

Abstract
Innate immunity factors such as conversion of the 26S proteasome to form the immunoproteasome and the Toll-like receptor signaling pathways are activated in chronic hepatitis induced by the carcinogenic drug DDC. Over time, preneoplastic hepatocyte phenotypes appear in the liver parenchyma. These changed hepatocytes expand in number because they have a growth advantage over normal hepatocytes when responding to chronic liver injury. The changed hepatocytes can be identified using immunofluorescent antibodies to preneoplastic cells e.g. FAT10/UbD, A2 macroglobulin, glutathione transpeptidase, alpha fetoprotein, glycipan 3, FAS, and gamma glutamyl transpeptidase. The formation of the preneoplastic cells occurs concomitant with activation of the Toll-like receptor signaling pathways and the transformation of the 26S proteasome to form the immunoproteasome. This transformation is in response to interferon stimulating response element on the promoter of the FAT10/UbD gene. NFκB, Erk, p38 and Jnk are also up regulated. Specific inhibitors block these responses in vitro in a mouse tumor cell line exposed to interferon gamma. Mallory-Denk bodies form in these preneoplastic cells, because of the depletion of the 26S proteasome due to formation of the immunoproteasome. Thus, MDB forming cells are also markers of the preneoplastic hepatocytes. The UbD positive preneoplastic cells regress when the liver injury induced chronic hepatitis subsides. When the drug DDC is refed to mice and chronic hepatitis is activated, the preneoplastic cell population expands and Mallory-Denk bodies rapidly reform. This response is remembered by the preneoplastic cells for at least four months indicating that an epigenetic cellular memory has formed in the preneoplastic cells. This proliferative response is prevented by feeding methyl donors such as S-adenosylmethionine or betaine. Drug feeding reduces the methylation of H3 K4, 9, and 27 and this response is prevented by feeding the methyl donors. After 8 to 15 months of drug withdrawal in mice the preneoplastic liver cells persist as single or small clusters of cells in the liver lobules. Multiple liver tumors form, some of which are hepatocellular carcinomas. The tumors immunostain positively for the same preneoplastic markers that the preneoplastic cells. Similar cells are identified in human cirrhosis and hepatocellular carcinoma indicating the relevance of the drug model described here to the preneoplastic changes associated with human chronic hepatitis and hepatocellular carcinoma.
Keywords: Mallory-Denk bodies, ubiquitin D, 26S proteasome, immunoproteasome, Toll-like receptors
INTRODUCTION
The review outline follows: 1) Role of the shift from 26S proteasome to immunoproteasome in the pathogenesis of preneoplasia in chronic drug hepatitis; 2) Role of the TLR signaling system in the pathogenesis of preneoplasia in the liver; 3) Role of NFκB p38, Src, JNK, Erk, STAT1, STAT3 and p53 in the pathogenesis of preneoplasia in the liver; 4) Role of epigenetics in the pathogenesis of preneoplasia in the liver; 5) Relevance of the preneoplastic liver cell in the mouse model to the pathogenesis of human hepatocellular carcinoma.
1. The role of the shift from the 26S proteasome to the immunoproteasome in the pathogenesis of preneoplasia in chronic drug hepatitis
Chronic hepatitis develops after 10 weeks of feeding diethyl 1, 4-dihydro-2, 4, 6-trimethyl-3, 5-pyridinedicarboxylate (DDC). At this time preneoplastic hepatocytes are identified, some of which have formed Mallory-Denk bodies (MDBs) (Nan et al., 2006a). After 1 to 4 months of withdrawal of the drug the liver returns largely to normal except for the persistence of scattered preneoplastic hepatocytes some of which have retained a few small MDBs (Nan et al., 2006b; Li et al., 2008). The preneoplastic liver cells forming MDBs immunostain positive for the catalytic subunits of the immunoproteasome (Bardag-Gorce et al., 2010a). There is at this time a reduction in 26S proteasome activity (Bardag-Gorce et al., 2010a). The immunoproteasome colocalizes with FATI0/UbD in the MDB forming hepatocytes (Bardag-Gorce et al., 2010a). The loss of the 26S proteasome activity in the MDB forming preneoplastic cells provides a mechanism for MDB formation. Aggresomes of keratin 8 and 18 form are the result of the decreased turnover of the aggregated proteins. These proteins are normally turned over by the 26S proteasome (Bardag-Gorce et al., 2010a). Quantitation of the immunoproteasome catalytic subunits LMP2, LMP 7 and MECL-1, both by Western blot and quantitative PCR, showed that the subunits were up regulated when DDC was refed for 6 days in mice withdrawn from the drug for 1 month (Bardag-Gorce et al., 2010a). The increase in the subunit proteins of the immunoproteasome was also found after the initial 10 week feeding of DDC (Bardag-Gorce et al., 2010a).
Interferon gamma (IFNg) and tumor necrosis factor have been shown to induce the formation of the immunoproteasome at the expense of the 26S proteasome (Kloetzel, 2001). Likewise, FAT10/UbD is over expressed along with the up regulation of LMP2 in response to IFNg and TNFa (Lukasiak et al., 2008). In the mouse DDC feeding model the receptors for both INFg and TNFa were up regulated and liver TNFa expression was also up regulated. INFg, but not TNFa, increased MDB formation in vitro when it was added to the tissue culture media of primary cultures of hepatocytes from mice refed DDC (Bardag-Gorce et al, 2010c). When IFNg was added to the media of mouse tumor Hepa 1-6 cell line cultures, UbD, LMP2, LMP7 and MECL-1 were all up regulated, but TNFa was not effective when added to the media (Oliva et al., 2010a). However, the combination of IFNg and TNFa was synergistic. IFNg activated the promoter for the UbD gene in vitro by binding to the interferon stimulated response element (ISRE) (Oliva et al., 2010a). When different truncated promoter of the UbD were transfected in Hepa 1-6 cells, only the D1 promoter region was activated by IFNg and TNFa, using the luciferase reporter gene. The promoter activation by IFNg was repressed by presence of p53 consensus sequence. The ISRE was shown to be located at the D1 promoter region. When the mouse tumor cell line Hepa 1-6 was incubated with both TNFa and IFNg added to the media, MDB like aggresomes (CK8 and UB positive) formed in vitro presumably by activating the immunoproteasome in vitro (Oliva et al., 2010a).
Human hepatocytes express immunoproteasome subunits in normal human liver in health and chronic hepatitis (Vasuri et al., 2010). Normal human hepatocytes express UbD/FAT10, which colocalizes with the subunits of the immunoproteasome in the cytoplasm of hepatocytes as well as in MDBs in mice refed DDC and in human hepatocytes and MDBs (French et al., 2011). Similarly, in humans, hepatocellular carcinomas that form MDBs also show colocalization of ubiquitin and the immunoproteasome subunits in the MDBs (French et al., 2011). Thus both mouse and human hepatocytes co express UbD/FAT10 and the immunoproteasome subunits when the markers of the preneoplastic phenotype are over expressed. These markers are also over expressed in HCCs in both human and mouse livers (French et al., 2011; Oliva et al., 2008).
INFg stimulates the up regulation of the expression of FAT10 and the 3 immunoproteasome subunits LMP2, LMP7 and MECL-1 at the expense of the 26S proteasome catalytic subunits. This causes a down regulation of the 26S proteasome activity, and consequently accumulation of proteins. These proteins aggregate and form Mallory Denk bodies (MDB) in the DDC refed mouse model of phenotypically switched hepatocytes (Bardag-Gorce et al., 2010a; Strehl et al., 2005; Schroder et al, 2004). At the same time, the expression of the INFg receptors (IFNgR1 and 2) were up regulated as were the TNFa receptors (TNFR21 and TNFRS21a). TNFa protein levels in the liver were also increased (Bardag-Gorce et al., 2010a). Hepatocytes from these DDC refed mice, isolated and grown in primary culture, increased the numbers of MDBs formed when IFNg, but not TNFa, was added to the media (Bardag-Gorce et al., 2010a). When INFg, but not TNFa, was added to the media of Hepa 1-6 mouse liver tumor cells, MDBs were also induced (Oliva et al., 2010a).
2. Role of the TLR signaling system in the pathogenesis of preneoplasia in the liver
Machida (Machida, 2010; Machida et al., 2009) has shown in mice transfected with HCV NS5A and fed ethanol chronically, develop cancer-initiating stem cells (CSC) which indicates that synergism between alcohol and HCV may lead to liver tumorigenesis through TLR signaling. By analogy we found that the TLR signaling system was also involved in the induction of the proliferation of preneoplastic cell phenotype, which forms tumors long after the drug has been withdrawn in the chronic mouse DDC drug-model of carcinogenesis (French et al., 2010a; French et al., 2010b). The TLR signaling pathway is an important part of the innate immune system (Szabo et al., 2007). It is involved in the development of cirrhosis and hepatocellular carcinogenesis in chronic alcoholic liver disease and hepatitis C (Szabo et al., 2007; Gao et al., 2011) where progenitor cells and cancer stem cells are found, both at the cirrhosis and carcinoma stages (Szabo et al., 2010; Oliva et al., 2010b; French 2010c). Because of this, TLR4 has been characterized as a putative proto-oncogene (Machida 2010).
Toll-like receptors 2 and 4 (TLR2, TLR4) were up regulated when DDC was refed for 7 days in the mouse model of preneoplasia and hepatocellular carcinogenesis (Nan et al., 2006a; Bardag-Gorce et al., 2010b). This was true for the expression of the genes measured by qRT-PCR and when TLR4 was measured by Western blot (Bardag-Gorce et al., 2010b). The up regulation of the expression of the two genes was completely blocked by feeding SAMe with DDC, probably as a result of the methylation of histones H3K9 and H3K27, which silences gene expression (Oliva et al., 2008; Bardag-Gorce et al., 2008).
TLR2 and 4 are up regulated by IFNg and TNFa (Oliva et al., 2008; Bardag-Gorce et al., 2008). This may be the mechanism for the up regulation of TLR2 and 4 in the DDC refed mouse model of preneoplasia and carcinogenesis, because IFNg up regulates the expression of the TLRs MyD88 and IRAK (Schroder et al., 2004; Franklin et al., 2009). CD14 is also up regulated in the DDC refed model which supports a role for the increased sensitivity of the TLR signaling response to LPS stimulation seen in alcoholic liver disease (Szabo et al., 2007). MyD88, downstream in the TLR signaling pathway, was also up regulated in the mouse model (Bardag-Gorce et al., 2010b). TRAF-6, down stream in the TLR signaling pathway, tended to be up regulated in the TLR signaling pathway in the DDC refeeding mouse model (Bardag-Gorce et al., 2010b). Evidence for activation of NFκB downstream from TRAF-6 leads to the up regulation of cytokines including IL-1. IL-1 is up regulated in the DDC refed mouse model (Bardag-Gorce et al., 2010b).
3. Role of NFκB, p38, SRC, JNK, ERK, STAT1 and 3 and p53 in the pathogenesis of preneoplasia in the liver
Evidence that NFκB activation plays an essential role down stream from the activation of the TLR signaling response is based on in vitro assays done on primary cell cultures of liver cells derived from the DDC mouse model in which DDC is fed for 10 weeks then withdrawn from DDC for 1 month (Nan et al., 2005). Using this model the preneoplastic liver cells spontaneously formed MDBs in vitro over a 6 day period (Nan et al., 2006a). Even 3 h after seeding the isolated hepatocytes from the one month drug withdrawn mice, the microarrays showed that IKK, ERK, JNK, MEKK1 and p38 were up regulated compared to liver cells from control mice (Nan et al., 2006b). FAT10/UbD positivity was increased beginning on the 2nd day of incubation at the same time as NFκB expression was up regulated and MDBs began to form in vitro (Nan et al., 2006b). NFκB was up regulated both in the livers of DDC refed mice in vivo (Yuan et al., 2000) and in the primary liver cell cultures in vitro (Nan et al., 2006a). When a NFκB inhibitor was added to the medium, no MDBs formed over 6 days of culture (Nan et al., 2006b). The NFκB inhibitor also blocked the up regulation of SRC, NFκB, p105, ERK, JNK, MEKK1 and the phosphorylation of JNK and ERK½ (Nan et al., 2006b). Using the mouse HCC cell line Hepa 1-6 model, previously used to study the immunoproteasome up regulation by IFNg (Oliva et al., 2010a), it was shown that IFNg plus TNFa up regulation of FAT10 expression was blocked by inhibitors of NFκB, JNK and p38 as well as by the presence of two p53 consensus sequences (Oliva et al., 2010a). Using this in vitro assay it was shown that IFNg treatment in vitro activated the phosphorylation of ERK and STAT1 and 3 (Oliva et al., 2010a). Activation of hepatocyte gene expression by IFNg requires the activation of both STAT 1 and IRF-1 transcription factors (Howas et al., 2011).
To further study p38 up regulation, the DDC withdrawn MDB formation 6 day in vitro assay was used. It showed that p38 phosphorylation was up regulated when MDBs formed in vitro and both the phosphorylation of p38 and MDB formation were blocked by an inhibitor of p38 phosphorylation (SB202190) (Nan et al., 2006b). Likewise, specific inhibitors of ERK and JNK phosphorylation inhibited MDB formation in the 6 day primary liver tissue culture assay (Nan et al., 2006b; Wu et al., 2005). ERK, MEKK1 and SRC were up regulated in vivo in the liver, after 10 months of DDC feeding and also after 7 days of DDC refeeding (Wu et al., 2005). TGFb1 and 2 and PKCb were up regulated in the livers in vivo, after 10 weeks of DDC feeding and also after 7 days of DDC refeeding (Nan et al., 2006b). In prior studies using Northern blots, where DDC was fed for 5 months, withdrawn for 1 month, and then refed for 7 days, c-myc and c-jun were up regulated and PPARα and RXRα were down regulated (Nagao et al., 1998). Gel retardation assays showed up regulation of AP-1, which correlated with the marked proliferation of the preneoplastic FAT10/MDB forming hepatocytes, as indicated by PCNA positive nuclear counts (Nagao et al., 1998; Oliva et al., 2008; Yuan et al., 1996). A similar proliferation response occurred when another liver toxin, thioacetamide was fed instead of DDC (Roomi et al., 2006), indicating that the preneoplastic cell phenotype has a growth advantage over the neighboring normal cells and is a nonspecific response. It also indicates that any kind of toxic liver cell injury will cause the FAT10 positive cells to proliferate.
4. Role of epigenetics in the pathogenesis of preneoplasia in the liver
The role of epigenetics in liver preneoplasia pathogenesis is important as indicated by the observation that SAMe or betaine prevented virtually all of the changes in gene expression that resulted from DDC refeeding. This included the prevention of the formation of the preneoplastic cell phenotype UbD/FAT10, MDB formation, as well as the up regulation of TLR2, TLR4, MyD88 and TRAF6, IFNg and TNFa receptors, CD14 and IL-1. The proliferation of the FAT10 positive liver cells was also prevented by SAMe and/or betaine (Bardag-Gorce et al., 2008; Oliva et al., 2009; Bardag-Gorce et al., 2010a; Oliva et al., 2008; Bardag-Gorce et al., 2010b). In addition, the expression of a large number of genes was changed in the liver after DDC refeeding and the changes were prevented by feeding SAMe or betaine with DDC (Li et al., 2008; Oliva et al., 2009). The cellular epigenetic memory was retained by the UbD/FAT10 preneoplastic hepatocytes, at least for 4 months after DDC was withdrawn (Li et al., 2008). UbD/FAT10 was expressed by individual hepatocytes and small groups of hepatocytes 8 to 15 months after DDC withdrawal when hepatocellular tumors developed (Oliva et al., 2008) indicating that the epigenetic memory of the phenotype change was retained through many generations of hepatocytes.
Many changes in acetylation and methylation of histones and methylation of DNA cytosine developed in the liver after DDC refeeding and these changes were prevented by feeding SAMe or betaine (Li et al., 2008; Bardag-Gorce et al., 2010a, Oliva et al., 2008; Bardag-Gorce et al., 2008). The expression of enzymes which methylate or demethylate DNA and histones; acetylate and deacetylate, or ubiquitinate histones were changed by DDC refeeding and SAMe or betaine prevented these changes (Bardag-Gorce et al., 2008, Bardag-Gorce et al, 2010b). The expression of the enzymes involved in SAMe, betaine and methionine metabolism were altered by DDC refeeding. Feeding SAMe or betaine prevented most of these changes, including changes in intermediates such as S-adenosylhomocysteine (Bardag-Gorce et al., 2008; Oliva et al., 2009). SAMe prevented MDB formation in 6 day cultures of primary liver cells (Li et al., 2008) as did the inhibition of deacetylation of histones by TSA (Oliva et al., 2008). Probably the most significant change induced in histone methylation was the decrease in H3K9me3 and H3K27 me3 (Bardag-Gorce et al., 2010b and 2008) induced by DDC refeeding and prevented by SAMe. A decrease in methylation of H3K9me3 and H3K27me3 would decrease the silencing of gene expression. Prevention of the demethylation of these histones would maintain the silencing of gene expression (Blackledge and Klose, 2010).
5. Relevance of the preneoplastic mouse liver cell model to the pathogenesis of human hepatocellular carcinoma
The preneoplastic cells of the DDC mouse model of chronic hepatitis stain positive for markers of progenitor cells seen in human cirrhosis and in preneoplastic foci including FAT10, MDB, AFP, OV6, CD133, A2M, GST, GGTP, HGF, CK19, H19, EGF, AIR, OV6, Nanog and FAS (Oliva et al., 2010b; Nan et al., 2006a; Li et al., 2008; French et al., 2011; French 2010c; Nan et al., 2005; Roomi et al., 2006; Nagao et al., 1999; Tazawa et al., 1983). Human cirrhosis and/or HCC progenitor/preneoplastic/cancer stem cell (CSC) markers, which stain positive immunohistochemically or by FISH, are similar to the mouse model studied here as follows: GP3, AFP, GST, MDBs, FAT10, OV6, Nanog, Oct 3, Oct4, CD49f, and CD133 (Oliva et al., 2010; Wang et al., 2008). The CSC markers which stain positive in HCC stem cells have been further extended to include CD44, ALDH and CD90 (Lingdala et al., 2010). The markers for CSC in the mouse model have been expanded to include YAP1, IMP3, (IGF2bp3) CD133 and Nanog in 29 mouse tumors and surrounding chronic hepatitis.
Figure 1 shows some histochemical double stained hepatocytes with antibodies to two different CSC markers in each double stain (FAT10, Nanog, CD133, Yap 1 and IMP3) from human cirrhosis and HCCs. These are compared with mouse liver tumors and chronic drug hepatitis from the DDC mouse model (Fig.2). Both single small cells representative of CSC and large differentiated hepatocytes stain positive for the CSC markers. The CSC cells present in cirrhotic livers, HCC, mouse liver tumors and mouse chronic hepatitis, stain positive in the cytoplasm in these cells. The nucleus rarely stained positive in these cells. The positive stained cells often showed colocalization of two CSC markers in the same cell when double stained with two CSC antibodies. Since the markers which stained positive in liver cells were present in both the tumors and the surrounding non-tumor liver tissue they must be present in both precursor progenitor cells as well as in CSC cells.
Fig 1.
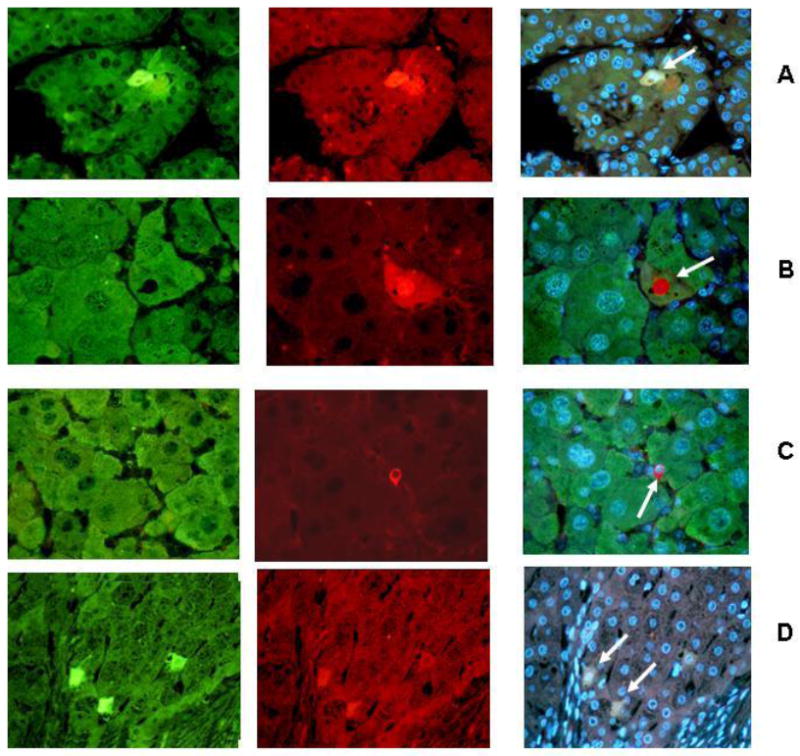
CSC markers were used (arrows) to identify progenitor /stem cells in human HCCs and cirrhosis, (1A-1D). A. Human HCC stained for Imp3 (green), Yap1 (red) and tricolor x 260. B. Human HCC stained for FAT10 (green), CD133 (red) and tricolor, x780. C. Human HCC stained for FAT10 (green), CD133 (red) and tricolor x520. D. Human cirrhosis stained for Imp3 (green), Yap1 (red) and tricolor x520.
Fig 2.
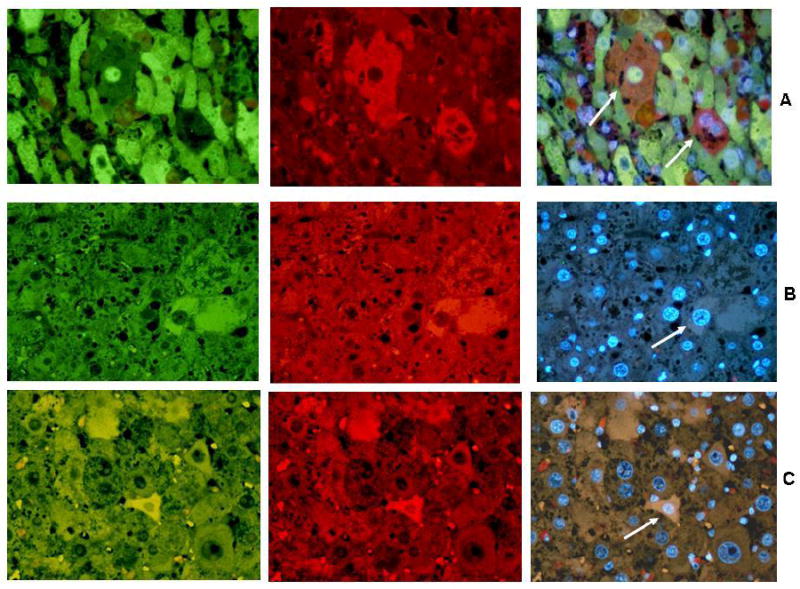
CSC markers were used to double stain mouse liver tumors and a non tumor area (arrows) (A-C). A. Mouse non tumor stained for FAT10 (green), CD133 (red) and tricolor x 780. B. Mouse liver non tumor stained for Nanog (green), FAT10 (red) and tricolor x 520. C. Mouse liver tumor stained for Yap1 (red), Imp3 (green) and tricolor x 520.
Nanog is a marker for pluripotential stem cells (Villasante et al., 2011). Its expression is regulated by Ezh2, in pluripotent stem cells (Villasante et al, 2011). TLR4 activation up regulates Nanog down stream, which leads to CD133-Nanog positive CSC cells present in liver tumors (Machida et al., 2009). When Nanog levels are high, CSC cell self renewal is supported. When Nanog levels are low, CSC cell differentiation is supported (Villasante et al., 2011). When Nanog levels are high, Imp3 and Yap1 are activated. Up regulation of Imp3 and Yap1 develops when Nanog is increased in pluripotential stem cells, in both human and mice livers (Fig.1 and 2). Ezh2 is a histone methyltransferase which regulates H3K27me3. When both Ezh2 and H3k27me3 are low, Nanog levels are high, and self renewal is supported. When the levels of H3K27me3 and Ezh2 are high at the Nanog promoter, cell differentiation is favored. Ezh2 activity is inhibited when phosphorylated by CDK1 and 2 (Chen et al., 2010) which allows the H3K27me3 epigenetic marker to persist through cell divisions (Zeng et al., 2011). In the mouse DDC refed model, H3K27me3 levels were significantly decreased, which would favor Nanog supported self renewal and the progenitor cells would proliferate (Bardag-Gorce et al., 2010b). Immunohistochemical staining of livers from mice refed DDC for both the phosphorylated and unphosphorylated Ezh2 showed that pEzh2 was sequestered within the MDBs formed by the progenitor cells (Fig. 3). This would favor self renewal. When pEzh2 levels are low Nanog levels are high, which supports cell proliferation. The results support the concept that the UbD/MDB progenitors, which proliferate in response to DDC refeeding, reduce the activity of EZH2 by phosphorylating it and sequestering the phosphorylated Ezh2 in MDBs in the phenotypically changed hepatocytes. These hepatocytes have depressed levels of H3K27me3 and reduced methylation of nuclear DNA methylcytosine (Oliva et al., 2008; Li et al., 2008). These changes would epigenetically be remembered in the phenotypically changed hepatocytes through the cell cycle when they proliferate in response to DDC refeeding. This response is completely prevented by feeding SAMe with DDC where the decrease in H3k27me3 is prevented by the methyl donor which methylates H3K27.
Fig 3.
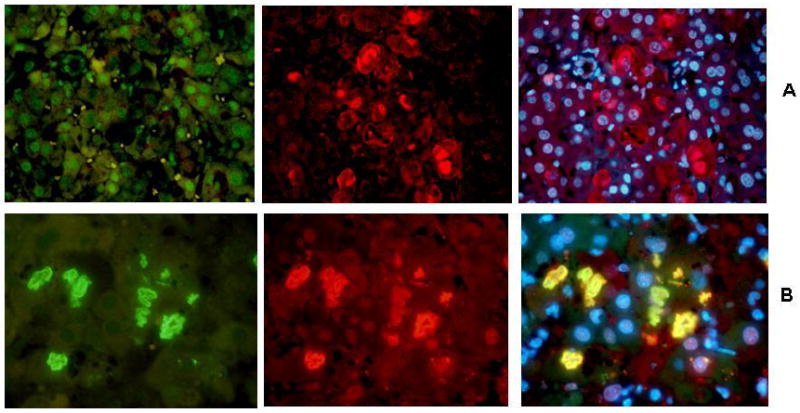
Livers of DDC refed mice double immunostained for EZH2 or phosphorylated EZH2 (pEZH2), ubiquitin and Dapi. A. Stained for EZH2 (green), ubiquitin (red) and tricolor x520. B. Stained for pEZH2 (green), ubiquitin (red) and tricolor x730.
The presence of Nanog positive, Yap1 positive, Imp3 (Igf2bp) positive CSCs in both human cirrhosis and HCC as well as DDC induced liver tumors, implicates TLR4 activation as the primary driving factor in the induction and maintenance of CSCs derived from precursor progenitor cells located within the chronic hepatitis and cirrhotic livers. Preliminary studies showed that a TLR4 knockout mice refed DDC showed a reduction in the proliferating FAT10 positive hepatocytes (Fig.4). This correlates with that SAMe or betaine prevents both TLR4 up regulation by DDC and ethanol feeding (Bardag-Gorce et al, 2010b; Oliva et al,2011), prevents the TLR4 up regulation by DDC refeeding, prevents FAT10/UbD and MDB formation by DDC refeeding (li et al 2008; Oliva et al 2008; Oliva et al, 2009). There was no reduction however, in the FAT10 positive cells in the DDC refed TLR2 mouse compared to the DDC refed wild type mouse (Fig 4).
Fig 4.
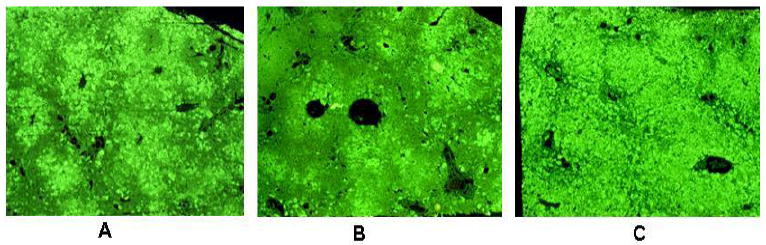
Livers from TLR4 and 2 knock out mice refed DDC showing FAT10 liver cell proliferation. A. Centrilobular proliferation of FAT10 positive cells is shown x 520. B. This shows a reduction in the proliferation of FAT10 positive liver cells in the liver of a TLR4 knock out mouse x 520. C. This shows an increase in the proliferation of FAT10 positive cells in the liver of a TLR2 knock out mouse. Almost all of the liver has been replaced by FAT10 positive liver cells x 520
Summary
TLR4 up regulation is triggered and maintained by interferon receptor IFNgR 1 and 2. NFκB, STAT 1 and 3, ERK, JNK and p38 phosphorylation are increased by INFg+TNFa up regulation (Fig 5 Inhibitors of NFκB prevented MDB formation, FAT10/UbD up regulation and the up regulation of SRC, NFκB, ERK, JNK, and MEKK1. They also prevented the phosphorylation of JNK and ERK. The increase in CD14 enhances the TLR4 up regulation by LPS. The increase in activation of NFκB increases the expression of cytokines such as IL1. INFg increases the activity of STAT1, 2 and 3. IFNg converts the 26S proteasome to the immunoproteasome which leads to 26S proteasome deficiency and MDB formation. The reduction of gene silencing by the low levels of H3K9me3 and H3k27me3 and the prevention of this change by feeding the methyl donors SAMe and betaine indicates that epigenetic mechanisms are involved. These epigenetic mechanisms include the maintenance of the oncogene Nanog self renewal of CSC in the DDC tumorigenic model of carcinogenesis, where multiple liver tumors develop (Fig 6).
Fig 5.
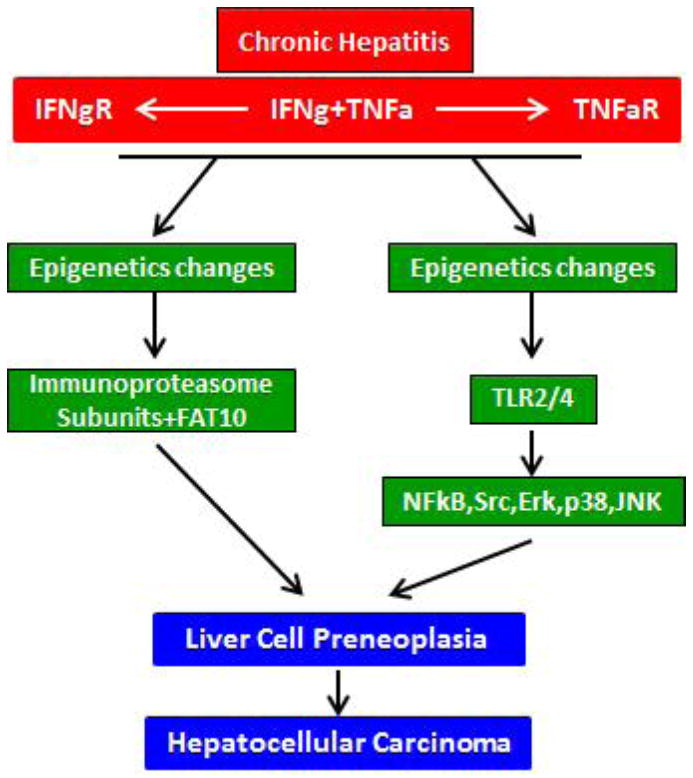
A scheme showing two signaling pathways converging to induce liver cell preneoplasia, which presumably, eventually form liver tumors. SAMe blocks both pathways by preventing epigenetic changes.
Fig 6.
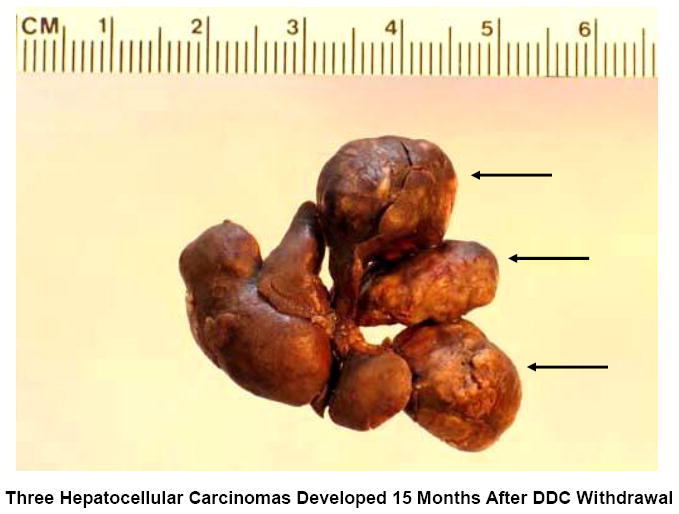
Example of multiple tumors forming in the liver of a mouse fed DDC for 10 months and then the DDC withdrawn for 15 months. Note that 3 HCCs were formed (arrows).
Acknowledgments
We thank Adriana Flores for typing the manuscript. The work was supported by Grants NIH/NIAAA 8116 and PA50-01999 Alcohol Center Grant on Liver and Pancreas Morphology Core.
Abbreviations
- DDC
diethyl-1, 4-dihydro-2, 4, 6-trimethyl-3, 5-pyridinecarboxylate
- MDBs
Mallory-Denk bodies
- UbD
ubiquitin D in mice, FAT10 in man
- ISRE
interferon stimulated response element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bardag-Gorce F, Oliva J, Villegas J, Fraley S, Amidi F, Li J, Dedes J, French B, French SW. Epigenetic mechanisms regulate Mallory Denk body formation in the livers of drug-primed mice. Exp Mol Pathol. 2008;94:113–121. doi: 10.1016/j.yexmp.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardag-Gorce F, Oliva J, French BA, Li J, Dedes J, French SW. SAMe prevents the induction of immunoproteasome and preserves the 26s proteasomes in the DDC induced MDB. Mouse Model Exp Mol Pathol. 2010a;88:352–62. doi: 10.1016/j.yexmp.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardag-Gorce F, Oliva J, Lin A, Li J, French BA, French SW. SAMe prevents the up regulation of toll-like receptor signaling in Mallory-Denk body forming hepatocytes. Exp Mol Pathol. 2010b;88:376–379. doi: 10.1016/j.yexmp.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardag-Gorce F, Oliva J, Li J, French BA, French SW. SAMe prevents the induction of the immunoproteasome and preserves the 26S proteasome in the DDC-induced MDB mouse model. Exp Mol Pathol. 2010 Jun;88(3):353–62. doi: 10.1016/j.yexmp.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Klose RJ. Histone lysine methylation: An epigenetic modification? Epigenetics. 2010;2:151–161. doi: 10.2217/epi.09.42. [DOI] [PubMed] [Google Scholar]
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of E2H2. Nature Cell Biol. 2010;121:1108–1114. doi: 10.1038/ncb2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Panoche P, Ataide MA, Lauw F, Ropert C, deOlivera RB, Pereira D, Tada MS, Noqueira P, daSilva LH, Bjorkbacka H, Golenbock DT, Gazzinelli RT. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci USA. 2009;106:5789–94. doi: 10.1073/pnas.0809742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SW, Bardag-Gorce F, Li J, French BA, Oliva J. Mallory-Denk body pathogenesis revisited. World J J Hepatol. 2010a;2:295–301. doi: 10.4254/wjh.v2.i8.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SW, Oliva J, French BA, Bardag-Gorce F. Alcohol Nutrion and Liver Cancer. Roll of toll-like receptor signaling. World J Gastroenterol. 2010b;16:1344–1348. doi: 10.3748/wjg.v16.i11.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SW. Molecular events in hepatic preneoplasia: A review. Exp Mol Pathol. 2010c;88:219–224. doi: 10.1016/j.yexmp.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French BA, Oliva J, Bardag-Gorce F, French SW. The immunoproteasome in steatohepatitis: It’s role in Mallory-Denk body formation. Exp Mol Pathol. 2011;90:252–256. doi: 10.1016/j.yexmp.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Seki E, Brenner DA, Friedman S, Cohen J, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldiwein RA, Liang MD, Andresen TK, Thomas KE, Marty AM, Costa N, Vogel SM, Fenton MJ. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J Leukoc Biol. 2003;74:277–286. doi: 10.1189/jlb.0103026. [DOI] [PubMed] [Google Scholar]
- Howas CJ, Lamb CL, Mitchell KA. Regulation of hepatocyte fate by interferon-γ cytokine. Growth Factor Reviews. 2011;22:35–43. doi: 10.1016/j.cytogfr.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetzel PM. Antigen processing by the proteasome. Nat Rev Mol Cell Biol. 2001;2:79–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- Lingala S, Cui Y-Y, Chen X, Ruebner BH, Qian X-F, Zern MA, Wu J. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp Mol Pathol. 2010;89:27–35. doi: 10.1016/j.yexmp.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bardag-Gorce F, Dedes J, French BA, Oliva J, Amidi F, French SW. S-adenosylmethionine prevents Mallory Denk body formation in drug-primed mice by inhibiting epigenetic memory. Hepatology. 2008;47:613–624. doi: 10.1002/hep.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiak S, Schiller C, Oehischaeger P, Schmidtke G, Krause P, Legler DF, Autobach F, Schmirmacher P, Breuhahn K, Groettrop M. Proinflammatory cytokines cause FAT10 up regulation in cancers of liver and colon. Oncogene. 2008;46:6068–6074. doi: 10.1038/onc.2008.201. [DOI] [PubMed] [Google Scholar]
- Machida K. TLRs, alcohol, HCV and tumorigenesis. Gastroenterol Res Practice Hindaur Publ Corp; New York: 2010. pp. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, Tsukamto H, Mkrkhiyan H, Duan L, Dynnyk A, Lin HM, Asanina K, Govindaragan S, Ray R, Ou J-H-J, Seki E, Deshaies R, Miyake K, Lai M. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. PNAS. 2009;106:1548–1553. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao Y, Yuan Q-X, Wan Y-JY, French BA, French SW. Pathogenesis of Mallory body formation: Studies using the drug-primed mouse model. Hepatol Res. 1998;13:42–64. [Google Scholar]
- Nagao Y, Wan Y-JY, Yuan QX, Kachi K, Marceau N, French SW. Mouse model of hepatocellular hyperplastic nodule formation characterization of mRNA expression. Hepatol Res. 1999;15:110–123. [Google Scholar]
- Nan L, Wu Y, Bardag-Gorce F, Li J, French BA, Wilson LT, French SW. The p105/50NF-κB pathway is essential for Mallory body formation. Exp Mol Pathol. 2005;78:198–206. doi: 10.1016/j.yexmp.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Nan L, Bardag-Gorce F, Wu Y, Li J, French BA, French SW. Mallory body forming cells express the preneoplastic hepatocyte phenotype. Exp Mol Path. 2006a;80:109–118. doi: 10.1016/j.yexmp.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Nan L, Dedes J, French BA, Bardag-Gorce F, Li J, Wu Y, French SW. Mallory body (cytokeratin aggresomes) formation is prevented in vitro by p38 inhibitor. Exp Mol Pathol. 2006b;80:228–240. doi: 10.1016/j.yexmp.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Oliva J, Bardag-Gorce F, French BA, Li J, McPhaul L, Amidi F, Dedes J, Habibi A, Nguyen S, French SW. FAT10 is an epigenetic marker for liver preneoplasia in a drug-primed mouse model of tumorigenesis. Exp Mol Pathol. 2008;84:107–112. doi: 10.1016/j.yexmp.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva J, Bardag-Gorce F, Li J, French BA, Nguyen SK, Lu SC, French SW. Betaine prevents Mallory-Denk body formation in drug-primed mice by epigenetic mechanisms. Exp Mol Pathol. 2009;86:77–86. doi: 10.1016/j.yexmp.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva J, Bardag-Gorce F, Lin A, French BA, French SW. The role of cytokines in UbD promoter regulation and Mallory-Denk body-like aggresomes. Exp Mol Pathol. 2010a;89:1–8. doi: 10.1016/j.yexmp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva J, French BA, Qing X, French SW. The identification of stem cells in human liver diseases and hepatocellular carcinoma. Exp Mol Pathol. 2010b;88:331–40. doi: 10.1016/j.yexmp.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva J, Bardag-Gorce F, Li J, French BA, French SW. S-adenosylmethionine prevents the up regulation of Toll-like receptor (TLR) signaling caused by chronic ethanol feeding in rats. Exp Mol Pathol. 2011 Jun;90(3):239–43. doi: 10.1016/j.yexmp.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roomi MW, Gaal K, Yuan QX, French BA, Fu P, Bardag-Gorce F, French SW. Preneoplastic liver cell foci induced by thioacetamide toxicity in drug primed liver. Exp Mol Pathol. 2006;81:8–14. doi: 10.1016/j.yexmp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Strehl B, Seifert U, Krüger E, Heink S, Kuckelkom U, Kloetzel PM. Interferon-gamma, the functional plasticity of the ubiquity-proteasome system, and MHC class 1 antigen processing. Immunol Rev. 2005;207:19–30. doi: 10.1111/j.0105-2896.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P, Dolganine A. Inmate immune response and hepatic inflammation. Seminars Liver Disease. 2007;27:339–350. doi: 10.1055/s-2007-991511. [DOI] [PubMed] [Google Scholar]
- Szabo G, Wands JR, Eken A, Osna NA, Weiman SA, Machida K, Wang HJ. Alcohol and hepatitis C virus-interactions in immune dysfunctions and liver damage. Alcoholism: Clin Exp Res. 2010;34:1675–1686. doi: 10.1111/j.1530-0277.2010.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazawa J, Irie T, French SW. Mallory body formation is linked to gamma glutamyl transferase induction in hepatocytes of griseofulvin-fed mice. Hepatology. 1983;3:989–1001. doi: 10.1002/hep.1840030617. [DOI] [PubMed] [Google Scholar]
- Vasuri F, Capizzi E, Bellavista E, Mishto M, Santoro A, Fiorentino M, Capri M, Cescon M, Grazi GL, Grigioni WF, D’Errico-Grigionni A, Franceschi C. Studies on immunoproteasome in human liver. Part 1: Absence in fetuses, presence in normal subjects, and increased levels in chronic active hepatitis and cirrhosis. Biochem Biophys Res Commun. 2010;397:301–306. doi: 10.1016/j.bbrc.2010.05.104. [DOI] [PubMed] [Google Scholar]
- Villasante A, Piazzolla D, Li H, Gomez-Lopez G, Djabali M, Serrano M. Epigenetic regulation of of Nanog expression by E2h2 in pluripotent stem cells. Cycle Cycle. 2011;110:1488–1498. doi: 10.4161/cc.10.9.15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Anatelli F, Zhai J, Adley B, Chuang S-T, Yang XJ. Glypican-3 as a useful diagnostic marker that distinguishes hepatocellular carcinoma from benign hepatocellular mass lesions. Arch Pathol Lab Med. 2008;132:1723–1728. doi: 10.5858/132.11.1723. [DOI] [PubMed] [Google Scholar]
- Wu Y, Nan L, Bardag-Gorce F, Li J, French BA, Wilson L-T, Debes J, French SW. The role of laminin-integrin signaling in triggering (MB) formation: An in vivo and in vitro study. Exp Mol Pathol. 2005;79:1–8. doi: 10.1016/j.yexmp.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Yuan QX, Marceau N, French BA, Fu P, French SW. Mallory body induction in drug primed mouse liver. Hepatology. 1996;24:603–612. doi: 10.1002/hep.510240324. [DOI] [PubMed] [Google Scholar]
- Yuan QX, Nagao Y, French BA, Wan Y-JY, French SW. Dexamethasone enhances Mallory body formation in drug-primed mouse liver. Exp Mol Pathol. 2000;69:202–210. doi: 10.1006/exmp.2000.2320. [DOI] [PubMed] [Google Scholar]
- Zeng XZ, Chen SA, Huang HJ. Phosphorylation of EZH2 by CDK1 and CDK2. A possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle. 2011;10:579–583. doi: 10.4161/cc.10.4.14722. [DOI] [PMC free article] [PubMed] [Google Scholar]