

Abstract
Vanadate is a potent modulator of a number of biological processes and has been shown by crystal structures and NMR to interact with numerous enzymes. Although these effects often occur under conditions where oligomeric forms dominate, crystal structures and NMR data suggest the inhibitory form is usually monomeric orthovanadate, a particularly good inhibitor of phosphatases due to its ability to form stable trigonal-bipyramidal complexes. We performed a computational analysis of a 1.14 Å structure of the phosphatase VHZ in complex with an unusual metavanadate species, and compared it with two classical trigonal-bipyramidal vanadate-phosphatase complexes. The results support extensive delocalized bonding to the apical ligands in the classical structures. In contrast, in the VHZ metavanadate complex the central, planar VO3 moiety has only one apical ligand, the nucleophilic cysteine-95, and a gap in electron density between vanadium and sulfur. A computational analysis shows the V-S interaction is primarily ionic. A mechanism is proposed to explain the formation of metavanadate in the active site from a dimeric vanadate species that previous crystallographic evidence shows can bind to the active sites of phosphatases related to VHZ. Together, the results show that the interaction of vanadate with biological systems is not solely reliant upon the prior formation of a particular inhibitory form in solution. The catalytic properties of an enzyme may act upon the oligomeric forms primarily present in solution to generate species such as the metavanadate ion observed in the VHZ structure.
Because of vanadate’s ability to modulate a number of biological processes there is considerable interest in the origin of the interactions of this simple inorganic species with proteins.1–8 Over 173 structures in the Protein Data Bank (PDB) display the interactions of different vanadate forms with a broad number of enzymes from multiple organisms.9–13. Vanadate is a potent inhibitor of many phosphatases, enzymes with key roles in biological signaling throughout the living world. In particular, the insulin mimetic effect of vanadate is associated with its inhibition of protein tyrosine phosphatases (PTPs).14,15 Compared to orthophosphate ion (PO43), orthovanadate ion (VO43−) is a more potent inhibitor of phosphatases with a Ki that is often several orders of magnitude lower. This difference is attributed to the ability of vanadate to form a trigonal bi-pyramidal complex at the active site, resembling the transition state for phosphoryl transfer.13,16–20 Experimental data with PTPs indicate that both formation and hydrolysis of the phosphoenzyme intermediate proceed via a loose transition state with low bond orders to the nucleophile and the departing leaving group,21–24 whereas crystal structures of trigonal bi-pyramidal vanadate complexes in enzymes are commonly modeled with full bonds to the apical ligands. Previous experimental and computational results suggest that such complexes resemble the transition state only in overall geometry and charge, whereas the bond orders between vanadium and the apical ligands are higher than those of the corresponding bonds in the transition state. 25,26
An understanding of the inhibitory effect of vanadate on phosphatases, and of its biological effects, is complicated by the tendency of vanadate to oligomerize in solution.27 These effects are frequently observed under conditions where vanadate is primarily oligomerized and the monomer is a minor form.3,27 Interestingly, even though crystallization conditions often require vanadate concentrations that would primarily result in oligomeric species, crystal structures almost exclusively show monomeric vanadate at the active site. This has been attributed to the facile interconvertability of different vanadate species in solution and the ability of the active site of phosphatases to selectively stabilize the monomeric form.28 Here, we report results indicating that the classical trigonal bi-pyramidal vanadate species is not the only form capable of binding to PTPs, and that other forms contribute to the inhibition of PTPs and potentially to other biological effects of vanadate.
VHZa is a recently described member of the PTP family of phosphatases.29 A recently obtained high-resolution structure of VHZ in complex with vanadate revealed what appeared to be an unusual “metavanadate” in the active site (Figure 1; PDB ID 4ERC). The VO3 moiety is coordinated to the sulfur atom of cysteine 95 as one apical ligand, with a 2.4 Å V-S distance. The opposite apical position is occupied by a nitrogen atom of the arginine 60 (RS60) side chain trapped in the active site from a symmetry-related VHZ molecule in the crystal (Figure 2A). The V-N distance of 3.2Å argues against a significant bonding interaction, nor would a significant interaction be expected with the positively charged guanidinium group. Although the V-S distance is typical of those commonly observed in trigonal bi-pyramidal vanadate-PTP complexes,17 a distinct electron density gap between the atoms is evident in the high resolution unbiased composite omit map (Figure 1). Furthermore, the VO3 moiety is nearly planar, while a tetrahedral geometry would be expected from a covalent V-S bond and the absence of an apical V-N bond.20,30 These observations suggest that the VO3 moiety is better described as an electrostatically stabilized metavanadate (VO3−) rather than a covalent thiovanadate adduct. A computational analysis of this structure was carried out to analyze the unusual bonding in this complex, compared with more typical, previously reported vanadate-PTP structures (Figure 2). The two more conventional structures chosen for comparison were the complex of PTP1B with a vanadate ester of the peptide DADEYL (Figure 2B, PDB ID 3I7Z), and PTP1B with orthovanadate (Figure 2C, PDB ID 3I80). Both of the latter structures are typical of the trigonal bi-pyramidal vanadate-PTP complexes well represented in the literature. The nature of the chemical bonding between the VO3 ion and the protein was assessed using quantum mechanical calculations. For this purpose portions of the proteins shown in Figure 2 were extracted directly from the crystal structures.
Figure 1.
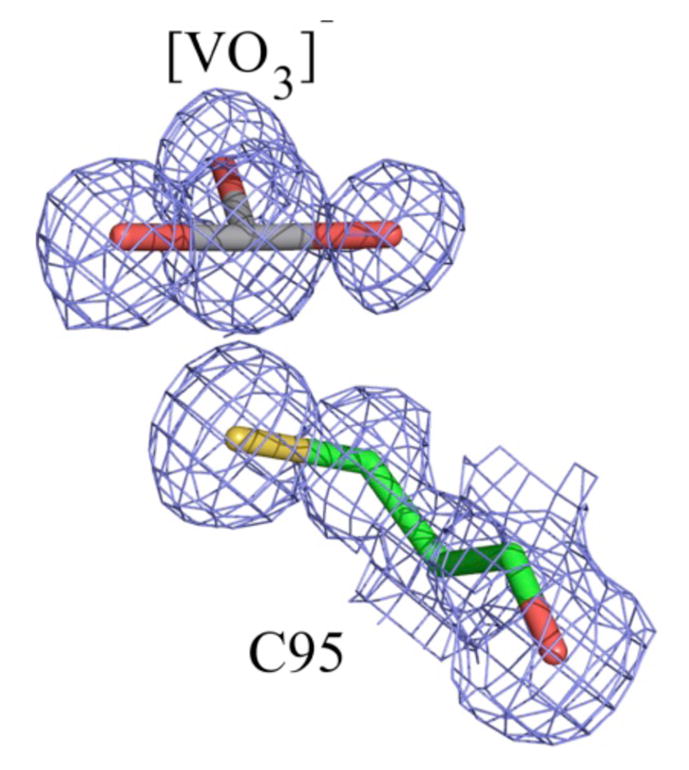
The electron density at the VHZ active site suggests a non-covalently bound VO3− ion. Shown is an unbiased composite omit map contoured at 1.5 σ in the refined model.
Figure 2.

The regions of the three vanadate complexes used in quantum mechanical calculations. (A) VHZ-VO3 (PDB 4ERC) (B) PTP1B-Tyr-VO4 (PDB 3I7Z), (C) PTP1B-VO4 (PDB 3I80). Hydrogens have been omitted from the picture for the sake of clarity. In the VHZ complex, the Rs60 residue in the active site comes from a symmetry-related VHZ molecule. DADEYL peptide was omitted with exception of Y side chain.
Truncation was done to include all residues interacting with vanadate; all residues with hydrogen bonds or pi-stacking to residues that interact with vanadate; and all groups that are polar or charged, and located in proximity to the vanadate ion even if they do not directly interact. Hydrogen atoms were added to satisfy all dangling valencies using the Chimera program.31 The standard protonation states of amino acids were assumed (deprotonated Asp, Glu, protonated Lys and Arg), except for the Tyr coordinating vanadium in Figure 2B, which was deprotonated. The nucleophilic cysteine was assumed to be deprotonated; this residue has a pKa in the range of 5.5 to 6 in this enzyme family.32 The truncated complexes effectively contained the first and the second coordination spheres of the vanadate ion, and consisted of 140–170 atoms (including hydrogens), depending on the complex. The spin states of the complexes were zero, assuming the empty d-shell in vanadium. The Natural Bond Orbital (NBO) analysis,33–36 at the B3LYP37–39/6-31G*40,41 level of theory, was used to elucidate the chemical bonds present in the complexes. This level of approximation, including the fairly modest size of the basis set, were considered adequate for the problem at hand, because NBO results in general are quite insensitive to the basis set used, and because only the qualitative assessment was required to differentiate the state of V in the complexes. Calculations were performed using the Gaussian 09 suite of programs42.
Table 1 contains the key electronic structure parameters characterizing the vanadium atom and its bonding environment in the three complexes. There is an apparent difference between the bonding exhibited by VHZ complex containing VO3 (Figure 2A) and the other two complexes. As expected from its distance of 3.2 Å, there is no significant interaction between the V and the apical N of Rs60. The partially populated lone pair (LP) on N of the distant apical Arg (1.66 |e|) donates electron density to the guanidinium group, participating in π-resonance, and is only weakly coordinated to V through Van der Waals interactions. One of the O atoms of the VO3 ion exhibits a pi bond with V, representing one of the resonance structures (the lowest energy one) possible in this π-delocalized VO3 ion. There is a clear 2 center – 2 electron (2c-2|e|) bond between V and SCys95. This bond is primarily ionic, with 79% of electron density residing on S and 21% on V. This is consistent with the electron density gap observed in the unbiased composite omit map between the C95 sulfur and V, which is indicative of ionic rather than covalent ligand stabilization. The bonding within the vanadate moiety and its interaction with VHZ differ in several ways from the more conventional PTP1B complexes. In contrast to the VHZ-VO3 complex 2A, in the PTP1B complexes 2B and 2C there is no π-bonding within the central VO3 part of the ion. This is reflected in different averaged V-O bond lengths in the x-ray structures, which are 1.70 Å for 2A, 1.89 Å for 2B, and 1.91 Å for 2C. One lone pair on SCys215 (LP3) and one on OTyr (LP3) are populated only by ca. 1.7 |e|, within the NBO localized picture. The low-populated lone pair on S (LP3) partially donates electron density to the d-atomic orbitals (AOs) of vanadium. The low-populated lone pair on O of Tyr (LP3) primarily donates electrons into 3-resonance within the Tyr phenyl ring. However, both LP2 and LP3 on OTyr also donate to the d-AOs on vanadium.
Table 1.
Electronic structure parameters characterizing the vanadium atom and its bonding in complexes 2A–C, calculated with NBO at B3LYP/6-31G*: Charges on atoms (Q), electronic populations of 2 center-2 electron (2c-2|e|) σ and bonds, and π electronic populations of lone pairs (LP). Atoms are labeled according to Figure 2.
| VO3 (2A) | VO4 (2B) | VO4 (2C) | |
|---|---|---|---|
|
| |||
| Q(V) | +1.09 | +1.34 | +1.33 |
| Q(SCys) | −0.31 | −0.42 | −0.42 |
| Q(N/Oapical) | −0.79a | −0.68 | −0.90 |
|
| |||
| 2c-2|e| bonds: | |||
| V-O1 | 1.99 |e| - σ | 1.99 |e| | 1.98 |e| |
| 1.96 |e| - π | none | none | |
| V-O2 | 1.98 |e| | 1.99 |e| | 1.99 |e| |
| V-O3 | 1.98 |e| | 1.99 |e| | 1.98 |e| |
| V-S | 1.95 |e| | none | none |
| Lone pairs: | |||
| LP1(S) | 1.94 |e| | 1.94 |e| | 1.94 |e| |
| LP2(S) | 1.87 |e| | 1.88 |e| | 1.88 |e| |
| LP3(S) | noneb | 1.73 |e| | 1.73 |e| |
| V-N or Oapical | none | none | none |
| LP1(N or Oapical) | n/ac | 1.91 |e| | 1.94 |e| |
| LP2(Oapical) | n/a | 1.86 |e| | 1.83 |e| |
| LP3(N or Oapical) | 1.66 |e| | 1.61 |e| | 1.65 |e| |
−0.79 is the atomic charge on the Rs60 nitrogen atom in an apical position to vanadium; the total charge on the guanidinium group of this arginine is +0.6.
“none” indicates that suitable orbitals are available, but not occupied.
“n/a” indicates that the lone pair orbitals are not present.
To summarize, in complexes 2B and 2C neither the V-SCys, nor the V-OTyr apical interactions are classic 2c-2|e| bonds. Bonding between vanadium and these apical ligands is significant and highly delocalized between SCys, V, and OTyr. The equatorial V-O interactions are single bonds. In contrast, the complex 2A can be described as a π-delocalized VO3 ion with only one apical interaction, an ionic bond between V and SCys. PTP-VO4 complex 2C.
The complex solution chemistry of vanadate must be considered together with the chemical properties of the enzymes it can associate with. Monomeric vanadate dominates at low concentrations, with dimeric forms becoming significant at concentrations above 0.2 mM, followed by higher oligomers.28 Interconversion is rapid, and it is logical that the various forms of vanadate should be similarly labile in an enzymatic active site. There is no reason, a priori, that an inhibiting form of vanadate must pre-form in solution before binding to the enzyme. While numerous oligomeric forms of vanadate have been documented, metavanadate has not been reported in solution, and presumably was generated by the enzyme from some other vanadate species present in solution, and subsequently stabilized by the active site environment. We propose a simple mechanism that can explain the formation of this species from divanadate, and possibly polyvanadate, forms that are well known to exist, and usually dominate, in solution. In light of the highly conserved nature of the active sites of PTPs, this mechanism would apply to this family as whole, and potentially to any similar enzyme with a general acid. This proposal (Figure 3) incorporates the general acid conserved in PTPs, and also explains why the first report of a divanadate species at the active site of a PTP was found in a YopH W354F mutant in which the flexible loop that bears the general acid is locked in a catalytically unproductive position.43 The crystal structure of this divanadate complex showed that dimeric, and possibly higher oligomeric species, are capable of binding to PTPs (Figure 3D). We believe it is no coincidence that this complex was identified in a mutant in which general acid catalysis is disabled. Presumably, this or other dimeric forms bind to the native enzyme, but are rapidly hydrolyzed to the monomer and not observed crystallographically.
FIGURE 3.

A proposed mechanism that explains the formation of a metavanadate species in the active site of VHZ. The vanadium species D, F and G have been observed in crystal structures. Protonation states of the vanadium species are not known from the crystal structures, but are depicted in states consistent with the pH at which crystals were grown. D) PDB ID 3F9B - divanadate bound in the active site of the YopH mutant W354F with catalytic WPD (acid)-loop locked in a half-open conformation; E) transient complex (not observed in the crystal form) showing the acid-loop closure which triggers the catalytic transformation of divanadate into metavanadate; VHZ has no mobile loop but its general acid occupies an analogous position. F) PDB ID 4ERC– metavanadate trapped in the active site of VHZ; G) PDB ID 3I80 – orthovanadadate in the active site of PTP1B.
According to the proposed mechanism, divanadate (Figure 3D) binds in the active site and the general acid protonates one of its bridge oxygens, triggering decomposition and formation of “metavanadate”, stabilized by the P-loop and electrostatic interaction with the nucleophilic thiolate. The resulting complex (Figure 3E) is the species trapped in the VHZ-VO3 crystal structure. The classically observed trigonal bi-pyramidal orthovanadate species can result from a process analogous to the second step of the PTP-catalyzed reaction, in which a nucleophilic water attacks with positioning assistance of Q-loop residues, assuming the other apical position. Such a process would explain the high affinity and selectivity in crystal structures of PTPs for monomeric orthovanadate under conditions where oligomeric polyvanadates dominate in solution. Recent work44 shows that vanadate speciation is sensitive to surfaces such as micelles, consistent with the idea that an enzyme active site could preferentially stabilize a species such as the dimer in 3D, or metavanadate.
We note that the reversible interconversion between F and G in Figure 3 provides an alternative pathway for enzyme-catalyzed metavanadate formation without invoking binding of a dimeric form. However, the VHZ crystals were grown in the presence of 10 mM vanadate, far above the 0.2 mM level at which dimeric and higher forms begin to dominate. Potent vanadate inhibition of many phosphatases is reported across wide ranges of pH and vanadate concentrations, supporting the notion that multiple forms are capable of binding, and that the interconvertability in the active site is similar to that in solution. If anything, the active site of an enzyme optimized to catalyze hydrolysis should facilitate such processes.
The metavanadate species observed here is not an anomalous structure only relevant to VHZ, or potentially other PTPs. A geometrically similar VO3 species has been previously observed in a crystal structure of phosphoglucomutase,45 an enzyme that has no similarity to PTPs. That structure also reveals a planar VO3 moiety with a single apical ligand (an aspartate carboxyl group) and a similar gap in the 2Fo-Fc electron density obtained from the Electron Density Server (EDS) observed at 1.0 σ.
In summary, this work shows that the interaction of vanadate with the active sites of PTPs in solution is more versatile than the simple trigonal bi-pyramidal model that dominates the RCSB. The inhibitory effect of vanadate can involve dimeric, and potentially higher oligomers, an important factor since such species often dominate in solution. The ability of the active site environment to electrostatically stabilize a metavanadate ion adds another facet to the interaction of vanadate with such enzymes. In the VHZ-VO3 complex there is only a single significant apical interaction, which is primarily ionic.
This complex is a good analogue of the loose metaphosphate-like transition state concluded from experimental studies. The average V-Oeq bond distance in the VHZ-VO3 structure is shorter compared to the commonly observed trigonal bi-pyramidal vanadate complexes, but in a good agreement with experimental values determined with Raman difference spectroscopy of vanadate at the active site of the related enzyme YopH.25 Hence, it is conceivable that such VO3 species may form more commonly than suspected in PTP-vanadate solutions, and the dominance among crystal structures of the trigonal bi-pyramidal structures (like 3E and 3F) arise from species that subsequently form and are more amenable to trapping. The rich speciation of vanadate in solution clearly extends to its ability to also adopt multiple species at enzymatic active sites.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM 47297 (ACH), and a DARPA Young Faculty Award N66001-11-1-4138 (ANA).
Footnotes
The name VHZ denotes VH1-like member Z, one of the group of small Vaccinia virus VH1-related dual specific phosphatases.
Supporting Information. The absolute energies (in Hartrees) and the atomic coordinates of the computationally analyzed structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bishayee A, Waghray A, Patel MA, Chatterjee M. Cancer Lett. 2010;294:1. doi: 10.1016/j.canlet.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 2.Cam MC, Brownsey RW, McNeill JH. Can J Physiol Pharmacol. 2000;78:829. [PubMed] [Google Scholar]
- 3.Crans DC, Smee JJ, Gaidamauskas E, Yang L. Chem Rev. 2004;104:849. doi: 10.1021/cr020607t. [DOI] [PubMed] [Google Scholar]
- 4.Evangelou AM. Crit Rev Oncol Hematol. 2002;42:249. doi: 10.1016/s1040-8428(01)00221-9. [DOI] [PubMed] [Google Scholar]
- 5.Heyliger CE, Tahiliani AG, McNeill JH. Science. 1985;227:1474. doi: 10.1126/science.3156405. [DOI] [PubMed] [Google Scholar]
- 6.Rubinson KA. Proc R Soc Lond B Biol Sci. 1981;212:65. doi: 10.1098/rspb.1981.0025. [DOI] [PubMed] [Google Scholar]
- 7.Tsiani E, Bogdanovic E, Sorisky A, Nagy L, Fantus IG. Diabetes. 1998;47:1676. doi: 10.2337/diabetes.47.11.1676. [DOI] [PubMed] [Google Scholar]
- 8.Tsiani E, Fantusa IG. Trends in Endocrinology and Metabolism. 1997;8:51. doi: 10.1016/s1043-2760(96)00262-7. [DOI] [PubMed] [Google Scholar]
- 9.Crans DC, Sudhakar K, Zamborelli TJ. Biochemistry. 1992;31:6812. doi: 10.1021/bi00144a023. [DOI] [PubMed] [Google Scholar]
- 10.Crans DC, Willging EM, Butler SR. JACS. 1989;112:427. [Google Scholar]
- 11.Messmore JM, Raines RT. Journal of the American Chemical Society. 2000;122:9911. doi: 10.1021/ja0021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Messmore JM, Raines RT. Archives of biochemistry and biophysics. 2000;381:25. doi: 10.1006/abbi.2000.1951. [DOI] [PubMed] [Google Scholar]
- 13.Stankiewicz PJ, Gresser MJ. Biochemistry. 1988;27:206. doi: 10.1021/bi00401a031. [DOI] [PubMed] [Google Scholar]
- 14.Shechter Y. Diabetes. 1990;39:1. doi: 10.2337/diacare.39.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharyya S, Tracey AS. J Inorg Biochem. 2001;85:9. doi: 10.1016/s0162-0134(00)00229-4. [DOI] [PubMed] [Google Scholar]
- 16.Zhang M, Zhou M, Van Etten RL, Stauffacher CV. Biochemistry. 1997;36:15. doi: 10.1021/bi961804n. [DOI] [PubMed] [Google Scholar]
- 17.Brandao TA, Hengge AC, Johnson SJ. J Biol Chem. 2010;285:15874. doi: 10.1074/jbc.M109.066951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies DR, Hol WG. FEBS Lett. 2004;577:315. doi: 10.1016/j.febslet.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 19.Stankiewicz PJ, Tracey AS, Crans DC. Met Ions Biol Syst. 1995;31:287. [PubMed] [Google Scholar]
- 20.Denu JM, Lohse DL, Vijayalakshmi J, Saper MA, Dixon JE. PNAS. 1996;93:2493. doi: 10.1073/pnas.93.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lassila JK, Zalatan JG, Herschlag D. Annu Rev Biochem. 2011;80:669. doi: 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hengge AC. Advances in Physical Organic Chemistry. 2005;40:49. [Google Scholar]
- 23.Hengge AC, Zhao Y, Wu L, Zhang ZY. Biochemistry. 1997;36:7928. doi: 10.1021/bi970364c. [DOI] [PubMed] [Google Scholar]
- 24.Hengge AC, Sowa GA, Wu L, Zhang ZY. Biochemistry. 1995;34:13982. doi: 10.1021/bi00043a003. [DOI] [PubMed] [Google Scholar]
- 25.Krauss M, Basch H. J Am Chem Soc. 1992;114:3630. [Google Scholar]
- 26.Deng HCR, Huang Z, Zhang ZY. Biochemistry. 2002;41:5865. doi: 10.1021/bi016097z. [DOI] [PubMed] [Google Scholar]
- 27.Crans DC, Bunch RL, Theisen LA. J Am Chem Soc. 1989;111:7597. [Google Scholar]
- 28.Crans DC, Rithner CD, Theisen LA. J Am Chem Soc. 1990;112:2901. [Google Scholar]
- 29.Alonso A, Burkhalter S, Sasin J, Tautz L, Bogetz J, Huynh H, Bremer MC, Holsinger LJ, Godzik A, Mustelin T. J Biol Chem. 2004;279:35768. doi: 10.1074/jbc.M403412200. [DOI] [PubMed] [Google Scholar]
- 30.Pannifer AD, Flint AJ, Tonks NK, Barford D. J Biol Chem. 1998;273:10454. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Denu JM, Dixon JE. Proc Natl Acad Sci U S A. 1995;92:5910. doi: 10.1073/pnas.92.13.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carpenter JE, Weinhold F. J Mol Struct (THEOCHEM) 1988;41–62:169. [Google Scholar]
- 34.Foster JP, Weinhold F. J Am Chem Soc. 1980;102:7211. [Google Scholar]
- 35.Reed AE, Weinhold F. J Chem Phys. 1983;78:4066. [Google Scholar]
- 36.Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899. [Google Scholar]
- 37.Parr RG, Yang W. Oxford Univ. Press; Oxford: 1989. [Google Scholar]
- 38.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 39.Perdew JP, Chevary JA, Vosko SH, Jackson KA, Pederson MR, Singh DJ, Fiolhais C. Phys Rev B. 1992;46:6671. doi: 10.1103/physrevb.46.6671. [DOI] [PubMed] [Google Scholar]
- 40.Clark TCJ, Spitznagel CW, Schleyer PvR. J Comput Chem. 1983;4:294. [Google Scholar]
- 41.Frisch MJPJA, Binkley JS. J Chem Phys. 1984;80:3265. [Google Scholar]
- 42.RA, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. In: Gaussian 09. Gaussian I, Wallingford CT, editors. 2009. [Google Scholar]
- 43.Brandao TA, Robinson H, Johnson SJ, Hengge AC. J Am Chem Soc. 2009;131:778. doi: 10.1021/ja807418b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crans DC, Baruah B, Ross A, Levinger NE. Coordin Chem Rev. 2009;253:2178. [Google Scholar]
- 45.Lu Z, Dunaway-Mariano D, Allen KN. Proc Natl Acad Sci U S A. 2008;105:5687. doi: 10.1073/pnas.0710800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.