

Abstract
Objective
Endothelium dysfunction is an initiating factor in atherosclerosis. ADAM15 is a multi-domain metalloprotease recently identified as a regulator of endothelial permeability. However, whether and how ADAM15 contributes to atherosclerosis remains unknown.
Methods and Results
Genetic ablation of ADAM15 in apolipoprotein E-deficient mice lead to a significant reduction in aortic atherosclerotic lesion size (by 52%), plaque macrophage infiltration (by 69%), and smooth muscle cell deposition (by 82%). In vitro studies implicated endothelial derived ADAM15 in barrier dysfunction and monocyte transmigration across mouse aortic and human umbilical vein endothelial cell monolayers. This role of ADAM15 depended on intact functioning of the cytoplasmic domain, as evidenced in experiments with site-directed mutagenesis targeting the metalloprotease active site (E349A), the disintegrin domain (RDG→TDD) or the cytoplasmic tail. Further investigations revealed that ADAM15-induced barrier dysfunction was concomitant with dissociation of endothelial adherens junctions (VE-cadherin/γ-catenin), an effect that was sensitive to Src family kinase (SFK) inhibition. Through siRNA-mediated knockdown of distinct SFK members, c-Src and c-Yes were identified as important mediators of these junctional effects of ADAM15.
Conclusions
These results suggest that endothelial cell-derived ADAM15, signaling through c-Src and c-Yes, contributes to atherosclerotic lesion development by disrupting adherence junction integrity and promoting monocyte transmigration.
Keywords: Vascular permeability, Metalloproteinase, Endothelial dysfunction, Intercellular junctions, Inflammation
Introduction
The development of atherosclerosis is partially attributed to endothelial dysfunction and deregulated monocyte transendothelial migration.1 The vascular endothelium provides a selective barrier that controls the traffic of plasma proteins and circulating cells across the blood vessel wall. A major determinant of endothelial paracellular permeability is the adherens junction composed of VE-cadherin, a transmembrane molecule that forms homophilic bonds on the cell surface with its intracellular segment facilitating connection to the actin-linking α, β, and γ-catenins and p120.2 This junctional structure can be degraded by proteases 2, 3 or undergo conformational changes in response to biological or physical signaling, 4–6 leading to increased paracellular permeability and leukocyte transmigration.
The ADAM (A Disintegrin And Metalloproteinase) molecules are a family of transmembrane glycoproteins with multiple extracellular domains including metalloprotease and disintegrin domains. Among the human ADAMs identified, ADAM15 is unique due to the presence of an RGD motif in its disintegrin domain that binds the αvβ3 and αvβ1 integrins.7 Moreover, ADAM15 is among the several members in this family exhibiting sheddase activity, conferred by zinc-binding protease active sites in their metalloprotease domain, enabling cleavage of cell surface proteins.8 In addition to these characteristic extracellular domains, the C-terminal cytoplasmic tail contains consensus recognition sites for protein kinases and the Src homology (SH2 and SH3) adaptors, suggesting a potential role in intracellular signaling.8, 9 Based on these structures, ADAM15 exerts diverse functions in various physiological and pathological processes including shedding, integrin-binding and/or cell signaling transduction.7, 8
Since the initial report of ADAM15 expression in human endothelial cells 10 and in animal micro/macrovessels,11 evidence has emerged for a role of this molecule in regulating cell-cell adhesion 12 and vascular functions. 8, 9, 11 In particular, ADAM15 supports lung cancer metastasis by promoting tumor cell migration and angiogenesis.9, 13 Additionally, increased ADAM15 is detected in cytokine-stimulated endothelial cells 14 and in tissues during atherosclerosis, arthritis, and inflammatory bowl disease.8, 15–17 While these observations have suggested a pathological function of ADAM15 in inflammation, direct evidence regarding a specific role and mechanism of action in disease processes is deficient. Recently, our own investigations have implicated ADAM15 in endothelial barrier dysfunction.18 In the present study, we provide novel evidence for a pathogenic role of ADAM15 in chronic vascular inflammation in a mouse model of atherosclerosis.
Methods
Additional details are available online as Supplemental Materials.
Apoe−/−Adam15−/− mouse generation and characterization of atherosclerosis
Apoe−/− mice (C57/BL6, Jackson Laboratory) were crossbred with Adam15−/− mice (C57/BL6/129S, from Dr. Carl P. Blobel of The Hospital for Special Surgery, New York), to generate Apoe−/− Adam15+/− mice, which were subsequently bred to obtain Apoe−/− Adam15−/− (n=10) and Apoe−/− Adam15+/+ (n=10) littermates (Supplemental Figure I). Male mice at 6 weeks of age were fed an atherogenic diet (Research Diet, New Brunswick, NJ) for 12 weeks, following which heart-aorta complexes were excised for assessment of lesion. Thoracic-abdominal aortas were fixed with 10% formalin, and aortic sinuses and arches were frozen and embedded for cryo-sectioning. Oil red O staining for lesion size, Picrosirius red staining for collagen, and immunohistochemical labeling for macrophages and smooth muscle cells were performed as previously described.19 All animal procedures were conducted in compliance with NIH guidelines and approved by the Institutional Animal Care and Use Committee.
Primary culture of aortic endothelial cells
Primary aortic endothelial cells (AEC) were isolated from mice using positive immuno-selection with a rat anti-mouse CD31 antibody. 19
Monocyte isolation and transmigration
Murine peripheral monocytes were separated via histopaque (Sigma, MO) gradient centrifugation followed by negative selection (Stem Cell, BC, Canada). Transendothelial migration was examined as previously described. 18
Albumin transendothelial flux
Albumin flux across endothelial monolayers in culture was measured as an indicator of barrier properties. 18
Construction of ADAM15 mutant cDNA and transfection of HUVECs
Plasmid pcDNA containing C-terminal hemagglutinin (HA)-tagged human wild type Adam15, 20 a gift from Dr. Mark L. Day of the University of Michigan, was used as template to generate ADAM15 mutants. This included substitution of alanine for glutamate at amino acid 349 (E349A) to render the metalloproteinase domain proteolytically dead, conversion of RGD motif at 484–486 to TDD to disrupt integrin binding, and truncation of the cytoplasmic tail at tyrosine 715 (CT) to disrupt intracellular signaling.
Statistical analysis
Animal experiments utilized 10 ApoE−/− Adam15−/− mice and 10 ApoE−/− Adam15+/+ littermates for all parameters analyzed. For in vitro studies, at least three independent experiments were performed. Data are presented as mean ± SE. Unpaired Student’s t-test was employed for comparisons between two groups while multi-group analyses were performed using one-way analysis of variance (ANOVA) with Neuman-Keuls post-hoc (GraphPad Prism). Statistical significance was defined as p≤0.05.
Results
Genetic ablation of ADAM15 reduces atherosclerosis and alters cell/collagen contents in the lesion of Apoe−/− mice
Apoe−/− Adam15−/− mice displayed no change in weight (data not shown) or in serum concentration of lipids, including triglyceride, total, LDL- and HDL-cholesterol when compared with Apoe−/− Adam15+/+ littermates (Supplemental Table I). In contrast, lesion area in the thoracic-abdominal aorta of Apoe−/− Adam15−/− mice was reduced by 52% in comparison to that observed in Apoe−/− Adam15+/+ littermate controls, as demonstrated by oil red O staining (Figure 1A). Similarly, ADAM15 deficiency led to a significant reduction in the intimal area of aortic arches (Figure 1B) and the aortic sinus (Figure 1C) in Apoe−/− mice. Consistent with the reduction in lesion size, plaque macrophage infiltration was observed to be decreased in ADAM15-deficient Apoe−/− mice (69% reduction, Figure 1D). Absence of ADAM15 also resulted in a lower smooth muscle cell content (by 82%, Figure 1E) and reduced collagen deposition (by 59%, Figure 1F) when compared to plaques of littermate control animals.
Figure 1. ADAM15 deficiency reduces atherosclerosis and alters macrophage, SMC and collagen deposition in atherosclerotic lesions of Apoe−/− mice.

After consuming an atherogenic diet for 12 weeks, aortas were dissected from Apoe−/−Adam15−/− mice (n=10) and their Apoe−/−Adam15+/+ littermates (n=10). A, en face staining of lesion with oil red O in thoracic-abdominal aorta (TA), *p<0.05 vs. Apoe−/−Adam15+/+ mice. B, C, longitudinal sections of aortic arches (B) and cross-sections of aortic sinuses (C) stained with oil red O for intimal area and lipid deposition. L, lumen; A, adventitia. D–F, cryosections of aortic were immunostained for macrophages (Mφ, D) and smooth muscle cells (SMC, E) with anti-Mac-3 and anti-α-actin, respectively. Collagen (F) was stained with picrosirius red. *p<0.05, **p<0.01 vs. Apoe−/− Adam15+/+ mice. L, lumen; A, adventitia. Shown right are representative photographs.
Endothelial ADAM15 mediates monocyte transmigration
The direct role of ADAM15 in endothelial cell function was examined in primary aortic ECs and peripheral monocytes isolated from Apoe−/− Adam15+/+ and Apoe−/− Adam15−/− mice. As indicated by Western blotting, ADAM15 protein was detected in abundance in aortic ECs; however, no significant expression was detected in monocytes (Figure 2A). Similar to mouse cells, human endothelial cells displayed a high level of ADAM15; the protein was not detectable in human monocytes (Figure 2B). A flow cytometric analysis for cell surface expression of ADAM15 further supports the lack of ADAM15 in monocytes. Moreover, there was no obvious expression of ADAM15 in other types of leukocytes including neutrophils and lymphocytes (data not shown). Functionally, ADAM15 deficiency in ECs significantly attenuated transendothelial migration of monocytes in response to an MCP-1 gradient (Figure 3A). Similarly, depletion of ADAM15 in HUVECs via siRNA significantly impaired monocyte transmigration across EC monolayers (Figure 3B, Supplemental Figure IIA-C). These observations could not be attributed to altered expression of cell adhesion molecules (CAMs) on the endothelial surface, as ICAM-1, Nectin2, PECAM-1, and VCAM-1 were expressed at comparable levels in Adam15−/− vs. Adam15+/+ mouse ECs (Figure 3C and D), as well as in control vs. ADAM15 knockdown endothelial cells (Supplemental Figure III). Also, depletion of ADAM15 in endothelial cells did not affect CAM integrin expression (data not shown).
Figure 2. ADAM15 is expressed on endothelial cells but not on circulating monocytes.

A, Western blotting detected ADAM15 expression in mouse primary ECs isolated from aorta (mAEC) but not in peripheral monocytes (mMC). The bands are from the same blot. The results shown are representatives of three independent experiments. B, Western blotting and flow cytometry detected ADAM15 expression in HUVECs but not in human monocytes (hMC). Shown are representative results from more than three different donors.
Figure 3. Endothelial ADAM15 contributes to monocyte transendothelial migration.
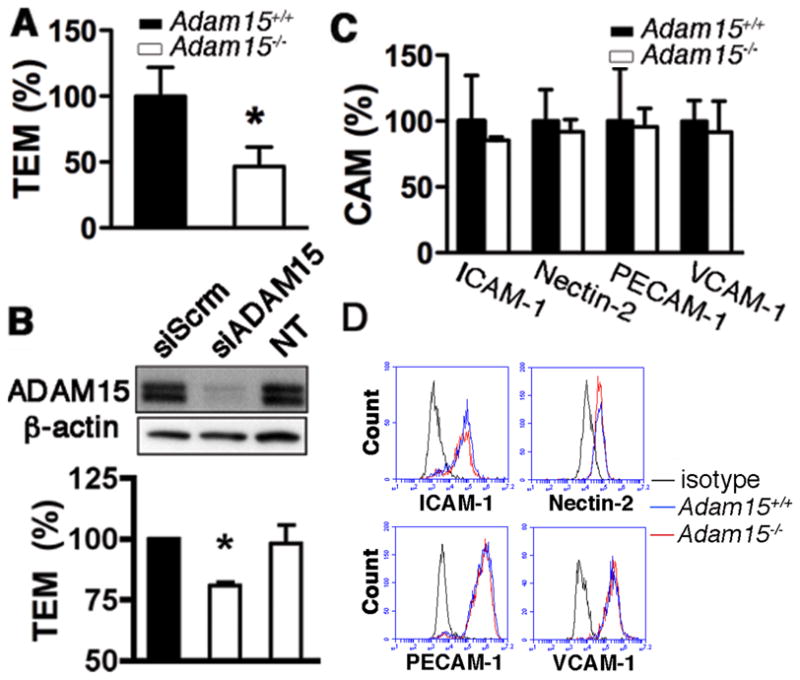
A, monocyte transmigration across Adam15−/− aortic ECs was significantly attenuated (n=4; *p<0.05 vs. Adam15+/+ ECs). B, depletion of ADAM15 with siRNA (siADAM15) in HUVECs attenuated monocyte transmigration across the HUVEC monolayer. Scrambled siRNA (siScrm) and non-transfected (NT) were used as negative controls (n=4; *p<0.05 vs. siScrm).C and D, ADAM15 deficiency in ECs did not affect surface expression of main Ig superfamily of cellular adhesion molecules (CAM), including ICAM-1, Nectin-2, PECAM-1, and VCAM-1. C, quantitative analysis of CAM expression (n=4); D, representative flow cytometric histograms.
The cytoplasmic domain is required for ADAM15-mediated monocyte transendothelial migration
With the aim of identifying the domains of ADAM15 important to facilitation of monocyte transmigration, an array of mutated cDNA constructs for human ADAM15 were generated. The E349A mutant, in which alanine was substituted for the conservative glutamic acid, had a catalytically dead metalloprotease active site. The RGD→TDD mutation rendered the disintegrin binding domain inactive and truncation of the cytoplasmic tail (CT) created a mutant deficient in intracellular signaling capabilities. Flow cytometry indicated a homogeneous increase of ADAM15 expression on the EC surface following transfection (Figure 4A, Supplemental Figure IID and 2E). Indeed, both total (Figure 4B) and cell surface expression of ADAM15 was comparable between WT and mutant constructs. Overexpression of ADAM15 WT markedly increased monocyte transmigration (Figure 4C). Interestingly, E349A and TDD mutants also increased monocyte transmigration albeit to a lesser degree (Figure 4C). In sharp contrast, overexpression of the CT mutant failed to affect monocyte transmigration across the endothelium (Figure 4C). Thus, while metalloprotease and integrin-binding activities may have a role in the full effect of ADAM15, a functional cytoplasmic C-terminus is an absolute requisite for the promotion of monocyte transendothelial migration.
Figure 4. The cytoplasmic domain of endothelial ADAM15 is important in monocyte transendothelial migration.

A and B, overexpression of ADAM15 WT and three mutants in HUVECs was detected by flow cytometry with an antibody targeting the extracellular domain (A) and western blotting with an antibody targeting HA-tag or amino acids 561–620 of ADAM15 (B). Truncation of cytoplasmic tail caused HA undetectable in CT overexpression. The arrowhead indicates the CT mutant with a reduced molecular size. The blots are representatives of three independent experiments. C, overexpression of ADAM15 WT, E349A or TDD mutant, but not CT mutant, increased monocyte transmigration across HUVEC monolayers (n=4; **p<0.01, *p<0.05 vs. mock; #p<0.05 vs. overexpression of WT). D and E, Adam15−/− mouse aortic ECs were transfected with Adam15 WT or mutant cDNA and detected with flow cytometry (D) and western blotting (E). The arrowhead indicates the CT mutant. F, rescue expression of ADAM15 in ADAM15-deficient ECs produced a similar effect to overexpression. CT mutant failed to reverse the impaired monocyte transmigration across Adam15−/− EC monolayers (n=4; *p<0.05 vs. mock; #p<0.05 vs. overexpression of WT).
To eliminate any possible confounding effects of endogenous ADAM15 in HUVECs, the activities of the WT and mutant constructs were investigated in ECs isolated from Adam15−/− mouse aorta. Due to lower transfection rates, homogenous overexpression was unobtainable in Adam15−/− aortic ECs (Figure 4D, Supplemental Figure IVA–C). In line with HUVECs, however, the mutations did not affect individual cellular transfection efficiency or total expression level of ADAM15 on the aortic EC surface (Supplemental Figure IVA–C, Figure 4E). Reflective of the findings in HUVECs, monocyte transmigration was significantly enhanced upon rescue of ADAM15 expression in aortic ECs. Again, an intact cytoplasmic domain was crucially involved (Figure 4F). Of note, the metalloprotease activity of ADAM15 appeared to play a more important role in transmigration in mouse ECs than in HUVECs (Figure 4F).
ADAM15 regulates junctional organization in a cytoplasmic domain-dependent manner
Monocyte transmigration is closely linked to endothelial barrier integrity. Consistent with our previous data,18 we report that overexpression of ADAM15 WT, E349A, or TDD constructs in HUVECs significantly increased the permeability coefficient of albumin Pa (Figure 5A), an indicator of barrier function. The essential role of the ADAM15 cytoplasmic domain in endothelial barrier dysfunction was reinforced by a blunting of the ADAM15-induced hyperpermeability in cells transfected with the cytoplasmic tail truncated mutant (Figure 5A).
Figure 5. ADAM15 cytoplasmic domain mediates junction protein dissociation.

A, overexpression of ADAM15 WT, E349A or TDD mutant, but not CT mutant, increases albumin permeability (Pa) across HUVEC monolayers (n=4; *p<0.05 vs. mock; #p<0.05 vs. overexpression of WT). B and C, immunoprecipitation assay indicated a decreased VE-cadherin/γ-catenin binding in ECs overexpressing ADAM15 WT, but not CT mutant. Transfected cell lysates were immunoprecipitated with an anti-VE-cadherin antibody followed by western analysis of VE-cadherin binding catenins. B, representative blots. C, quantification of catenins that bind VE-cadherin (n=4; **p<0.01, *p<0.05 vs. mock). D–F, knockdown of γ-catenin (siγ-Cat) in HUVECs increased albumin permeability (E) and monocyte transmigration (F). Scrambled siRNA (siScrm) served as negative control (n=4; **p<0.01 vs. siScrm).
The characterization of ADAM15 as an adherens junction molecule12 is supported by our imaging data showing both cytoplasmic and cell membrane distribution of ADAM15 in HUVECs (Supplemental Figure V). Co-localization with VE-cadherin at cell-cell junctions suggests potential interactions between ADAM15 and the adherens junction complex. With this in mind, the hypothesis that ADAM15 regulates junction integrity through alteration of VE-cadherin/catenin binding dynamics was tested. ADAM15 overexpression led to marked dissociation of γ–catenins from VE-cadherin, as demonstrated by immunoprecipitation (Figure 5B and 5C) and image analyses (Supplemental Figure VI). Morphologically, ADAM15 overexpression caused cell-cell junction disorganization evidenced by intercellular slits or gaps. The dissociation of γ–catenin from VE-cadherin was not observed in cells overexpressing ADAM15 CT mutant (Figure 5B and 5C), further supporting the importance of the cytoplasmic domain in barrier regulation. The critical role of γ–catenin in preservation of barrier integrity was further confirmed by evidence that siRNA-mediated knockdown of γ–catenin in HUVECs resulted in increased permeability to albumin (Figure 5E) and monocyte transmigration (Figure 5F).
Given that ADAM15 had no shedding effect on VE-cadherin,18 we sought to determine a signaling role in tyrosine phosphorylation of junction proteins. Western blotting showed significantly increased phosphorylation of VE-cadherin (at Y658) following ADAM15 overexpression in HUVECs (Supplemental Figure VIA) or rescue expression in Adam15−/− mouse aortic ECs (Supplemental Figure VIB). These findings were not recapitulated following ADAM15 CT mutant overexpression (Supplemental Figure VIA and VIB).
ADAM15 signals through c-Src and c-Yes to mediate junction disassociation and monocyte migration
We previously reported that Src family kinase (SFK) and MAPK signaling may be involved in the endothelial hyperpermeability response to ADAM15.18 In the present study, however, the SFK inhibitor PP2, but not the MAPK inhibitor U0126, prevented both the dissociation of γ-catenin from VE-cadherin (Figure 6A) and tyrosine phosphorylation of VE-cadherin (Supplemental Figure VIID) in response to ADAM15 upregulation. Consistently, PP2 treatment of ECs also blocked ADAM15-induced monocyte transendothelial migration (Fig. 6B). This data reveals a requirement for SFK activity in ADAM15-mediated endothelial junction responses and monocyte transmigration.
Figure 6. C-Src and c-Yes are required for ADAM15-induced VE-cadherin/γ-catenin disassociation and monocyte transmigration.

A, pretreatment of cells with 5 μmol/L SFK inhibitor (PP2) but not MEK1/2 inhibitor (U0126) for 2 hours reversed VE-cadherin/γ-catenin disassociation caused by ADAM15 overexpression (n=4; *p<0.05 vs. mock; #p<0.05 vs. ADAM15-overexpressed cells treated with vehicle.) B, pretreatment with PP2 inhibited monocyte transendothelial migration promoted by ADAM15 overexpression in HUVECs (n=4, **p<0.01 vs. ADAM15-overexpressed cells treated with vehicle.) C. HUVECs were co-transfected with ADAM15 WT cDNA and siRNA targeting Fyn, c-Src and c-Yes. Empty vector and scrambled siRNA were used as respective controls. Western blotting indicated successful knockdown of each Src family member and overexpression of ADAM15. D, knockdown of c-Src or c-Yes but not Fyn significantly decreased monocyte transmigration across ADAM15-overexpressed HUVECs (n=4; *p<0.05, **p<0.01 vs. cells transfected with mock and scrambled siRNA; #p<0.05, ## p<0.01 vs. cells transfected with ADAM15 cDNA and scrambled siRNA.) E, knockdown of c-Src or c-Yes but not Fyn significantly restored VE-cadherin/γ-catenin association in ADAM15-overexpressed HUVECs (n=3; *p<0.05, **p<0.01 vs. cells transfected with mock and scrambled siRNA; #p<0.05 vs. cells transfected with ADAM15 cDNA and scrambled siRNA).
To identify specific SFK family members involved in these effects of ADAM15, Fyn, c-Src or c-Yes expression was depleted in HUVECs via siRNA (Figure 6C). Basal junction protein association was unaffected by all SFK member-specific siRNAs (Supplemental Figure VIIC). However, knockdown of c-Src or c-Yes but not Fyn reversed ADAM15-induced VE-cadherin/γ-catenin disassociation (Figure 6E) and monocyte transmigration (Figure 6D). In accord, c-Src or c-Yes depleted HUVECs showed attenuated VE-cadherin phosphorylation in response to ADAM15 overexpression (Supplemental Figure VIID). Interestingly, knockdown of Fyn also decreased VE-cadherin phosphorylation in ADAM15-overespressed HUVECs.
Discussion
ADAM15 has been characterized as an adherens junction molecule 12 and implicated in cancer and chronic inflammatory disorders.8, 15–17 While correlative analyses showing increased abundance in inflammatory tissues and atherosclerotic lesions support its involvement in inflammation, the direct role of ADAM15 and its mechanistic contributions to particular disease processes remain to be evaluated. We demonstrate that ADAM15 deficiency is associated with attenuated vascular lesions, intimal hyperplasia, and macrophage infiltration in atherosclerotic mice. To the best of our knowledge, this constitutes the first line of evidence for a pathological role of ADAM15 in atherosclerosis. Consistent with these in vivo observations, cell experiments showed that overexpression of ADAM15 increased protein permeability and monocyte transmigration across endothelial monolayers, whereas depletion of ADAM15 resulted in the opposite. Furthermore, we report a novel signaling mechanism of ADAM15-induced endothelial barrier dysfunction mediated by its cytoplasmic domain and involving specific Src-family kinase (c-Src and c-Yes) activities.
The structural domains of ADAM15 confer diverse biological functions, including sheddase activity, integrin interactions, and cell signal transduction. The presence of a zinc-binding protease site in the extracellular metalloprotease domain renders endopeptidase activity. ADAM10 and ADAM17, close relatives of ADAM15, are known to cleave adhesion molecules involved in leukocyte transmigration, including VE-cadherin, VCAM-1, and Nectins.3, 21–23 While several substrates have been characterized as targets of ADAM15 sheddase activity,13, 20, 24 our previous study indicated that ADAM15 did not cleave VE-cadherin.18 In the present study, we further explored the sheddase activity of ADAM15 on the Ig superfamily CAMs (ICAM-1, Nectin-2, PECAM-1, and VCAM-1) with respect to their potential involvement in regulating monocyte-endothelial cell interactions during atherosclerosis development. The results showed that the presence or absence of ADAM15 in vivo did not alter the cell surface expression of these adhesion molecules, and that in vitro overexpression or knockdown of ADAM15 in endothelial cells did not affect the level of their soluble fragments (shedding products) in culture medium. These results, in combination with the finding that proteolytically dead ADAM15 increased endothelial permeability and monocyte transendothelial migration, suggest a role of ADAM15 in regulating monocyte-endothelium interactions independent of its shedding activity.
Integrin signaling in cell-matrix focal adhesions regulates the barrier property of the vascular endothelium that controls the outward flux of plasma proteins (e.g., LDL) and circulating leukocytes (e.g. monocytes), cellular processes known to contribute to atherosclerotic plaque formation. ADAMs have the capability to interact with integrins through their disintegrin domain.25 In particular, human ADAM15 contains an RGD motif that binds αvβ3 and αvβ1 integrins thereby competitively inhibiting their binding to matrix substrates.7 To investigate the specific contribution of this motif to barrier dysfunction, RGD was replaced with TDD in the disintegrin domain and this mutant form of ADAM15 still increased endothelial permeability and monocyte transmigration in the same pattern as WT ADAM15. This corroborates the previous observation that mutation of murine ADAM15 in the same manner (RDG→TDD) did not affect binding to integrins.26 On the other hand, it has been reported that RGD-blocking peptides inhibited ADAM15 binding to integrins in hematopoietic cells 27 and that RGD mutation decreased T cell adhesion to intestinal epithelial cells.28 The discrepancies may be due to different experimental conditions employed in those studies. Alternatively, ADAM15 may interact with integrins in a manner that does not require the RGD motif. 27, 29
Interestingly, truncation of the cytoplasmic tail blunted ADAM15-induced monocyte transmigration. We therefore pursued an alternative mechanism focusing on the cytoplasmic domain of ADAM15, which contains protein kinase binding sequences involved in signal transduction. The endothelial adherens junction responds to a variety of physical forces and biological factors. Under inflammatory conditions, junction responses are characterized by weakened cell-cell adhesion and intercellular gap formation.5 Rather than depending on junction protein degradation, this process can be a dynamic interaction triggered by intracellular signaling events.30 In this regard, tyrosine phosphorylation of VE-cadherin and/or catenins is viewed as an important event upstream of junction dissociation upon stimulation by cytokines and neutrophils.6, 31, 32 The current finding that ADAM15 phosphorylates and dissociates VE-cadherin/catenin complexes is indicative of a regulation of junction dynamics in a fashion similar to inflammatory mediators. A previous study suggested that dissociation of β-catenin from VE-cadherin is a key triggering event in junction signaling,2 however we found that γ-catenin/VE-cadherin dissociation appeared to be a dominant event in the ADAM15-mediated junction response. Depletion of γ-catenin led to hyperpermeability and enhanced monocyte transmigration. The importance of γ-catenin in preserving barrier function is supported by an in vitro study showing that stabilization of endothelial junctions in the presence of tyrosine phosphatase required γ-catenin, but not β-catenin. 33 Also, an in vivo study demonstrated that γ-catenin is necessary for maintenance of endothelial barrier integrity.34
Our experiments with PP2, a pan inhibitor of Src-family kinases (SFK), indicated that ADAM15-induced VE-cadherin/γ-catenin dissociation is dependent upon the tyrosine kinase activity of this family. SFK-mediated protein tyrosine phosphorylation has been implicated in angiogenesis and inflammation,31 where a functional link between the tyrosine kinase activity and endothelial hyperpermeability has been established.5, 30–32, 35 The cytoplasmic C-terminus of ADAM15 bears putative recognition sites for tyrosine kinases and Src-homology (SH3/SH2) binding sequences. 10 In hematopoietic cells, ADAM15 C-terminus binds to SFK in a phosphorylation-dependent manner.36 In cancer cells, splice variants of ADAM15 containing Src-binding sequences were associated with enhanced catalytic activity and malignant tumor behavior.24, 37 We propose that the ADAM15 cytoplasmic tail serves as a scaffold to recruit SFK into close proximity with junctional structures, where SFK-triggered signal transduction leads to VE-cadherin/γ-catenin dissociation. This hypothesis is supported by evidence of co-localization between ADAM15 and VE-cadherin observed in the present and previous studies.12
Three SFK members, Fyn, c-Src and c-Yes, have been identified in endothelial cells.38 The present finding that depletion of c-Src and c-Yes, but not Fyn, attenuated ADAM15-induced junction disruption and monocyte transmigration suggests that distinct SFKs have diverse roles in cellular events. In line with this, while VEGF-mediated angiogenesis required SFK activity in general, VEGF-induced endothelial hyperpermeability was dependent on c-Src and c-Yes but not Fyn.39 Furthermore, it might be well recognized that tyrosine kinase activity plays a critical role in endothelial hyperpermeability, but whether adherens junction opening is consequential to tyrosine phosphorylation of VE-cadherin, and indeed which specific phosphorylation sites are involved, remains controversial.2, 40 Given that knockdown of Fyn decreased VE-cadherin phosphorylation without recovering VE-cadherin/γ-catenin association in ADAM15-overexpressing endothelial cells, it is unlikely that VE-cadherin phosphorylation at tyrosine 658 is a requisite of endothelial barrier disruption. In further support of this, knockdown of c-Yes decreased VE-cadherin phosphorylation to below basal levels (Supplemental Figure VIID) without totally recovering VE-cadherin/γ-catenin association (Figure 6E).
In an effort to discern the relative importance of endothelial vs. monocyte-derived ADAM15 in junction dissociation and monocyte transmigration, we compared protein levels as well as cell surface expression of ADAM15 between endothelial cells and leukocyte subpopulations from both human and mouse blood. While abundant ADAM15 was detected in endothelial cells, it was hardly detectable in monocytes or neutrophils, indicating a minimal contribution of leukocytic ADAM15 to the observed inflammatory response. Based on the absence of ADAM15 in monocytes and the data from in vitro experiments with endothelial cell-specific ADAM15 knockdown, we suggest that endothelium-derived ADAM15 plays an essential role in mediating endothelial barrier dysfunction and leukocyte infiltration during atherosclerosis.
It is noteworthy that along with decreased macrophage infiltration, deficiency of ADAM15 also resulted in reduced SMC migration and collagen deposition into the lesion, which might be attributable to the fact that as a metalloprotease, ADAM15 is capable of digestion of type IV collagen 41 and regulation of cell migration.13 These suggest that ADAM15 may also contribute to stenosis of the artery lumen during atherosclerosis.
In conclusion, we provide novel evidence that ADAM15 contributes to atherosclerosis at least in part by promoting endothelial barrier dysfunction and monocyte transmigration. Disruption of endothelial barrier integrity by ADAM15 involves induction of VE-cadherin phosphorylation coupled with VE-cadherin/γ-catenin dissociation. The junction response to ADAM15 is mediated by its cytoplasmic domain and requires c-Src and c-Yes activity. While the current study contributes to a better understanding of both the molecular biology of ADAM15 and the pathophysiology of atherosclerosis, further investigation in this area is warranted to aid the identification of therapeutic targets for effective treatment and prevention of vascular inflammation.
Supplementary Material
Acknowledgments
We thank Dr. Danielle McLean from University of South Florida for excellent assistance in manuscript preparation. We also thank Mr. Chris Pivetti and Mr. Bert Frederich from UC Davis for animal handling, and Dr. Scott Simon and Mr. Greg Foster from UC Davis for assistance in flow cytometric analysis of leukocytes.
Sources of funding
This work was supported by the NIH grants GM97270, HL61507, HL84542 and HL96640.
Footnotes
Disclosures: none
References
- 1.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 3.Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ Res. 2008;102:1192–1201. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo M, Breslin JW, Wu MH, Gottardi CJ, Yuan SY. VE-cadherin and beta-catenin binding dynamics during histamine-induced endothelial hyperpermeability. Am J Physiol Cell Physiol. 2008;294:C977–984. doi: 10.1152/ajpcell.90607.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med. 2009;11:e19. doi: 10.1017/S1462399409001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuan SY. Signal transduction pathways in enhanced microvascular permeability. Microcirculation. 2000;7:395–403. [PubMed] [Google Scholar]
- 7.Kratzschmar J, Lum L, Blobel CP. Metargidin, a membrane-anchored metalloprotease-disintegrin protein with an RGD integrin binding sequence. J Biol Chem. 1996;271:4593–4596. doi: 10.1074/jbc.271.9.4593. [DOI] [PubMed] [Google Scholar]
- 8.Charrier-Hisamuddin L, Laboisse CL, Merlin D. ADAM-15: a metalloprotease that mediates inflammation. FASEB J. 2008;22:641–653. doi: 10.1096/fj.07-8876rev. [DOI] [PubMed] [Google Scholar]
- 9.Lucas N, Najy AJ, Day ML. The therapeutic potential of ADAM15. Curr Pharm Des. 2009;15:2311–2318. doi: 10.2174/138161209788682370. [DOI] [PubMed] [Google Scholar]
- 10.Herren B, Raines EW, Ross R. Expression of a disintegrin-like protein in cultured human vascular cells and in vivo. FASEB J. 1997;11:173–180. doi: 10.1096/fasebj.11.2.9039960. [DOI] [PubMed] [Google Scholar]
- 11.Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, Brodie TA, Ludwig T, Chiusaroli R, Baron R, Preissner KT, Manova K, Blobel CP. Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol. 2003;23:5614–5624. doi: 10.1128/MCB.23.16.5614-5624.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ham C, Levkau B, Raines EW, Herren B. ADAM15 is an adherens junction molecule whose surface expression can be driven by VE-cadherin. Exp Cell Res. 2002;279:239–247. doi: 10.1006/excr.2002.5606. [DOI] [PubMed] [Google Scholar]
- 13.Najy AJ, Day KC, Day ML. ADAM15 supports prostate cancer metastasis by modulating tumor cell-endothelial cell interaction. Cancer Res. 2008;68:1092–1099. doi: 10.1158/0008-5472.CAN-07-2432. [DOI] [PubMed] [Google Scholar]
- 14.Langer H, May AE, Bultmann A, Gawaz M. ADAM 15 is an adhesion receptor for platelet GPIIb-IIIa and induces platelet activation. Thromb Haemost. 2005;94:555–561. doi: 10.1160/TH04-12-0784. [DOI] [PubMed] [Google Scholar]
- 15.Al-Fakhri N, Wilhelm J, Hahn M, Heidt M, Hehrlein FW, Endisch AM, Hupp T, Cherian SM, Bobryshev YV, Lord RS, Katz N. Increased expression of disintegrin-metalloproteinases ADAM-15 and ADAM-9 following upregulation of integrins alpha5beta1 and alphavbeta3 in atherosclerosis. J Cell Biochem. 2003;89:808–823. doi: 10.1002/jcb.10550. [DOI] [PubMed] [Google Scholar]
- 16.Bohm BB, Aigner T, Blobel CP, Kalden JR, Burkhardt H. Highly enhanced expression of the disintegrin metalloproteinase MDC15 (metargidin) in rheumatoid synovial tissue. Arthritis Rheum. 2001;44:2046–2054. doi: 10.1002/1529-0131(200109)44:9<2046::AID-ART354>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Charrier L, Yan Y, Driss A, Laboisse CL, Sitaraman SV, Merlin D. ADAM-15 inhibits wound healing in human intestinal epithelial cell monolayers. Am J Physiol Gastrointest Liver Physiol. 2005;288:G346–353. doi: 10.1152/ajpgi.00262.2004. [DOI] [PubMed] [Google Scholar]
- 18.Sun C, Wu MH, Guo M, Day ML, Lee ES, Yuan SY. ADAM15 regulates endothelial permeability and neutrophil migration via Src/ERK1/2 signalling. Cardiovasc Res. 2010;87:348–355. doi: 10.1093/cvr/cvq060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun C, Wu MH, Yuan SY. Nonmuscle Myosin Light-Chain Kinase Deficiency Attenuates Atherosclerosis in Apolipoprotein E-Deficient Mice via Reduced Endothelial Barrier Dysfunction and Monocyte Migration. Circulation. 124:48–57. doi: 10.1161/CIRCULATIONAHA.110.988915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Najy AJ, Day KC, Day ML. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem. 2008;283:18393–18401. doi: 10.1074/jbc.M801329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim J, Lilliehook C, Dudak A, Prox J, Saftig P, Federoff HJ, Lim ST. Activity-dependent alpha-cleavage of nectin-1 is mediated by a disintegrin and metalloprotease 10 (ADAM10) J Biol Chem. 285:22919–22926. doi: 10.1074/jbc.M110.126649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fabre-Lafay S, Garrido-Urbani S, Reymond N, Goncalves A, Dubreuil P, Lopez M. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J Biol Chem. 2005;280:19543–19550. doi: 10.1074/jbc.M410943200. [DOI] [PubMed] [Google Scholar]
- 23.Singh RJ, Mason JC, Lidington EA, Edwards DR, Nuttall RK, Khokha R, Knauper V, Murphy G, Gavrilovic J. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. 2005;67:39–49. doi: 10.1016/j.cardiores.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 24.Maretzky T, Yang G, Ouerfelli O, Overall CM, Worpenberg S, Hassiepen U, Eder J, Blobel CP. Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem J. 2009;420:105–113. doi: 10.1042/BJ20082127. [DOI] [PubMed] [Google Scholar]
- 25.Wu MH. Endothelial focal adhesions and barrier function. J Physiol. 2005;569:359–366. doi: 10.1113/jphysiol.2005.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lum L, Reid MS, Blobel CP. Intracellular maturation of the mouse metalloprotease disintegrin MDC15. J Biol Chem. 1998;273:26236–26247. doi: 10.1074/jbc.273.40.26236. [DOI] [PubMed] [Google Scholar]
- 27.Nath D, Slocombe PM, Stephens PE, Warn A, Hutchinson GR, Yamada KM, Docherty AJ, Murphy G. Interaction of metargidin (ADAM-15) with alphavbeta3 and alpha5beta1 integrins on different haemopoietic cells. J Cell Sci. 1999;112:579–587. doi: 10.1242/jcs.112.4.579. [DOI] [PubMed] [Google Scholar]
- 28.Charrier L, Yan Y, Nguyen HT, Dalmasso G, Laboisse CL, Gewirtz AT, Sitaraman SV, Merlin D. ADAM-15/metargidin mediates homotypic aggregation of human T lymphocytes and heterotypic interactions of T lymphocytes with intestinal epithelial cells. J Biol Chem. 2007;282:16948–16958. doi: 10.1074/jbc.M700158200. [DOI] [PubMed] [Google Scholar]
- 29.Eto K, Puzon-McLaughlin W, Sheppard D, Sehara-Fujisawa A, Zhang XP, Takada Y. RGD-independent binding of integrin alpha9beta1 to the ADAM-12 and -15 disintegrin domains mediates cell-cell interaction. J Biol Chem. 2000;275:34922–34930. doi: 10.1074/jbc.M001953200. [DOI] [PubMed] [Google Scholar]
- 30.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2002;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 31.Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY. Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem. 1999;274:24930–24934. doi: 10.1074/jbc.274.35.24930. [DOI] [PubMed] [Google Scholar]
- 32.Tinsley JH, Ustinova EE, Xu W, Yuan SY. Src-dependent, neutrophil-mediated vascular hyperpermeability and beta-catenin modification. Am J Physiol Cell Physiol. 2002;283:C1745–1751. doi: 10.1152/ajpcell.00230.2002. [DOI] [PubMed] [Google Scholar]
- 33.Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, Filippova K, Lyck R, Engelhardt B, Kamenyeva O, Bixel MG, Butz S, Vestweber D. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. 2008;205:2929–2945. doi: 10.1084/jem.20080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin ED, Moriarty MA, Byrnes L, Grealy M. Plakoglobin has both structural and signalling roles in zebrafish development. Dev Biol. 2009;327:83–96. doi: 10.1016/j.ydbio.2008.11.036. [DOI] [PubMed] [Google Scholar]
- 35.Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol. 2004;167:223–229. doi: 10.1083/jcb.200408130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poghosyan Z, Robbins SM, Houslay MD, Webster A, Murphy G, Edwards DR. Phosphorylation-dependent interactions between ADAM15 cytoplasmic domain and Src family protein-tyrosine kinases. J Biol Chem. 2002;277:4999–5007. doi: 10.1074/jbc.M107430200. [DOI] [PubMed] [Google Scholar]
- 37.Zhong JL, Poghosyan Z, Pennington CJ, Scott X, Handsley MM, Warn A, Gavrilovic J, Honert K, Kruger A, Span PN, Sweep FC, Edwards DR. Distinct functions of natural ADAM-15 cytoplasmic domain variants in human mammary carcinoma. Mol Cancer Res. 2008;6:383–394. doi: 10.1158/1541-7786.MCR-07-2028. [DOI] [PubMed] [Google Scholar]
- 38.Bull HA, Brickell PM, Dowd PM. Src-related protein tyrosine kinases are physically associated with the surface antigen CD36 in human dermal microvascular endothelial cells. FEBS Lett. 1994;351:41–44. doi: 10.1016/0014-5793(94)00814-0. [DOI] [PubMed] [Google Scholar]
- 39.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 40.Adam AP, Sharenko AL, Pumiglia K, Vincent PA. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem. 2010;285:7045–7055. doi: 10.1074/jbc.M109.079277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin J, Eynstone LV, Davies M, Williams JD, Steadman R. The role of ADAM 15 in glomerular mesangial cell migration. J Biol Chem. 2002;277:33683–33689. doi: 10.1074/jbc.M200988200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.