.3 | 189 ± 5 nm | ND | 3.8 ± 0.3 | ND |
| GCaMP5L | A52V, T302L, R303P | 17.7 ± 0.3 | ND | ND | ND | 390 ± 18 nm | ND | ND | ND |
| GCaMP2-LIA | E61IA | 31.2 ± 0.8 | ND | ND | ND | ND | ND | ND | ND |
| GCaMP3-KF | L59K, E60F | 9.2 ± 0.4 | ND | ND | ND | ND | ND | ND | ND |
aGCaMP5s also contain the mutations R2 deletion, M65K, T115V, and N362D listed for GCaMP3.
bFbase, Fmax, and Ratio are from the acetylcholine assay.
cValues ± SEM, baseline brightness of GCaMP3 set to 100%.
dFirst values are measured at a calcium concentration of 670 nm, second at a calcium concentration of 8.52 μm. ND, not determined.
In our previous analysis of the GCaMP2 structure (Akerboom et al., 2009), we predicted that the chemical environment and solvent accessibility of the cpGFP chromophore would be modulated in the Ca2+-bound state by the interlobe linker of CaM (residues 377–383 of GCaMP3). Here, we tested a small number of mutations and found that Asp380Tyr increases the Ca2+-bound fluorescence and dynamic range (ΔF/F)max of GCaMP3 (1.5- and 1.4-fold; Table 1; GCaMP3-Asp380Tyr = “GCaMP5A,” GCaMP3-Thr302Leu, Arg303Pro, and Asp380Tyr = “GCaMP5G”). Comparison of the crystal structures of GCaMP2, GCaMP5A, and GCaMP5G confirm that Tyr380 is oriented toward the cpGFP chromophore; indeed this mutation reorganizes nearby side chains and the water network in close proximity to the cpGFP chromophore, resulting in an apparent decrease in solvent accessibility of the cpGFP barrel (Fig. 1B). Consequently, the Asp380Tyr mutation raises the brightness of the calcium-bound state of GCaMP3 for both GCaMP5A and GCaMP5G; in addition, calcium affinity and cooperativity (Hill coefficient) are increased by ∼25% for GCaMP5A (Table 1).
The structure of the M13pep-to-cpGFP linker (“linker 1”) is known to be critical to sensor function (Nakai et al., 2001). Crystal structure analysis of Ca2+-bound and Ca2+-free GCaMP2 (Wang et al., 2008; Akerboom et al., 2009) revealed that both amino acids of linker 1 (Leu60 and Glu61) make significant contacts to cpGFP and CaM and stabilize both the apo and Ca2+-bound conformations. After screening mutagenic libraries at the linker 1 positions (59–60), we found a number of variants that substantially increased the dynamic range of GCaMP3 in E. coli lysates (Table 1; Fig. 1C). Mutation of linker 1 from Leu-Glu (GCaMP3) to His-Pro (GCaMP5B) or Gln-Pro (GCaMP5C) approximately doubles or triples the (ΔF/F)max of GCaMP3, respectively (Table 1; Fig. 1C).
Mutation of the cpGFP-to-CaM linker (“linker 2”) has also been shown to affect sensor function (Souslova et al., 2007). Accordingly, we also screened targeted libraries at the linker 2 positions (302–303) and identified variants with increased dynamic range in E. coli lysates (Table 1; Fig. 1C). Most prominent was Leu-Pro (GCaMP5D), which has a (ΔF/F)max approximately twice that of GCaMP3, but with decreased affinity (Table 1).
Presuming that the improvements to sensor function resulting from the individual mutation of the cpGFP/CaM interface and linker 1 (L1) and linker 2 (L2) might be additive, different combinations of these (in the GCaMP3 scaffold, Fig. 1A) named GCaMP5E-J, were characterized extensively in vitro (Table 1). Compared with GCaMP3, all these GCaMP5 variants have significantly higher ΔF/F (Table 1). The Arg380Tyr mutation not only increases the brightness of both the saturated (sat) and apo states in the GCaMP3 background (GCaMP5A), it also increases the brightness of both states for several of the other sensors with modified linkers, e.g., 5G/D and 5H/F. Linker mutations L1-QP (GCaMP5C), L1-HP (GCaMP5B), and L2-LP (GCaMP5D) decrease the fluorescence of the apo state, and their combination has a cumulative effect: GCaMP5E, GCaMP5F, GCaMP5H, and GCaMP5I are all exceptionally dim in the apo state. This effect is strongest for GCaMP5F and GCaMP5H, both containing combinations of L1-QP and L2-LP, leading to an ∼160-fold fluorescence increase upon calcium binding in vitro (Table 1).
A GCaMP5 sensor incorporating the CaM mutation Arg90Gly (Sorensen and Shea, 1996) was constructed in the background of GCaMP5A, to further increase Ca2+ affinity (GCaMP numbering Arg392Gly, GCaMP5K). While GCaMP5K, as intended, has a higher affinity (and Hill coefficient) for Ca2+ than GCaMP3 (Kd = 190 nm, n = 3.8), it comes at a cost of (ΔF/F)max (Table 1). Another gain in affinity was achieved by mutation of the M13 peptide, Ala52Val, in a “hydrophobic anchor” position (Hultschig et al., 2004) and similar to the “D2” M13 variant (Palmer et al., 2006). This mutation, acquired serendipitously in GCaMP5D (resulting in GCaMP5L), increases calcium affinity ∼2-fold while preserving (ΔF/F)max (Table 1).
We used an in-depth biophysical characterization of this panel of variants to reduce the number taken forward for in vivo testing, and identified some with particularly useful characteristics. GCaMP5G has a significantly higher apo pKa (Table 1), indicating that the Ca2+-free state is more likely to have a protonated, dim chromophore at physiological pH. Apparent rates of fluorescence conversion were determined by stopped-flow mixing, beginning at 0 [Ca2+] and stepping to 250 nm–10 μm. Initial rates (k1) of fluorescence after steps to 10 μm [Ca2+] were significantly faster for 5D and 5G than for GCaMP3 (Table 1), although rates after steps to 500 nm [Ca2+] were comparable. Peak brightness and QY of GCaMP5s tested were comparable to GCaMP3 (Table 3). One- and two-photon excited brightness spectra (Mütze et al., 2012) of equimolar (1 μm) amounts GCaMP3 and GCaMP5G revealed that a larger fraction of molecules is in the fluorescent, deprotonated state for GCaMP5G compared with GCaMP3, and ΔF/F for GCaMP5G was >2-fold improved under one- and two-photon excitation (Fig. 1D). The GCaMP5 variants retain the slight red shift of GCaMP3 relative to EGFP (Fig. 1D).
Table 3.
Biophysical characterization of GCaMPs
| ϵa (M−1cm−1) |
QYb |
τc (ns) |
ΔF/Fod pH 7.25 | QY*σ2e pH 9.5 (+ Ca2+) | Peak brightnessf (kcpsm) pH 9.5 (+Ca2+) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 7.25 (+Ca2+) | pH 9.5 (+Ca2+) | pH 7.25 (+Ca2+) | pH 9.5 (+Ca2+) | pH 7.25 (+Ca2+) | pH 7.25 (−Ca2+) | pH 9.5 (+Ca2+) | pH 9.5 (−Ca2+) | ||||
| EGFP | 56,000 | 57,800 | 0.73 | 0.70 | 2.6 | 2.6 | 2.6 | 2.6 | 1 | 13.4 | |
| GCaMP2 | 38,100 | 54,800 (60,600) | 0.53 | 0.55 | 2.0 | 2.1 | 2.0 | 2.1 | 5.7 ± 1.4 | 0.80 | 6.7 |
| GCaMP3 | 37,700 | 56,300 (66,500) | 0.65 | 0.67 | 2.8 | 2.7 | 2.7 | 2.6 | 10.6 ± 1.2 | 0.83 | 9.5 |
| GCaMP5A | 56,100 | 58,100 (60,900) | 0.65 | 0.62 | 2.7 | 2.8 | 2.6 | 2.7 | 17 ± 4.2 | 0.89 | 9.9 |
| GCaMP5D | 25,300 | 48,100 (58,300) | 0.67 | 0.63 | 2.7 | 2.7 | 2.7 | 2.8 | 18 ± 3.7 | 0.78 | 9.0 |
| GCaMP5G | 49,300 | 58,500 (61,300) | 0.67 | 0.67 | 2.7 | 2.8 | 2.7 | 2.7 | 31 ± 9.4 | 0.96 | 9.3 |
aPeak extinction coefficient at 497 nm (485 nm for GCaMP2, 487 nm for EGFP), based on concentration determined by alkali denaturation: 0.1 m NaOH, ϵ = 44,000 m−1cm−1 at 447 nm. Values in parentheses are based on concentration measurements obtained by two-photon FCS at pH 9.5.
bQuantum yield (QY) using fluorescein (QY = 0.93) in pH 11/H2O as a standard.
cFluorescence lifetime under two-photon excitation (960 nm, 2 mW), fit to a single-exponential decay.
dCalcium-dependent contrast ΔF/F in pH 7.25 buffer, for 940 nm excitation; SD calculated for eight or more measurements.
eTwo-photon action cross section (QY × two-photon cross section σ2 per molecule) at 940 nm excitation, relative to EGFP, in pH 9.5 buffer containing 1 mm CaCl2.
fPeak brightness per emitting fluorophore, in kilocounts per second per molecule (kcpsm), defined as the maximum fluorescence rate divided by the number of emitting fluorophores in the excitation beam volume as determined from FCS measurements, as the laser power is scanned from 1–80 mW at an excitation wavelength of 940 nm.
Characterization in HEK293 cells and cultured neurons
We next characterized the GCaMP5 variants in HEK293 cells in response to acetylcholine-induced Ca2+ mobilization (Tian et al., 2009). The ratio of peak-to-baseline fluorescence was increased for most variants, by up to ∼17 fold for GCaMP5H (Table 1). This is the direct result of lower baseline fluorescence for all variants except GCaMP5A, in agreement with in vitro data (Table 1). GCaMP5s containing L1-QP (GCaMP5C, GCaMP5F, and GCaMP5H) were not selected for further analysis in cultured neurons, due to the low baseline brightness of variants containing this linker. GCaMP5K was directly tested in primary rat hippocampal neurons due to its high affinity; the HEK293 cell testing was omitted for this variant.
A selection of eight GCaMP5 variants was expressed in primary rat hippocampal neurons by lentivirus-mediated gene transfer. After 16–18 d, infected neurons appeared healthy (Fig. 2A); most GCaMP5s were dimmer than GCaMP3 at baseline, except GCaMP5K (“5K”), which was approximately twice as bright before stimulation (Table 1). APs were elicited at 83 Hz via a custom-built extracellular field stimulator (1 AP per stimulus, data not shown) and imaged at 34.8 Hz with an EMCCD camera. In parallel experiments we incubated neurons with cell-permeable versions (acetoxymethyl ether, “AM” derivatives) of the small molecule calcium dyes OGB-1 or Fluo-4. Robust fluorescence responses were seen from 1 to 160 field stimuli (Fig. 2B–D). We extracted fluorescence from individual neurons and computed background-subtracted fluorescence transients. Single field stimuli, corresponding to one AP, evoked fluorescence transients that were detectable in single trials (Fig. 2B). Peak ΔF/F (Fig. 2C) and signal-to-noise ratio (SNR; Fig. 2D) were significantly improved for most GCaMP5 variants compared with GCaMP3, over at least part of the stimulus regime (Fig. 2C,D; ΔF/F and SNR for each variant including GCaMP3, OGB-1, Fluo-4, and G-GECO1.2).
Figure 2.
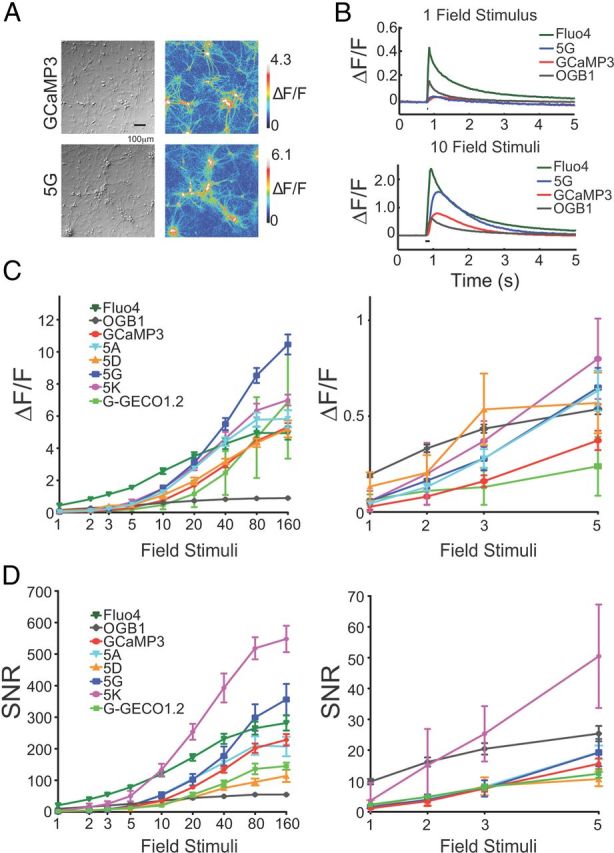
Neuronal testing of GCaMP5s. A, GCaMP3 and 5G responses in neurons. DIC (left) and false-colored image of fluorescence response to 40 field stimuli (right). B, Trial-averaged responses of GCaMP3 and 5G, and OGB-1 and Fluo-4, to 1 and 10 field stimuli. C, Peak ΔF/F versus stimuli. Error bars indicate SEM. Right, blow-up of 1–5 stimuli. D, SNR including SEM. SNR was computed as the ratio between the peak fluorescence response amplitude (ΔF) and the SD of the fluorescence trace before stimulus onset. Fluo-4 was omitted from the blow-ups.
Fusion-directed localization in neurons and astrocytes
GCaMP5G (“5G”), which showed the highest response at maximum stimulation in the cultured neuron screen, was selected for testing in the context of specific protein fusions. Variant 5G was fused to synaptophysin (Dreosti et al., 2009) creating “SyGCaMP5G” (targeted to the outside of synaptic vesicles), and to the Lck domain (Shigetomi et al., 2010b) (“Lck-GCaMP5G”; inside of the plasma membrane). SyGCaMP constructs were transfected into hippocampal neurons, and boutons were imaged (Fig. 3A); the response of SyGCaMP5G (“Sy5G”) to small field stimuli was significantly higher compared with SyGCaMP2 and SyGCaMP3 (Fig. 3B).
Figure 3.

Targeted GCaMP5s and astrocyte testing. A, Heat map showing peak ΔF/F of hippocampal neurons transfected with SyGCaMP5s following 10 field stimuli (20 Hz). B, Fluorescence response versus field stimuli for SyGCaMPs. C–E, Performance of LckGCaMPs in astrocytes. C, Top, Baseline fluorescence of LckGCaMP3 and 5G. Middle, Spotty calcium signals. Bottom, Quantified baseline fluorescence: red, GCaMP3; blue, GCaMP5G. D, Spotty calcium transients for LckGCaMP3 and Lck-GCaMP5G, respectively. E, Peak ΔF/F for spontaneous (left) and ATP-evoked (right) responses in astrocytes. F, Peak ΔF/F for neuronal AP-evoked astrocyte responses. Error bars indicate SEM. G, Fluorescence micrograph of GFAP-GCaMP5G-transfected astrocytes and fluorescence response of GFAP-GCaMP5G to field stimulation (30 Hz) of cocultured neurons; 1–120 field stimulations. Raw traces in gray, trial-average in blue.
5G also proved superior to GCaMP3 for detection of Ca2+ hotspots in astrocytes. The membrane-targeted Lck-GCaMP5G (“Lck-5G”) detected spontaneous Ca2+ transients (“spotty calcium” signals) (Shigetomi et al., 2010a) and ATP-induced responses with ∼2-fold greater response magnitude than Lck-GCaMP3 in astrocytes; the 5G-determined responses also had more apparent local structure, significantly increasing the resolution of spotty calcium signal detection (Fig. 3C–E). Furthermore, the threshold of detection was lowered from ∼10 field stimuli for Lck-GCaMP3 to ∼2 field stimuli for Lck-5G for transfected astrocytes co-cultured with neurons, indicating that both dynamic range and sensitivity have been substantially improved for membrane-targeted 5G compared with GCaMP3 (Fig. 3F). SNR was slightly improved for most of the stimulus range, although variability was higher due to lower baseline fluorescence (Fig. 3F).
In parallel, membrane-targeted 5G and GCaMP3 were expressed in astrocytes (under control of the glial fibrillary acidic protein (GFAP) promoter) cocultured with neurons. Fluorescence changes in astrocytes following neural activity evoked by electrical field stimulation were easily recorded using GFAP-5G (Fig. 3G). These results demonstrate that the GCaMP5 sensors will be useful to study Ca2+ dynamics in a range of cell types, and that these indicators are suited for membrane targeting and protein fusions.
Characterization in mouse retina
GCaMP5D (“5D”) displayed large (ΔF/F)max to 1–3 field stimuli in cultured neurons (Fig. 2D), and was selected for further testing in in vitro mouse retina, as before (Borghuis et al., 2011). We introduced 5D into AAV under the neuron-specific hsyn1 promoter and infected mouse retinas in vivo (see Materials and Methods)(Borghuis et al., 2011). Retinal ganglion cells (RGCs) were bright and appeared healthy, with nuclear-excluded fluorescence (Fig. 4A, top). We recorded light-evoked responses from 5D-expressing RGCs in dissociated retinas (Fig. 4A, bottom). RGCs showed fluorescence changes both to the onset of the infrared scan laser (910 nm; 20 mW after the objective)(Borghuis et al., 2011) and to a flash of visible light (420 nm) delivered with an LED 2 s after scan onset (Fig. 4B). Time course, amplitude, and polarity of the responses varied across the labeled population, consistent with the known diversity of RGC types (e.g., ON and OFF, brisk and sustained). The peak amplitude (ΔF/F)max of the evoked fluorescence response was 1.6 ± 1.3 (n = 55; mean ± SEM; range 0.0–4.5) (Fig. 4C). Of all 5D-expressing cells, 65% responded with (ΔF/F)max > 0.6 (mean 2.3 ± 1.0). For comparison, GCaMP3 and OGB-1 produced distributions of (ΔF/F)max with peaks around 0.2, with only ∼30% of GCaMP3- and OGB-labeled RGCs showing (ΔF/F)max > 0.15 (Borghuis et al., 2011) (Fig. 4C). SNR for GCaMP5D was significantly improved compared with GCaMP3 (GCaMP3: SNR average = 8.97 ± 1.08, n = 187; GCaMP5D: SNR average 13.43 ± 1.16; n = 64; p = 0.025) (Fig. 4D).
Figure 4.
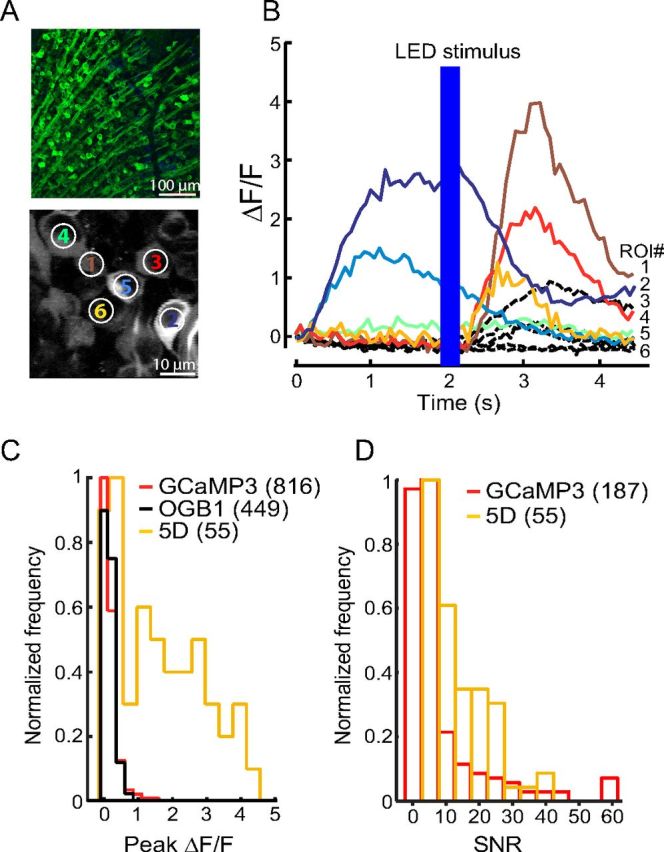
GCaMP5D in mouse retina. A, Top, Fluorescence micrograph of retinal ganglion cells (RGCs) expressing GCaMP5D. Bottom, Two-photon fluorescence image of RGCs expressing GCaMP5D. B, Fluorescence response of six indicated RGCs to infrared scan laser onset and to full-field blue LED flash. Black dashed lines show responses of a representative population of GCaMP3-labeled RGCs recorded under identical stimulus conditions (data replotted from Borghuis et al., 2011). C, Peak ΔF/F distribution for 55 imaged RGCs. Corresponding data for GCaMP3 and OGB-1 labeled neuron populations are shown in red and black, respectively (data reproduced from Borghuis et al., 2011). D, SNR distribution of GCaMP3 and GCaMP5D.
Imaging sensory-evoked Ca2+ transients in worms
To test the in vivo performance of GCaMP5 in worms, we selected GCaMP3, 5A, and 5G for analysis. We monitored the activity of the C. elegans AWCon chemosensory neuron, which responds to odor presentation with graded calcium decreases and to odor removal with graded calcium increases (Tian et al., 2009) (Fig. 5A). All GCaMPs were expressed from the same promoter; in all cases, no defect was observed in AWC-dependent spontaneous Ω turning frequency was unaffected by the transgene (Tian et al., 2009) (Fig. 5B). Individual worms were imaged in a microfluidic chamber during an odor addition–removal sequence with the odor isoamyl alcohol (IAA) (Fig. 5C,D). All GCaMPs detected the known decrease in AWC calcium upon odor addition and the calcium increase upon odor removal (Tian et al., 2009). GCaMP5G performed comparably to GCaMP3 for odor presentation and for odor removal, based on total fluorescence change and SNR. GCaMP5A showed a strong suppression in baseline fluorescence upon odor addition, and a large increase after odor removal. In addition, the higher baseline fluorescence of 5A resulted in a threefold improvement in SNR for odor presentation, and a twofold increase in SNR for odor removal, over GCaMP3. However, the 5A fluorescence plateaued near peak, suggesting a truncation of the response to the highest calcium levels, unlike GCaMP3. These results are consistent with the higher affinity and cooperativity of 5A Ca2+ binding compared to GCaMP3 (Table 1). Many neurons in C. elegans exhibit graded responses to stimuli, and 5A extends the observable range to calcium fluctuations too low for GCaMP3 to detect reliably. These results indicate that 5A may be an improved indicator for C. elegans neurons, with the choice between 5A and GCaMP3 dictated by the specific application.
Figure 5.

GCaMPs in C. elegans AWCon neuron in response to odor addition and removal. A, Schematic of AWC neuron location, and fluorescence micrograph of corresponding view. Scale bar, 10 μm. B, Omega bending frequency of wild-type C. elegans (N2) and 5A and 5G animals. C, Odor addition-evoked Ca2+ transients. Top, ΔF/F (%). Bottom, SNR. D, Odor removal-evoked Ca2+ transients. For both C and D, trial-averaged responses are colored red, blue, and cyan for GCaMP3, GCaMP5G, and 5A, respectively. Shaded area represents odor presence. Error traces indicate SEM.
Characterization of GCaMP5s in larval Drosophila
Both GCaMP3 and 5G were expressed in Drosophila melanogaster larvae using a motor-neuron promoter (OK6-Gal4). Type 1b NMJ boutons of third instar larvae were imaged following activity evoked from electrically stimulating motor neuron axons using a suction electrode (Macleod et al., 2002) (Fig. 6A). Larval NMJs showed robust fluorescence changes to evoked APs across a range of stimulation frequencies. GCaMP5G showed a threefold increase in (ΔF/F)max over GCaMP3, saturating at ∼8. Single APs were clearly detectable in trial-averaged 5G responses but only rarely in single trial responses (Fig. 6B). The shapes of the ΔF/Fmax and SNRmax curves were similar for both indicators, but responses with 5G were significantly greater than those with GCaMP3 across all stimuli frequencies (n = 6, 10 NMJs/GCaMP, 7 larvae, for electrical stimuli at 1, 5, 10, 20, 40, 80, and 160 Hz, p values at ΔF/Fmax were 0.0006, 0.01, 0.002, 4.8e-06, 6.6e-10, 1.5e-11, and 1.3e-12, respectively, and p values at SNRmax were 6.6e-06, 0.006, 0.001, 4.7e-06, 1.2e-09, 9.7e-10, and 1.2e-06, respectively).
Figure 6.
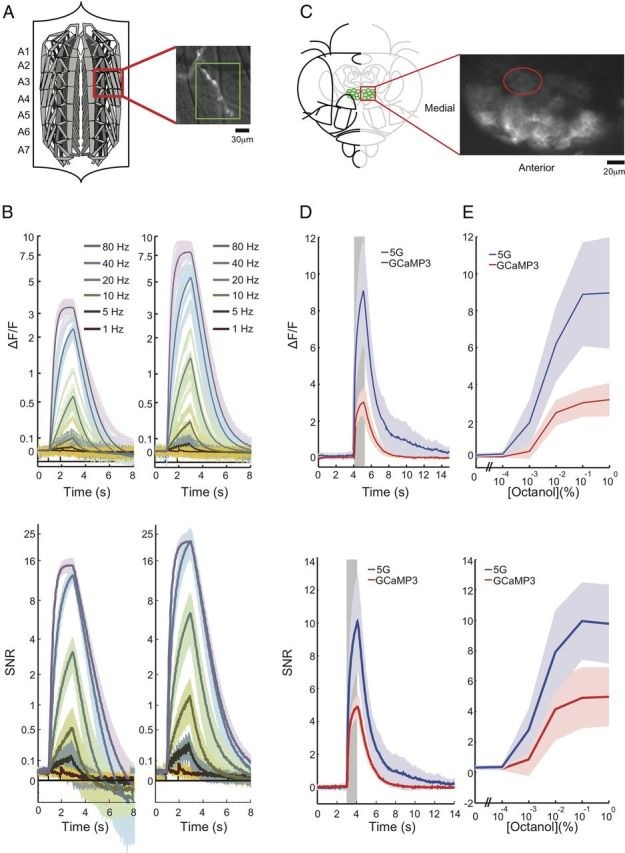
GCaMPs in Drosophila. A, Schematic of larval NMJ preparation, and close-up of Type 1b boutons from muscle 13 (segments A3–A5) used for wide-field imaging. Scale bar, 30 μm. B, Single trials of electrically evoked Ca2+ transients from wide-field imaging in the Drosophila larval NMJ. Top: Fluorescence changes (ΔF/F) traces from presynaptic terminals obtained by delivering 2 s of electrical stimulus at different frequencies. Bottom: SNR of the same data. Left, GCaMP3. Right, GCaMP5G. C, Two-photon imaging frame scan of PNs innervating the DC1 glomerulus in the adult fly AL (dorsal view) Scale bar, 20 μm. D, The mean of five replicate stimulations from six ALs (5 animals) is shown along with the SD (between AL means). Response to a 0.1% octanol, 1 s odor pulse from DC1 PNs. E, Mean octanol response from PNs from DC1 glomerulus (averaged over 5 flies) to increasing concentration. All panels show mean ± SD.
Imaging sensory-evoked Ca2+ transients in adult Drosophila
In parallel we crossed UAS:GCaMP Drosophila flies with a Gal4 fly line (GH146-Gal4) expressing broadly in the olfactory projection neurons, and imaged calcium changes in the AL (Jayaraman and Laurent, 2007), in the DC1 glomerulus, in response to presentations of octanol (Fig. 6C). For 1% octanol (the highest intensity stimulus we presented) we observed: 5G, 8.96 ± 3.02 (ΔF/F)max (range 6.11–14.08, n = 6 ALs); GCaMP3, 3.18 ± 0.90 (ΔF/F)max (range 2.02–4.72, n = 6 ALs), a threefold improvement (Fig. 6D) in dynamic range. We presented a range of concentrations of octanol to obtain a tuning curve with GCaMP3 and 5G (Fig. 6E). The shapes of the ΔF/Fmax and SNRmax curves were similar for both indicators, but responses with 5G were significantly greater than those with GCaMP3 across all concentrations except 0.01% octanol (n = 6 ALs/GCaMP, five flies, for concentrations of 0.0001, 0.001, 0.01, 0.1, and 1% octanol, p values for ΔF/Fmax were 0.007, 0.005, 0.07, 0.0003, 0.0002, and 0.0003, respectively, and p values for SNRmax were 0.002, 0.001, 0.11, 0.004, 0.002, and 0.001, respectively).
In vivo imaging of evoked and spontaneous APs in zebrafish
We tested GCaMP2, GCaMP3, 5A, and 5G in vivo in zebrafish (Danio rerio), at three different locations in the visual pathway (Nevin et al., 2010). An overview of the fish visual system for each of the three imaging locations is shown (Fig. 7A,C,G). GCaMP2 and 5G were first expressed in sensory neuron ribbon synapses, under control of the Ribeye-A promoter. In Ribeye-A GCaMP fish (Fig. 7A), 5G showed a ∼2.5 fold larger (ΔF/F)max compared with GCaMP2 in bipolar cell terminals in the inner plexiform layer following a visual stimulus (Dreosti et al., 2009) (Fig. 7B), which might be related to Ca2+ spiking in some of these terminals (Dreosti et al., 2011). GCaMP3, 5A, and 5G were next expressed pan-neuronally, using the elavl3 (HuC) promoter. In RGC axons and tectal dendrites (Fig. 7C), 5A and 5G responses to visual stimuli were greater compared with GCaMP3 (Fig. 7D) in both average and maximum peak response (Fig. 7E). SNR was improved for both 5A and 5G. Interestingly, as for the worm odor addition/removal imaging, 5A outperformed GCaMP3 and 5G (Fig. 7D–F). In tectal neuron somata (Fig. 7G), (ΔF/F)max for 5G was over threefold improved compared with GCaMP3 (Fig. 7H,I). More importantly, the number of cells with detectable visual responses increased by sixfold (35% of the total number of cells) for 5G compared with GCaMP3 (Fig. 7H). SNR was fourfold improved for 5G compared with GCaMP3 (Fig. 7J).
Figure 7.
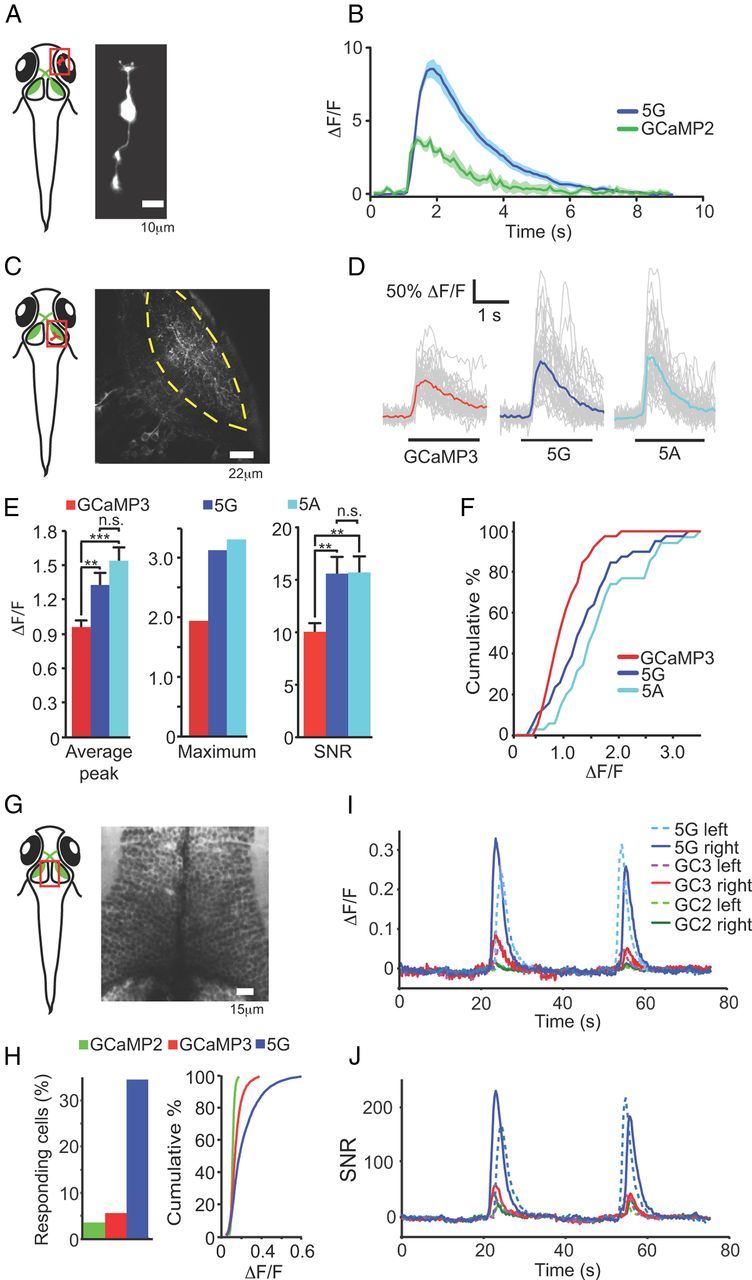
In vivo imaging in zebrafish. A, Schematic representation of area imaged (red square; retinal bipolar cell terminals) including fluorescence micrograph of bipolar cell. B, Two-photon imaging of calcium spikes in axon terminals of retinal bipolar cells in Tg(Ribeye-A:GCaMP2) (green line) and Tg(Ribeye-A:GCaMP5G) (blue line) fish. Mean (± SEM) of 20 spontaneous calcium spikes plotted. C, Schematic of tectal neuropil imaged in zebrafish (red square). Micrograph with dashed yellow lines marking the borders of the tectal neuropil. D, Imaging Ca2+ transients in RGC axons and tectal neuron dendrites in GCaMP3, 5A, and 5G fish. Single-trial (gray) and trial-average (GCaMP3, red; 5A, cyan; 5G, blue) ΔF/F traces recorded during 2 s visual stimulation to contralateral eye (black bars below traces); stimulus bar moves through the receptive field of the imaged neurons, and is unlikely to be visible to the imaged neuron for the entire 2 s. E, Histograms depicting average (ΔF/F)max values (left), maximum (middle) (ΔF/F)max values, and SNR (right), over the neurons analyzed. Error bars indicate SEM, **p < 0.01, ***p < 0.001, n.s., not significant; two-tailed t test. F, Cumulative distributions of amplitudes of visually evoked calcium transients: p = 0.03 for 5G, p < 0.001 for 5A (compared with GCaMP3); two-sample Kolmogorov–Smirnov test. For all experiments n ≥ 35 calcium transients recorded in ≥3 larvae. G, Schematic of two-photon imaged location [optic tectum somata of Tg(elavl3:GCaMP2, 3, and 5G) fish] including micrograph. H, Left, Fraction of cells with detectable response (for 5G vs GCaMP3: paired t test, one-tailed p < 0.01). Right, Cumulative histogram of peak ΔF/F values. I, Trial-averaged (n = 11, 9, and 9 animals for GCaMP2, GCaMP3, and GCaMP5G, respectively) responses to visual stimuli of top 50% of responding cells, ranked by peak ΔF/F. J, SNR of trial-averaged responses. Cell traces were divided by the SD of 10 s preceding visual response. The average of this SNR trace for each indicator is plotted.
In vivo imaging of visual stimulus-evoked activity in mouse cortex
L2/3 neurons in the mouse primary visual cortex (V1) show a broad distribution of spike rates in response to visual stimulation (0–20 Hz; median 4 Hz) (Niell and Stryker, 2008). In contrast to other sensory areas in the rodent (O'Connor et al., 2010), the majority of V1 neurons can be driven to spike. L2/3 in V1 thus provides an ideal system to assay the sensitivity of different calcium indicators in vivo. A larger fraction of responding neurons in V1 indicates higher sensitivity for detecting APs.
We thus tested GCaMPs in V1 under similar experimental conditions used in published studies (Niell and Stryker, 2008; Kerlin et al., 2010; Zariwala et al., 2012). Both 5G and GCaMP5K (“5K”) were tested in vivo because of their superb SNR in cultured neurons (Figs. 2D, 4). Both GCaMP5s and GCaMP3 were delivered by AAV-hsyn1 viral infection. For comparison, the synthetic indicator OGB-1-AM was bulk-loaded into V1 (Stosiek et al., 2003; Ohki et al., 2005). Three weeks after AAV infection, robust GCaMP fluorescence was observed in layer 2/3 neurons (Fig. 8A, 5G). We imaged many in vivo fields of view in mice ∼3–4 weeks after infection with GCaMP5G or GCaMP3. Nuclear filling, a correlate of cytomorbidity (Tian et al., 2009), was seen in only a small fraction of cells and was comparable to GCaMP3 (Fig. 8A,B).
Figure 8.

Comparing 5G and 5K with GCaMP3 and OGB-1 in mouse visual cortex. A, Left, Schematic showing experimental setup. Right, GCaMP5G expression in layer 2/3 neurons of V1 3 weeks following AAV injection. B, Normalized fluorescence intensity along a line through the center of a cell (red line in A, right). Mean in red and standard deviation in gray. C, Responses of three cells to eight oriented moving grating stimuli; gray, single trials, blue, trial-average. D, Visual responses ((ΔF/F)max) of 438 responsive cells, rank ordered by signal level, to eight orientations aligned in columns starting with the preferred orientation. E, Fraction of visually responsive neurons (GCaMP3, 10.2%; 5G, 21.5%; 5K, 20.6%; OGB-1, 36.5%; neuropil compensation factor r = 0.7). F, Fraction of responsive neurons as a function of the strength of neuropil compensation applied. G, Averaged visually evoked calcium transients of the 10% most responsive cells at their preferred orientations, normalized to the end of the stimulus period. H, Average ΔF/F at the preferred orientation for low responder (50–80th percentile), mid responder (80–97th percentile), and high responder (>97th percentile) cells. I, Fluorescence half-decay time after stimulus offset (quantified for the 10% most responsive cells at preferred orientation). GCaMP3 (520 ± 430 ms, median ± SD), 5G (510 ± 500 ms), 5K (570 ± 580 ms), OGB-1 (1510 ± 460 ms); p = 0.73 (3 vs 5G), 0.045 (3 vs 5K), 0.083 (5G vs 5K), (p < 10−23, GCaMPs vs OGB-1; Wilcoxon signed rank sum test). J, OSI. GCaMP3, 0.87 ± 0.05; 5G, 0.76 ± 0.06; 5K, 0.82 ± 0.11; OGB-1, 0.75 ± 0.08; ANOVA1, p = 0.03. K, Tuning width. GCaMP3, 23 ± 1; 5G, 25 ± 2; 5K, 24 ± 1; OGB-1, 25 ± 1; ANOVA1, p = 0.11. L, DSI. GCaMP3, 0.35 ± 0.03; 5G, 0.33 ± 0.04; 5K, 0.4 ± 0.1; OGB, 0.3 ± 0.02; ANOVA1, p = 0.15. Error bars indicate SEM.
To elicit neuronal activity, moving gratings were presented in eight orientations to the contralateral eye of lightly anesthetized mice (Niell and Stryker, 2008; Kerlin et al., 2010). Two-photon imaging revealed visual stimulus-evoked GCaMP responses in subsets of layer 2/3 neurons (Fig. 8C,D, 5G; 438/2041 cells in five mice; ANOVA, p < 0.01). Many of these cells were orientation-selective (e.g., cells 1, 2; Fig. 8C) or direction-selective (cell 3; Fig. 8C), consistent with the known properties of these neurons (Mangini and Pearlman, 1980; Sohya et al., 2007; Niell and Stryker, 2008; Kerlin et al., 2010; Zariwala et al., 2011). Of the responsive neurons, 56% (244/438) were orientation-selective (ANOVA across eight orientations, p < 0.01), and 24% (105/438) were direction-selective (DSI > 0.5; Fig. 8D). For 5G, the average OSI of visually responsive neurons was 0.75 ± 0.24 (mean ± SD); the DSI averaged 0.32 ± 0.26. The orientation/direction selectivity was comparable to published reports based on electrophysiology (Niell and Stryker, 2008), suggesting that expression of 5G does not compromise the tuning properties of mouse cortical neurons.
Identical experiments were performed with GCaMP3 (eight mice, 3910 cells) and OGB-1 (three mice, 3606 cells). Both 5G and 5K allowed the detection of visual responses in more than twice as many neurons as GCaMP3, although sensitivity still lagged OGB-1 (Fig. 8E). This improvement did not depend on compensation for the neuropil signal (Fig. 8F; see Materials and Methods) (Kerr et al., 2005). The fluorescence half-decay time after stimulus offset was not significantly different between 5G, 5K, and GCaMP3; all three were significantly faster than OGB-1 (Fig. 8G,I). The average 5G response amplitude (peak ΔF/F) at the preferred orientation was ∼2-fold greater than GCaMP3 among low responders (50–80th percentile of all neurons), mid-responders (80–97th percentile) and high responders (>97th percentile); for 5K the low and mid-responders were ∼2-fold greater, whereas the high responders were comparable to GCaMP3 (Fig. 8H). This indicates both an improved sensitivity and dynamic range for both GCaMP5s compared with GCaMP3. Average GCaMP5G and OGB-1 OSI, DSI, and tuning width were almost indistinguishable (Fig. 8J–L) and similar to published studies based on electrophysiology. These data show that long-term expression of GCaMPs does not perturb the synaptic circuits underlying orientation and direction tuning.
The relationship between fluorescence dynamics and spiking
To characterize the relationship between spiking and GCaMP signals in vivo, we performed simultaneous cell-attached recording (Sato et al., 2007; Tian et al., 2009) and Ca2+ imaging during visual stimulation (Fig. 9A) for GCaMP5K. The goal of these experiments was twofold. First, we wanted to directly relate the fluorescence change of one of the most promising GECIs, GCaMP5K, to spiking activity. Second, in our in vivo experiments using GCaMP3 we have noticed large trial-to-trial fluctuations in the fluorescence signal (Huber et al., 2012).We thus tried to determine if the variability in fluorescence is explained by variability in neural activity coupled to the nonlinear dynamics of the sensor, or if other noise sources need to be considered.
Visually evoked spikes were tightly correlated with increases in 5K fluorescence (Fig. 9B). Single spike-induced fluorescence events were occasionally detected (e.g., Fig. 9A, arrow). In simple cells, the phasic modulation of spike rate at the temporal frequency of the drifting grating (2 Hz in this set of experiments) was reflected in a step-like change in the fluorescence signal (Fig. 9A). The orientation selectivity of the spiking responses was reflected in the 5K responses (Fig. 9B).
The relationship between fluorescence change and spike rate within the stimulus period was supralinear (Fig. 9C–E). This supralinear relationship was also evident for spontaneously occurring spikes (Fig. 9G). The amplitude of 5K signal for single APs and bursts of two or three APs was 3.6 ± 5.7, 9.1 ± 7.7, and 18.5 ± 9.7% (mean± SD), respectively. For GCaMP3, the fluorescence response to 1–3 physiological APs was barely detectable in vivo (Tian et al., 2009). The half-rise time and half-decay time for single AP-induced signals were 28 ± 5 and 268 ± 20 s, respectively (mean ± SEM). We quantified the spike detection efficiency of 5K under our imaging conditions. The detection efficiency was 29.3% for 1 AP, 63.0% for 2 APs, and 95.2% for 3 APs, at a 5% false positive rate (Fig. 9F).
The supralinear relationship between spiking and GCaMP5K fluorescence has to be taken into account in the interpretation of GCaMP-based calcium imaging. This supralinearity provides superb SNR for imaging highly active neurons; however, the supralinear relationship also emphasizes differences in spike rate across trials, leading to a larger trial-to-trial variability. Indeed, trial-to-trial variability of GCaMP5K responses was larger compared with spikes (Fig. 9I) and OGB-1 responses (Fig. 9J) during repeated presentation of preferred stimuli.
This difference in variability is completely explained by a quantitative model relating 5K signals and spiking activity. For synthetic calcium indicators convolving spikes with a single exponential kernel accounts well for the signal of several synthetic calcium indicators (Yaksi and Friedrich, 2006; Greenberg and Kerr, 2009; Kerlin et al., 2010; Komiyama et al., 2010; Sato and Svoboda, 2010). When applied to GCaMP5K, however, a single exponential model underestimates the response during strong activity (Fig. 9H, arrows), and overestimates the response during weak activity (Fig. 9H, arrowheads). Adding a simple supralinearity after the linear convolution (see Materials and Methods) greatly improves the fit (Fig. 9H, green curve). The nonlinear model reproduces the higher trial-to-trial variation of the GCaMP5K response compared with spikes (Fig. 9I). Furthermore, removing the nonlinear fit from the data, and replacing it with a linear fit, brought the trial-to-trial variability back to a similar level as estimated by spikes.
Discussion
GCaMP3 has been widely used in diverse model organisms, facilitating a large number of new neuroscience applications. However, GCaMP3 has remained lacking in detection of sparse spiking activity. All of the GCaMP5s described showed improved dynamic range compared with GCaMP3. Several GCaMP5 indicators (5A, 5D, 5G, and 5K) were further characterized in a wide variety of neuronal and astrocyte activity imaging preparations, and in several model organisms in vivo. Depending on the application, the user will have the option to pick the ideal GECI.
GCaMP5s were produced from the GCaMP3 scaffold by targeted library screening at the cpGFP/CaM proto-interface and the two interdomain linkers. Functional analysis of GCaMP variants has largely validated the conjecture that crystal structures provide snapshots of conformational states that are directly relevant to the sensing mechanism. The primary utility of GCaMP crystal structures has been to delineate the protein domain proto-interfaces and the GFP chromophore environment, which guided targeted library mutagenesis, while also supporting some atomic-level predictions. The critical importance of linker length and composition to GCaMP function is consistent with recent results in other sensor classes(Horikawa et al., 2010; Alicea et al., 2011; Marvin et al., 2011). In GCaMP and other sensors based on circularly permuted fluorescent proteins, the linkers are in close proximity to the FP chromophore, and may both directly modulate fluorescence and contribute to the efficiency of larger scale ligand-dependent domain rearrangements. We have consistently found proline to be enriched in the interdomain linkers of high-SNR variants of cpGFP-based indicators (Alicea et al., 2011; Marvin et al., 2011); the increased rigidity of the proline polypeptide backbone may limit interdomain conformational sampling to states that differ more widely in fluorescence.
Some GCaMP5 variants show a >150-fold increase in fluorescence upon calcium binding in vitro; this is approximately equivalent to the best small-molecule probes (but both have low baseline fluorescence). This 10-fold improvement did not translate into increased performance for these specific GCaMPs in more demanding preparations because of the low baseline fluorescence, underlining the importance of these medium-throughput cell-based assays in biosensor development. Performance in various in vivo and reduced preparations in a variety of cell types shows that GCaMP5A, GCaMP5G, and GCaMP5K variants consistently outperform GCaMP3. This level of improvement was also consistent for GCaMP5G fused to synaptic vesicles and the plasma membrane. These results suggest that the improvements are “intrinsic” to the sensor, rather than due to “extrinsic” factors such as [Ca2+] levels in specific cells or subcellular locations, temperature, expression level, or vagaries of the particular system tested. Biophysical characterization showed that several factors contribute to this improvement: lower Ca2+-free fluorescence, higher Ca2+-bound fluorescence, and higher Ca2+ affinity.
GCaMP5G showed the largest responses to maximal stimulation when expressed in cultured neurons, and was therefore tested in all model organisms described. Comparative experiments with GCaMP5A and GCaMP5G in zebrafish and worm, and GCaMP5G and GCaMP5K in mouse, showed that all these GCaMP5s outperformed GCaMP3 in ΔF/F and/or SNR. In worm and zebrafish, GCaMP5A showed the largest (ΔF/F)max and SNR, indicating that for some preparations GCaMP5A might be the preferred version over GCaMP5G and GCaMP3. In mouse, GCaMP5G and GCaMP5K performed similarly well.
A recent report describes variants of GCaMP3, termed “G-GECOs”(Zhao et al., 2011), optimized by selecting for maximum ΔF/F in E. coli colonies expressing random mutagenic libraries. Each of the G-GECO variants described is significantly dimmer than GCaMP3 in both the Ca2+-free and Ca2+-bound states, with a greater decrease in the former leading to a higher ΔF/F. Although G-GECO1 displayed improved KCl-evoked signal change in dissociated rat hippocampal neurons at maximum stimulation, an intrinsically dimmer sensor may complicate imaging in more complex preparations. Indeed, SNR of G-GECO1.2 was lower than GCaMP3 over the complete range of field stimuli (Fig. 2). In vitro (ΔF/F)max for the best G-GECOs are ∼2-fold higher than GCaMP3, whereas some GCaMP5 variants show a 14-fold increase in the (ΔF/F)max of GCaMP3 in vitro. GCaMP5G, the most consistently high-performing variant across in vivo assays, outperforms G-GECOs in terms of (ΔF/F)max in vitro. The improved performance of the GCaMP5s versus the G-GECOs supports the strategy of structure-guided engineering as an efficient way to improve sensors, rather than random mutagenesis.
We compared the in vivo responses of mouse visual cortical neurons labeled with two of the best GCaMP5s (GCaMP5G and GCaMP5K), GCaMP3, and OGB-1, upon presentation of visual stimuli to the mouse. The GCaMP5 variants and OGB-1 showed similar fluorescence responses for most cells at the optimal stimulus orientation; GCaMP3 fluorescence responses were significantly lower. For all GCaMPs, after stimulation, fluorescence intensity returned to baseline level significantly faster compared with OGB-1. Annotation of the total fraction of visually responsive cells by GCaMP5 was twofold improved over GCaMP3, although still trailing OGB-1. This is in agreement with the zebrafish imaging, where GCaMP5G resulted in a larger fraction of visually responsive cells as well. These results are consistent with simultaneous imaging/cell-attached recordings; GCaMP5K detects single APs and bursts of 2–3 APs much better than GCaMP3 (Tian et al., 2009), although less well than OGB-1. Detection of larger bursts of activity is also much improved for GCaMP5s compared with GCaMP3 and OGB-1, increasing the effective dynamic range of imaging. A nonlinear model of fluorescence dependence on spike rate accurately fits the in vivo data, and reduces trial-to-trial variability. Together these results show that GECIs are approaching small molecule indicators in terms of detection of sparse activity and neural activity quantification.
With its improved performance, GCaMP5 will directly enable more experiments in neuroscience and other fields of biology. Improvements in GECI transgene delivery will also contribute to increased utility. We have recently published a Cre-dependent GCaMP3 reporter mouse, allowing stable long-term expression in genetically defined neurons (Zariwala et al., 2012). Trans-synaptic delivery of calcium indicators is possible using rabies virus (Osakada et al., 2011). Zinc finger nucleases have facilitated chromosomal knock-ins in a variety of organisms, including those with few established genetic resources.
Although the GCaMP5s are currently the best GECIs for single-wavelength calcium monitoring, FRET-based sensors offer the advantage of easy donor/acceptor ratioing, primarily for motion artifact control. Several versions of the FRET-based GECI Yellow Cameleon, including YC2.6, YC3.6 (Nagai et al., 2004), and the high-affinity YC-Nano (Horikawa et al., 2010), have been reported to detect sparse neural activity in various in vivo preparations (Grewe et al., 2010). Additionally, mutants of GCaMP3, GEX-GECO1, and GEM-GECO1 (Zhao et al., 2011), offer excitation- and emission-based ratioing from a single FP chromophore. Variants of GCaMP5 harboring similar mutations, fusion to a second FP, or stoichiometric expression using viral 2A peptides may offer increased performance levels with a ratiometric output.
Although we have shown that the GCaMP5 variants constitute a significant improvement over G-GECO and GCaMP3, further GECI engineering remains. Detection of sparse spiking activity should be brought in line with the best small molecule indicators. Improvements in rise and decay kinetics are required to precisely monitor spike number and time. Long-term overexpression artifacts, such as the cytomorbid nuclear-filling phenotype, must be understood and eliminated, either through protein engineering or fine-tuned control of expression by promoter and enhancer adaptation. Improved GECIs, in combination with recent advances in light delivery and collection, fast scanning, image analysis, and behavioral paradigms, are setting the stage for chronic neural activity imaging to address fundamental questions in learning and memory, development, and the neural basis of behavior.
Notes
Supplemental material consisting of 32 indexed supporting figures is available at http://www.janelia.org/lab/looger-lab. This material has not been peer reviewed.
Footnotes
A.F. has been supported by a European Molecular Biology Organization long-term fellowship. Work in H.B.'s laboratory was funded by the National Institutes of Health (NIH) Nanomedicine Development Center “Optical Control of Biological Function,” and work in S.S.-H.W.'s laboratory was funded by NIH R01 NS045193. We thank the Janelia Farm Shared Resources for critical support of this project: A. Arnold for imaging; A. Hu and B. Shields for histology; M. Ramirez, K. McGowan, and the Molecular Biology Shared Resource for molecular biology and virus production; H. White, D. Murphy, and the Cell Culture Shared Resource for cell culturing; Instrumentation Design and Fabrication for technical development and assistance; T. Laverty and the Fly Core for fly crosses and stock maintenance; R. Simmons, R. Rogall, and H. Naz for media prep; and C. Kimmel and J. Cox for mouse support. We thank R. Behnam and A. Karpova for help in establishing lentiviral production, and Kendra Morris for virus injections. We also thank D. Rinberg and T. Tabachnik for olfactometer design and construction, and S. Hampel and J. Simpson for cloning. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. We thank Greg MacLeod for generous assistance in establishing the Drosophila NMJ preparation. H.B. and A.F. thank J. Donovan, T. Thiele, and A. Tran for technical help, and C. Niell for helping with data analysis.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Akerboom J, Rivera JD, Guilbe MM, Malavé EC, Hernandez HH, Tian L, Hires SA, Marvin JS, Looger LL, Schreiter ER. Crystal structures of the GCaMP calcium sensor reveal the mechanism of fluorescence signal change and aid rational design. J Biol Chem. 2009;284:6455–6464. doi: 10.1074/jbc.M807657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alicea I, Marvin JS, Miklos AE, Ellington AD, Looger LL, Schreiter ER. Structure of the Escherichia coli phosphonate binding protein PhnD and rationally optimized phosphonate biosensors. J Mol Biol. 2011;414:356–369. doi: 10.1016/j.jmb.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghuis BG, Tian L, Xu Y, Nikonov SS, Vardi N, Zemelman BV, Looger LL. Imaging light responses of targeted neuron populations in the rodent retina. J Neurosci. 2011;31:2855–2867. doi: 10.1523/JNEUROSCI.6064-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainard DH. The Psychophysics Toolbox. Spat Vis. 1997;10:433–436. [PubMed] [Google Scholar]
- Brand AH, Manoukian AS, Perrimon N. Ectopic expression in Drosophila. Methods Cell Biol. 1994;44:635–654. doi: 10.1016/s0091-679x(08)60936-x. [DOI] [PubMed] [Google Scholar]
- Brenner JF, Dew BS, Horton JB, King T, Neurath PW, Selles WD. An automated microscope for cytologic research a preliminary evaluation. J Histochem Cytochem. 1976;24:100–111. doi: 10.1177/24.1.1254907. [DOI] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappe ME, Seelig JD, Reiser MB, Jayaraman V. Walking modulates speed sensitivity in Drosophila motion vision. Curr Biol. 2010;20:1470–1475. doi: 10.1016/j.cub.2010.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Zhang J, Zinsmaier KE, Macleod GT. Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J Neurosci. 2010;30:1869–1881. doi: 10.1523/JNEUROSCI.4701-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold PH, Rink TJ. Fluorescence and bioluminescence measurement of cytoplasmic free calcium. Biochem J. 1987;248:313–328. doi: 10.1042/bj2480313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Del Bene F, Wyart C, Robles E, Tran A, Looger L, Scott EK, Isacoff EY, Baier H. Filtering of visual information in the tectum by an identified neural circuit. Science. 2010;330:669–673. doi: 10.1126/science.1192949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 2007;56:43–57. doi: 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Harvey CD, Tian L, Looger LL, Tank DW. Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat Neurosci. 2010;13:1433–1440. doi: 10.1038/nn.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorostkar MM, Dreosti E, Odermatt B, Lagnado L. Computational processing of optical measurements of neuronal and synaptic activity in networks. J Neurosci Methods. 2010;188:141–150. doi: 10.1016/j.jneumeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreosti E, Odermatt B, Dorostkar MM, Lagnado L. A genetically encoded reporter of synaptic activity in vivo. Nat Methods. 2009;6:883–889. doi: 10.1038/nmeth.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreosti E, Esposti F, Baden T, Lagnado L. In vivo evidence that retinal bipolar cells generate spikes modulated by light. Nat Neurosci. 2011;14:951–952. doi: 10.1038/nn.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobizhev M, Makarov NS, Tillo SE, Hughes TE, Rebane A. Two-photon absorption properties of fluorescent proteins. Nat Methods. 2011;8:393–399. doi: 10.1038/nmeth.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Greenberg DS, Kerr JN. Automated correction of fast motion artifacts for two-photon imaging of awake animals. J Neurosci Methods. 2009;176:1–15. doi: 10.1016/j.jneumeth.2008.08.020. [DOI] [PubMed] [Google Scholar]
- Grewe BF, Langer D, Kasper H, Kampa BM, Helmchen F. High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat Methods. 2010;7:399–405. doi: 10.1038/nmeth.1453. [DOI] [PubMed] [Google Scholar]
- Guizar-Sicairos M, Thurman ST, Fienup JR. Efficient subpixel image registration algorithms. Opt Lett. 2008;33:156–158. doi: 10.1364/ol.33.000156. [DOI] [PubMed] [Google Scholar]
- Horikawa K, Yamada Y, Matsuda T, Kobayashi K, Hashimoto M, Matsu-ura T, Miyawaki A, Michikawa T, Mikoshiba K, Nagai T. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow Cameleon-Nano. Nat Methods. 2010;7:729–732. doi: 10.1038/nmeth.1488. [DOI] [PubMed] [Google Scholar]
- Huber D, Gutnisky DA, Peron S, O'Connor DH, Wiegert JS, Tian L, Oertner TG, Looger LL, Svoboda K. Multiple dynamic representations in the motor cortex during sensorimotor learning. Nature. 2012;484:473–478. doi: 10.1038/nature11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultschig C, Hecht HJ, Frank R. Systematic delineation of a calmodulin peptide interaction. J Mol Biol. 2004;343:559–568. doi: 10.1016/j.jmb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Properties of larval neuromuscular junction in Drosophila melanogaster. J Physiol. 1976;262:189–214. doi: 10.1113/jphysiol.1976.sp011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman V, Laurent G. Evaluating a genetically encoded optical sensor of neural activity using electrophysiology in intact adult fruit flies. Front Neural Circuits. 2007;1:3. doi: 10.3389/neuro.04.003.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerlin AM, Andermann ML, Berezovskii VK, Reid RC. Broadly tuned response properties of diverse inhibitory neuron subtypes in mouse visual cortex. Neuron. 2010;67:858–871. doi: 10.1016/j.neuron.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, Helmchen F. Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A. 2005;102:14063–14068. doi: 10.1073/pnas.0506029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama T, Sato TR, O'Connor DH, Zhang YX, Huber D, Hooks BM, Gabitto M, Svoboda K. Learning-related fine-scale specificity imaged in motor cortex circuits of behaving mice. Nature. 2010;464:1182–1186. doi: 10.1038/nature08897. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography. 1992 [Google Scholar]
- Lister JA, Robertson CP, Lepage T, Johnson SL, Raible DW. nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development. 1999;126:3757–3767. doi: 10.1242/dev.126.17.3757. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Hegström-Wojtowicz M, Charlton MP, Atwood HL. Fast calcium signals in Drosophila motor neuron terminals. J Neurophysiol. 2002;88:2659–2663. doi: 10.1152/jn.00515.2002. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Marin L, Charlton MP, Atwood HL. Synaptic vesicles: test for a role in presynaptic calcium regulation. J Neurosci. 2004;24:2496–2505. doi: 10.1523/JNEUROSCI.5372-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde D, Wong R, Seybold PG. Fluorescence quantum yields and their relation to lifetimes of rhodamine 6G and fluorescein in nine solvents: improved absolute standards for quantum yields. Photochem Photobiol. 2002;75:327–334. doi: 10.1562/0031-8655(2002)075<0327:fqyatr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Mangini NJ, Pearlman AL. Laminar distribution of receptive field properties in the primary visual cortex of the mouse. J Comp Neurol. 1980;193:203–222. doi: 10.1002/cne.901930114. [DOI] [PubMed] [Google Scholar]
- Mank M, Griesbeck O. Genetically encoded calcium indicators. Chem Rev. 2008;108:1550–1564. doi: 10.1021/cr078213v. [DOI] [PubMed] [Google Scholar]
- Mank M, Santos AF, Direnberger S, Mrsic-Flogel TD, Hofer SB, Stein V, Hendel T, Reiff DF, Levelt C, Borst A, Bonhoeffer T, Hübener M, Griesbeck O. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat Methods. 2008;5:805–811. doi: 10.1038/nmeth.1243. [DOI] [PubMed] [Google Scholar]
- Mao T, O'Connor DH, Scheuss V, Nakai J, Svoboda K. Characterization and subcellular targeting of GCaMP-type genetically-encoded calcium indicators. PLoS ONE. 2008;3:e1796. doi: 10.1371/journal.pone.0001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Schreiter ER, Echevarría IM, Looger LL. A genetically encoded, high-signal-to-noise maltose sensor. Proteins. 2011;79:3025–3036. doi: 10.1002/prot.23118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Wallace DJ, Czubayko U, Herb JT, Schaefer AT, Looger LL, Denk W, Kerr JN. Two-photon calcium imaging of evoked activity from L5 somatosensory neurons in vivo. Nat Neurosci. 2011;14:1089–1093. doi: 10.1038/nn.2879. [DOI] [PubMed] [Google Scholar]
- Muto A, Ohkura M, Kotani T, Higashijima S, Nakai J, Kawakami K. Genetic visualization with an improved GCaMP calcium indicator reveals spatiotemporal activation of the spinal motor neurons in zebrafish. Proc Natl Acad Sci U S A. 2011;108:5425–5430. doi: 10.1073/pnas.1000887108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mütze J, Iyer V, Macklin JJ, Colonell J, Karsh B, Petrášek Z, Schwille P, Looger LL, Lavis LD, Harris TD. Excitation spectra and brightness optimization of two-photon excited probes. Biophys J. 2012;102:934–944. doi: 10.1016/j.bpj.2011.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Nevin LM, Robles E, Baier H, Scott EK. Focusing on optic tectum circuitry through the lens of genetics. BMC Biol. 2010;8:126. doi: 10.1186/1741-7007-8-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niell CM, Stryker MP. Highly selective receptive fields in mouse visual cortex. J Neurosci. 2008;28:7520–7536. doi: 10.1523/JNEUROSCI.0623-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor DH, Peron SP, Huber D, Svoboda K. Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron. 2010;67:1048–1061. doi: 10.1016/j.neuron.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Ohki K, Chung S, Ch'ng YH, Kara P, Reid RC. Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature. 2005;433:597–603. doi: 10.1038/nature03274. [DOI] [PubMed] [Google Scholar]
- Ohkura M, Matsuzaki M, Kasai H, Imoto K, Nakai J. Genetically encoded bright Ca2+ probe applicable for dynamic Ca2+ imaging of dendritic spines. Anal Chem. 2005;77:5861–5869. doi: 10.1021/ac0506837. [DOI] [PubMed] [Google Scholar]
- Osakada F, Mori T, Cetin AH, Marshel JH, Virgen B, Callaway EM. New rabies virus variants for monitoring and manipulating activity and gene expression in defined neural circuits. Neuron. 2011;71:617–631. doi: 10.1016/j.neuron.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol. 2006;13:521–530. doi: 10.1016/j.chembiol.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Pelli DG. The VideoToolbox software for visual psychophysics: transforming numbers into movies. Spat Vis. 1997;10:437–442. [PubMed] [Google Scholar]
- Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, Rubin GM. Refinement of tools for targeted gene expression in Drosophila. Genetics. 2010;186:735–755. doi: 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser-scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez Guilbe MM, Alfaro Malavé EC, Akerboom J, Marvin JS, Looger LL, Schreiter ER. Crystallization and preliminary X-ray characterization of the genetically encoded fluorescent calcium indicator protein GCaMP2. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:629–631. doi: 10.1107/S1744309108016059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TR, Svoboda K. The functional properties of barrel cortex neurons projecting to the primary motor cortex. J Neurosci. 2010;30:4256–4260. doi: 10.1523/JNEUROSCI.3774-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato TR, Gray NW, Mainen ZF, Svoboda K. The functional microarchitecture of the mouse barrel cortex. PLoS Biol. 2007;5:e189. doi: 10.1371/journal.pbio.0050189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P, Haupts U, Maiti S, Webb WW. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys J. 1999;77:2251–2265. doi: 10.1016/S0006-3495(99)77065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig JD, Chiappe ME, Lott GK, Dutta A, Osborne JE, Reiser MB, Jayaraman V. Two-photon calcium imaging from head-fixed Drosophila during optomotor walking behavior. Nat Methods. 2010;7:535–540. doi: 10.1038/nmeth.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Kracun S, Khakh BS. Monitoring astrocyte calcium microdomains with improved membrane targeted GCaMP reporters. Neuron Glia Biol. 2010a;16:1–9. doi: 10.1017/S1740925X10000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Kracun S, Sofroniew MV, Khakh BS. A genetically targeted optical sensor to monitor calcium signals in astrocyte processes. Nat Neurosci. 2010b;13:759–766. doi: 10.1038/nn.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo A, Hara Y, Yamamoto TS, Ohkura M, Nakai J, Ueno N. Tissue-tissue interaction-triggered calcium elevation is required for cell polarization during Xenopus gastrulation. PLoS One. 2010;5:e8897. doi: 10.1371/journal.pone.0008897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohya K, Kameyama K, Yanagawa Y, Obata K, Tsumoto T. GABAergic neurons are less selective to stimulus orientation than excitatory neurons in layer II/III of visual cortex, as revealed by in vivo functional Ca2+ imaging in transgenic mice. J Neurosci. 2007;27:2145–2149. doi: 10.1523/JNEUROSCI.4641-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen BR, Shea MA. Calcium binding decreases the stokes radius of calmodulin and mutants R74A, R90A, and R90G. Biophys J. 1996;71:3407–3420. doi: 10.1016/S0006-3495(96)79535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souslova EA, Belousov VV, Lock JG, Strömblad S, Kasparov S, Bolshakov AP, Pinelis VG, Labas YA, Lukyanov S, Mayr LM, Chudakov DM. Single fluorescent protein-based Ca2+ sensors with increased dynamic range. BMC Biotechnol. 2007;7:37. doi: 10.1186/1472-6750-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straw AD. Vision egg: an open-source library for realtime visual stimulus generation. Front Neuroinform. 2008;2:4. doi: 10.3389/neuro.11.004.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Suter BA, O'Connor T, Iyer V, Petreanu LT, Hooks BM, Kiritani T, Svoboda K, Shepherd GMG. Ephus: multipurpose data acquisition software for neuroscience experiments. Front Neurosci Methods. 2010;4:1–12. doi: 10.3389/fncir.2010.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R, Lee J, Plan P, Wilson J, Xin HB, Sanbe A, Gulick J, Mathai J, Robbins J, Salama G, Nakai J, Kotlikoff MI. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci U S A. 2006;103:4753–4758. doi: 10.1073/pnas.0509378103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shui B, Kotlikoff MI, Sondermann H. Structural basis for calcium sensing by GCaMP2. Structure. 2008;16:1817–1827. doi: 10.1016/j.str.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WW. Biochemical and physical properties of green fluorescent protein. In: Chalfie M, Kain SR, editors. Methods of biochemical analysis, green fluorescent protein: properties, applications, and protocols. Ed 2. Hoboken, NJ: Wiley; 2005. p. 56. [Google Scholar]
- Xu C, Webb WW. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J Opt Soc Am B. 1996;13:481–491. [Google Scholar]
- Xu N, O'Connor DH, Clack NG, Tian L, Myers EW, Looger L, Svoboda K, Magee JC. Distal dendritic activity of layer 5 pyramidal neurons in awake behaving animals signals active tactile object detection. Soc Neurosci Abstracts. 2010;36:587.1. [Google Scholar]
- Yaksi E, Friedrich RW. Reconstruction of firing rate changes across neuronal populations by temporally deconvolved Ca2+ imaging. Nat Methods. 2006;3:377–383. doi: 10.1038/nmeth874. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Zariwala HA, Madisen L, Ahrens KF, Bernard A, Lein ES, Jones AR, Zeng H. Visual tuning properties of genetically identified layer 2/3 neuronal types in the primary visual cortex of cre-transgenic mice. Front Syst Neurosci. 2011;4:162. doi: 10.3389/fnsys.2010.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zariwala HA, Borghuis BG, Hoogland TM, Madisen L, Tian L, De Zeeuw CI, Zeng H, Looger LL, Svoboda K, Chen TW. A Cre-dependent GCaMP3 reporter mouse for neuronal imaging in vivo. J Neurosci. 2012;32:3131–3141. doi: 10.1523/JNEUROSCI.4469-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca2+ indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]