

Abstract
The discovery of “benzodiazepine receptors” provided the impetus to discover and develop anxioselective anxiolytics (“Valium® without the side effects”). The market potential for a GABAA receptor-based anxioselective resulted in multiple compounds entering clinical trials. In contrast to the anxioselective profile displayed in preclinical models, compounds such as bretazenil, TPA023, and MRK 409 produced benzodiazepine-like side effects (sedation, dizziness) in Phase I studies, whereas alpidem and ocinaplon exhibited many of the characteristics of an anxioselective in the clinic. Alpidem was briefly marketed for the treatment of anxiety, but withdrawn because of liver toxicity. Reversible elevations in liver enzymes halted development of ocinaplon in Phase III. The clinical profiles of these two molecules demonstrate that it is possible to develop GABAA receptor-based anxioselectives. However, despite the formidable molecular toolbox at our disposal, we are no better informed about the GABAA receptors responsible for an anxioselective profile in the clinic. Here, I discuss the evolution of a quest, spanning 4 decades, for molecules that retain the rapid and robust anti-anxiety actions of benzodiazepines absent the side effects that limit their usefulness.
Anxiolytics: past, present, and the need for anxioselective agents
Benzodiazepines (BZs) have been used to treat anxiety disorders for more than 50 years, and the commercial success of chlordiazepoxide (Librium®) and diazepam (Valium®) led to the introduction of more than a dozen analogs by the early 1980s. BZs remain in widespread use [1] despite a shift in prescribing practices, with most authorities [2] favoring serotonin-specific reuptake inhibitors (SSRIs) as first-line treatment for generalized anxiety disorder (GAD). The adoption of serotonin-based therapies to treat GAD, the most prevalent among the anxiety disorders [3], is attributable to safety concerns with long term use of BZs, primarily the potential for a discontinuation syndrome and abuse liability. The onset of symptom relief with serotonin-based therapies (such as SSRIs and buspirone) is slow; four or more weeks of treatment with an SSRI [4] are often required for meaningful symptom relief. By contrast, BZs have a significant advantage with respect to speed of onset and, at least initially, efficacy [5,6]. Moreover, patients prescribed aSSRI may experience an initial increase in anxiety symptoms, and BZs are often prescribed during this “cover” period.
Fifteen years elapsed between the commercialization of chlordiazepoxide (1961) and the first report indicating that BZs augment the effects of γ-aminobutyric acid (GABA), the principal inhibitory transmitter of the mammalian central nervous system (CNS) [6]. The identification of high affinity, saturable and stereoselective recognition sites for BZs (initially termed “benzodiazepine receptors”)1 in 1977, and the demonstration that the anxiolytic and anticonvulsant potencies of a series of 1,4-BZs were highly correlated with potencies to displace [3H]diazepam from brain tissue [10,11] suggested these sites were pharmacologically relevant, and provided a means to interrogate large numbers of structurally unrelated compounds for potential BZ-like properties.
CL 218,872 (a triazolopyridazine), the first non-benzodiazepine (Figure 1) described [12] following the identification of BZ receptors, was used to demonstrate that these receptors were heterogeneous. Perhaps most striking were significant regional differences in the apparent affinity of CL 218,872, which are not apparent with 1,4-BZ. Thus, CL 218,872 was most potent in displacing [3H]BZ from cerebellum, significantly less potent in hippocampus, and exhibited an intermediate potency in cortex. Like 1,4-BZ, CL 218,872 exhibited potent anticonflict actions [12–14] in preclinical models [15] predictive of anxiolytic activity. However, in contrast to BZ, much higher doses of CL 218,872 were required to produce sedation and muscle relaxation [12–14]. The unique pharmacological profile of this molecule2 provided the impetus for many companies to develop compounds with a similar profile, screening libraries based using the displacement of [3H]BZ from brain tissue as the starting point.
Fig. 1.
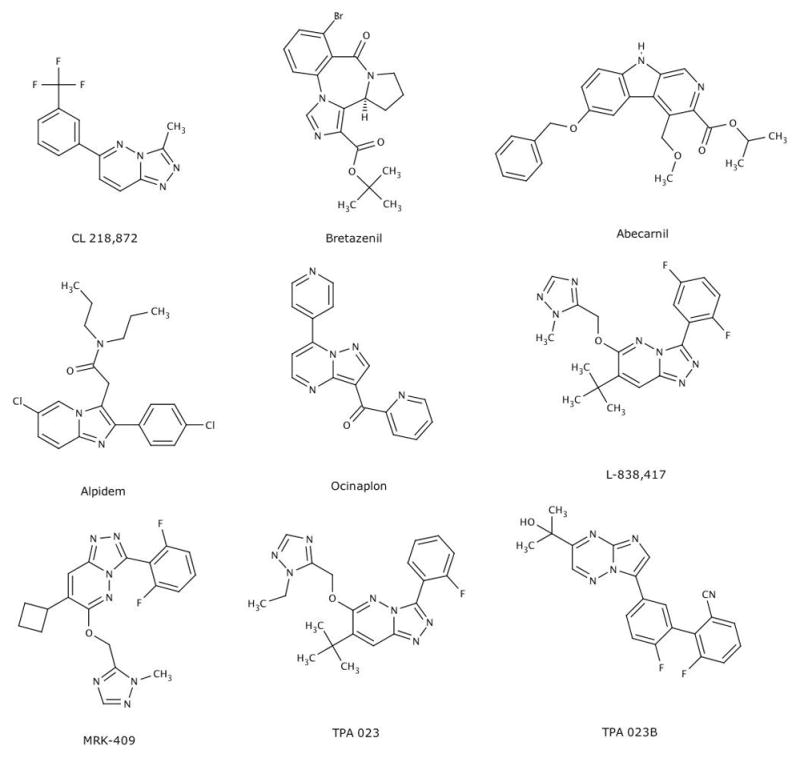
Representative molecules exhibiting anxioselective profiles in preclinical models. Among the nine compounds illustrated here, six (bretazenil, abecarnil, alpidem, ocinaplon, MRK 409, TPA023, TPA 023B) were advanced to the clinic.
Most simply stated, the product profile of an anxioselective agent – “Valium® without side effects” – is compelling. There is little dose separation among the pharmacological properties of BZs. For example, under double-blind, placebo controlled conditions, doses of a BZ that produce a robust reduction in anxiety are also sedating in about half the patients [5]. The ability to rapidly and effectively relieve anxiety without compromising daily activities (e.g., driving, operating machinery), eliminating the potential for falls (hip fractures are especially problematic in the elderly), a reduced potential for abuse, and lack of a discontinuation syndrome are all therapeutic attributes that would greatly benefit both the patient and prescriber. From a commercial perspective, both the widespread use of benzodiazepines (in 1980, it was estimated that 8000 tons of benzodiazepines were used in the U.S. [17]) and patent expirations on commercially successful benzodiazepines (Valium® and Librium®) provided a strong incentive to develop anxioselective agents. It could be argued that the use of serotonin-based therapeutics3 has reduced the saliency of such a compound. However, a compelling product profile (the rapid and robust anti-anxiety effects of a BZ absent the side effects) and the prospect of a large economic upside (in 2010, sales estimates of anxiolytic agents in the US was estimated in excess of $11 billion [18]) sustained research and development efforts in this arena well into this millennium [9, 19, 20].
An Overview of GABAA Receptors
Beginning in the late 1980s [21,22], multiple cDNAs were cloned that encode GABAA receptor subunits. Based on sequence homology, there are eight subunit families (α, β, γ, δ, ε, θ, π, ρ) comprising 20 distinct gene products [7]. The overwhelming majority of GABAA receptors in the mammalian central nervous system are heteropentamers, composed of two α, two β, and one γ subunit in a γ-β-α-β-α arrangement [23], with the second transmembrane domain of each subunit forming the pore of a GABA-gated chloride channel [24] (Figure 2). Although immunoprecipitation studies indicate about half of all GABAA receptors in the adult CNS are composed of α1β2γ2 subunits [25], there is a remarkable potential for receptor heterogeneity. Even by constraining the number of heteropentamers to receptors containing only α, β, and γ subunits and imposing a rule of within subunit homogeneity (e.g., only one type of α subunit permitted in a receptor), there is the potential for (6α · 3β · 3γ) 54 receptor isoforms. Nonetheless, anatomically restricted subunit expression [24, 26] markedly limits the potential number of αnβnγn GABAA receptors; immunoprecipitation studies suggest the presence of perhaps 10 distinct αnβnγn heteropentamers [25]. Many drugs, including barbiturates, volatile anesthetics, convulsants (e.g. pentylenetetrazole), neurosteroids, and alcohols (ethanol) modulate GABAA receptor function [24]. However, an α1, 2, 3, or 5 subunit and a γ subunit are required for high affinity binding of 1,4-BZs (and the other structurally distinct molecules discussed in this review)1. Hence, GABAA receptors containing an α1,2,3, or 5 subunit have been referred to as “diazepam-sensitive” (DS), whereas receptor isoforms bearing an α4 or α6 subunit are referred to as “diazepam-insensitive” (DI) [8].
Fig.2.

Schematic representation of a GABAA receptor. This GABAA receptor is constituted as an αβγ heteropentamer with 3 different subunits. Left panel: side view in a postsynaptic membrane. Right panel: top view. The second transmembrane domain (TM2) of each subunit forms the lumen of the GABA-gated chloride channel. The binding site for BZs and the other molecules highlighted in Fig. 1 is formed by the extracellular domains of the α and γ subunits *8,24+. This figure is adapted from Berezhnoy, et al. [24].
A histidine residue at position 101 of the α1 subunit (and homologous residues in the α2,3 and 5 subunits) is crucial for high affinity binding of BZs and the other molecules discussed in this review; point mutation of this residue to arginine, the corresponding amino acid in the α4 and α6 subunits, results in a dramatic reduction in affinity of these molecules [27]. Subsequent studies have demonstrated that multiple amino acid residues on both the α and γ subunits can influence both the potency and efficacy of these molecules [8]. However, the ability to dramatically reduce the affinity of BZs by this His→Arg mutation was critical for a second wave of efforts to develop anxioselective agents. Thus, the application of “knock-in” technology resulted in the creation of mice [28] containing the His→Arg mutation in either the α1, 2, 3, or 5 subunit. Mice produced by this approach have a dramatic loss in affinity for BZs only at GABAA receptors containing the mutated subunit. The loss of a pharmacological action (e.g., an anxiolytic effect) would then link the GABAA receptor bearing this mutated subunit to this pharmacological action, providing evidence that should converge with studies using subtype selective compounds in both knock-in and wild-type mice.
Partial agonists, subtype selective molecules, and hybrid solutions
The commercial interest in developing anxioselectives, together with a technology enabling the interrogation of large numbers of molecules [10, 11] and advances in the molecular biology of GABAA receptors [7,8, 28], resulted in the identification of more than a dozen molecules (Figure 1) [7,8, 29] with preclinical profiles consistent with anxioselectivity. A subset of these compounds was developed further, and the clinical effects of at least eight molecules have been reported in peer-reviewed journals. Four of these molecules (bretazenil, abecarnil, alpidem, and ocinaplon) were in development prior to the widespread use of recombinant GABAA receptors. These compounds, initially identified by radioreceptor assays (using [3H]BZs and native receptors derived from rodent brain), exhibited anxiolytic-like actions in multiple models, but were either devoid of BZ-like “side effects” (e.g. sedation, muscle relaxation, ataxia, amnesia) or produced these effects only at doses many fold higher. By contrast, separation between the anxiolytic and “side effect” doses of BZs are typically very modest in both preclinical models and in the clinic [5, 30, 31].
Bretazenil, developed at Hoffman-LaRoche, was 3–4 orders of magnitude more potent in preclinical measures of anxiolysis in both rodents and primates compared to doses producing “side effects” [32]. In addition to an apparent lack of BZ-like side effects, bretazenil was reported to antagonize the effects of diazepam-induced motor impairment (e.g., on the horizontal wire test) and to produce a much smaller potentiation of ethanol-induced sedation than diazepam [32]. In the clinic, bretazenil (0.5–4 mg) reduced GAD symptoms in a placebo-controlled, double-blind study using diazepam as a comparator [32]. A smaller study also demonstrated that, like BZs, bretazenil reduced the incidence and severity of panic attacks [32]. Despite the remarkable dissociation among pharmacological actions described in preclinical studies, bretazenil proved to be profoundly sedating at anxiolytic doses [33]. In normal volunteers, sedation was noted at doses as low as 0.2 mg and at 0.5 mg, most subjects were sedated to the point that they were unable to adequately perform eye tracking and movement tests [33]. Moreover, a profound reduction in performance was noted when bretazenil was combined with alcohol, greater than observed when alcohol and diazepam were administered [33].
A large separation between doses producing anxiolytic-like actions and side effects such as ataxia and myorelaxation were also reported for the β-carboline, abecarnil (Fig. 1), developed by Schering AG [34]. In Phase I studies [35], the most frequently noted adverse events were dizziness, unsteady gate, and lack of concentration. Despite adverse events that likely reflect ataxia and sedation, these normal volunteers did not rate abecarnil as sedating using a visual analog rating scale (VAS). VAS scores are used to evaluate a particular dimension in either a subject’s baseline or response to a treatment. The subject is asked to rate a dimension (in this case, sedation) by placing a tick mark on a 100 mm line, with 0 mm indicating no sedation and 100 mm as highly sedating. Abecarnil was advanced into efficacy trials despite the apparent disconnect between the preclinical profile and the adverse event profile in Phase I studies suggestive of BZ-like side effects. The efficacy and side effect profile of abecarnil was evaluated in over 450 patients with a diagnosis of GAD in double-blind, placebo-controlled trials that also included an active BZ comparator. The most prominent side effects for both abecarnil (dose range, 7.5–30 mg) and the BZ comparators were drowsiness and dizziness; the frequency of side effects produced by abecarnil fell within in the range of the BZ comparators, with the incidence in all active groups well above placebo. The efficacy of abecarnil in reducing scores on the Hamilton Scale for Anxiety(HAM-A scores), the “gold standard” used in clinical trials for evaluating anxiety,, was variable; in 4 of 5 studies, BZs produced a rapid (one week) and sustained separation from placebo. Abecarnil exhibited a BZ-like pattern in only one of five studies; in the other studies, abecarnil failed to show a consistent separation from placebo over the four-week study period [36]. Pollack et al. [37] compared the efficacy of two dose levels of abecarnil in a double-blind, placebo-controlled trial. Buspirone, a 5HT1A partial agonist approved for the treatment of GAD, was used as a comparator in this 6 week study. Both the low (3–9 mg/day) and high (7.5–22.5 mg/day) dose abecarnil groups experienced a rapid reduction in HAM-A scores, reaching statistical significance after one week of dosing, the earliest time point measured. Anxiolytic effects in the high, but not the low dose group, were maintained during the six-week trial. Consistent with the delayed onset of other serotonin-based anxiolytics [4], a statistically significant separation from placebo was not observed in the buspirone cohort until the sixth week of treatment. The side effect profile of abecarnil was strikingly similar to a BZ, with significant increases in the incidence of drowsiness (45% in the high dose abecarnil group compared to 13% in placebo), dizziness, fatigue, lack of concentration, and ataxia compared to the placebo group. The clinical profile of abecarnil, in particular the side effect profile so reminiscent of BZs, ultimately led to the decision to halt further development in the mid 1990s.
Ocinaplon (CL 273,547), a pyrazolopyrimidine, was developed from the Lederle chemistry platform that produced CL 218,872 [12]. The preclinical profile of ocinaplon (Figure 3) resembles other molecules described in this review: BZ-like effects in preclinical tests predictive of an anxiolytic action, with side effects manifested only at much higher doses [30, 38]. In the “thirsty rat conflict test” *15], perhaps the most widely used procedure to evaluate potential anti-anxiety actions, ocinaplon and diazepam were equipotent (minimum effective dose [MED] ~3.1 mg/kg) in increasing punished responding (that is, thirsty rats increase the number of licks taken from a water spout despite the contingency of receiving an unpleasant shock; this increase does not reflect analgesia because opiates are not active in this paradigm). These effects were flumazenil (Ro 15–1788) sensitive, indicative of a GABAA receptor mediated action (Figure 3). However, in tests designed [31] to assess BZ-like side effects, ocinaplon was far less potent than diazepam. For example, the ED50 for reducing motor activity (often used as a surrogate for sedation) was ~17.5 and 82 mg/kg for diazepam and ocinaplon, respectively. In the inclined screen and rod walking tests, used to assess muscle relaxation and ataxia respectively, ocinaplon was ~11 and ~7-fold less potent than diazepam [30]. In a primate conflict model, ocinaplon and diazepam increased punished responding with MED values of 4 and 0.5 mg/kg, respectively. However, diazepam significantly reduced non-punished responding (indicating a nonspecific reduction in behavior) at 4 mg/kg; a profound sedation was observed in these animals. Ocinaplon significantly reduced non-punished responding only at doses of 128 mg/kg, and no overt signs of sedation were noted [30].
Figure 3.

Comparison of the pharmacological properties of ocinaplon and diazepam: Adult, male CD rats (Charles River) were administered vehicle, ocinaplon or diazepam by gavage. Panel A: Anticonflict actions of ocinaplon and diazepam: blockade by flumazenil. Animals were evaluated 60 min. later in the “thirsty rat conflict” test, essentially as described [31]). In brief, rats were water deprived for 48 hours and food deprived for 24 hours prior to testing. Rats were placed in the test chamber, and a 10% glucose solution made available through a stainless steel spout. After locating the spout, the rat was allowed 25 seconds of free (no shock) drinking. A circuit was then activated which applied a mild electric shock through the spout in a 5-seconds “on” 5-seconds “off” schedule for 5 minutes. The number of shocks delivered (the animal must be in contact with the spout to receive a shock) is recorded. Parallel groups received flumazenil (12.5 mg/kg, i.p.) 30 min. prior to testing. This dose of flumazenil does not affect performance in the thirsty rat conflict test (data not shown). The minimum effective dose (MED; the first dose producing a statistically significant difference from vehicle treated rats) of ocinaplon and diazepam was 3.1 mg/kg. Values represent the mean (n ≥ 8 animals/dose) % increase in punished responding compared to vehicle treated rats. Symbols: open triangles, ocinaplon; closed squares, diazepam; closed triangles, ocinaplon+flumazenil; open squares squares: diazepam+flumazenil.
Panel B: Effects of ocinaplon and diazepam on motor activity in rats. Compounds were evaluated 60 minutes after oral administration. Values represent the mean % decrease in motor activity of 12–24 rats/dose compared to vehicle treated animals. The ED50 of diazepam and ocinaplon was 17.5 and 81.7 mg/kg, respectively. Symbols: Open triangles, ocinaplon; closed squares, diazepam.
Panel C: Effects of ocinaplon and diazepam on the inclined screen: The effect of diazepam and ocinaplon was evaluated on the ability of rats to remain on an inclined (60°) screen for 30 min. The ED50 of diazepam and ocinaplon was 15.5 (3.5–24.9, 95% C.I.) and 172.2 (123.3–244.5, 95% C.I.) mg/kg, respectively. Symbols as in Panel B.
Panel D: Effects of ocinaplon and diazepam on rod walking: Animals were trained to traverse a rod inclined at 17°. Values represent the mean of 10 rats/dose. The ED50 of diazepam and ocinaplon was 13.8 (2.7–20.4, 95% C.I.) and 92 (68–124, 95% C.I.) mg/kg, respectively. Symbols: as in Panel B. These data are redrawn from Lippa, et al. [30].
A conservative dose ranging strategy was adopted in Phase I studies based on the potency of ocinaplon in preclinical models [30]. However, in both single and multiple ascending dose studies, ocinaplon was safe and well-tolerated at single doses of up to 90 mg and total daily doses of up to 270 mg. No BZ-like adverse events were noted at these doses. A four-week, double-blind placebo-controlled trial using a 270 mg dose (90 mg, three times daily) of ocinaplon demonstrated a rapid improvement in HAM-A scores sustained throughout the trial [19]. Ocinaplon reduced HAM-A scores by 12.9 points at the conclusion of the trial compared to 5.4 points in the placebo group. By comparison, four week trials of anxiolytics generally report changes in HAM-A scores of 2–3.5 point differences from placebo [19]. Consistent with data obtained in healthy volunteers, there was no evidence of BZ-like side effects [19]. This trial was terminated early because of one case of jaundice; the patient fully recovered and was found to have a pre-existing condition that may have contributed to this serious adverse event. A second, shorter trial of ocinaplon in GAD patients demonstrated anxiolysis at 180–240 mg; separation from placebo was apparent as early as the first week of treatment [30]. The anxiolytic effect of ocinaplon in this study was robust: despite a high placebo response (a 9.7 point decline in the HAM-A over two weeks), the reductions in HAM-A scores were 4.4 and 5.6 points greater than the placebo cohort in the 180 and 240 mg arms, respectively. The rate and incidence of adverse events in this study yielded no hints of BZ-like side effects. These results triggered a larger (373 patient), four-week study comparing 60 and 120 mg of ocinaplon to placebo in GAD patients in order to establish a MED. This trial was halted after approximately 200 subjects had been randomized because elevations in liver function tests were seen in a small number of patients; one patient exhibited marked elevations in liver enzymes that ultimately normalized. Given the regulatory hurdles triggered by these events, development of ocinaplon was discontinued, despite preclinical extensive toxicology studies that did not yield premonitory signs of liver damage.
Although the number of subjects dosed with ocinaplon was arguably too small to unequivocally demonstrate an anxioselective profile, the approval of alpidem for GAD demonstrates the feasibility of developing an efficacious, rapid acting anxiolytic with a reduced incidence of BZ-like side effects. Alpidem (Figure 1) is a structural analog of zolpidem, a hypnotic marketed for more than twenty years. Despite this structural similarity, the pharmacological profile of alpidem is remarkably different from both zolpidem and BZs. Alpidem was active in several preclinical measures predictive of anxiolytic activity (the thirsty rat conflict test, marble burying) but inactive in others (e.g. four plate test, inhibition of foot shock-induced fighting) sensitive to BZs [39,40]. The anticonflict actions of alpidem were antagonized by flumazenil, indicating these effects are mediated by GABAA receptors. Although alpidem reduced exploratory activity in mice at doses within the anxiolytic range [39, 41], deficits in rotarod performance (generally used to model ataxia) and muscle strength were manifested only at doses >20 fold higher than those active in anticonflict tests [39]. Moreover, no evidence of amnestic effects was noted [42].
Based on studies in more 1,500 patients suffering from various anxiety disorders, alpidem was marketed in France (Ananxyl®) for the treatment of GAD in 1991. In the clinical trials leading to registration, the reductions in HAM-A scores produced by alpidem were significantly greater than placebo. For example, in a 3 week trial comparing 150 mg of alpidem (50 mg three times daily) to placebo, the separation of HAM-A scores between the two groups was highly significant (6.1 points; p<0.0001) [43]. At a total daily dose of 75–150 mg administered 2–3 times daily (i.e., single doses of 25–75 mg), reductions in HAM-A scores were equivalent to either flexible or fixed dosing of BZs such as lorazepam, chlorazepate and diazepam. There was no evidence of either withdrawal symptoms or rebound anxiety in patients who discontinued alpidem after periods of up to one year.
Healthy volunteers are generally very sensitive to the side effects of BZs – dizziness, muscle weakness, fatigue and sleepiness are often reported as adverse events at or below effective anxiolytic doses, and yet these were not prominent side effects with alpidem. For example, in healthy volunteers, doses of 50–100 mg (the anxiolytic dose range: 75–150 mg administered in two or three daily doses - i.e., single doses of 25–75 mg) did not impair alertness, nor did it affect psychomotor performance. At a higher dose (200 mg), alpidem did impair vigilance and psychomotor performance equal to 10–15 mg of diazepam [44]. Nonetheless, in some measures, including a VAS rating of “clearheadedness”, no dose of alpidem reduced ratings, whereas diazepam produced a dose dependent (10–15 mg) reduction in this measure [44]. In another study, doses of 25–50 mg of alpidem did not produce any significant psychomotor impairment, but a dose of 100 mg did, albeit to a much lower extent than a standard dose of lorazepam (2 mg) in healthy volunteers. In GAD patients, a comparison of alpidem with lorazepam demonstrated no effects on memory with alpidem, whereas lorazepam produced significant decrements in recall. Moreover, reaction time was slowed by lorazepam but not by alpidem. No rebound anxiety or withdrawal symptoms were manifested following abrupt withdrawal from alpidem; the study arm looking at withdrawal reactions to abrupt termination of lorazepam had to be terminated for ethical reasons [45]. Remarkably, alpidem (50 mg) significantly antagonized the amnestic effects of lorazepam (2 mg), and produced similar trends on other cognitive parameters, resulting in less impairment than after lorazepam alone [46]. In sum, alpidem proved to be an effective anxiolytic, as effective as standard BZs in direct comparison trials, producing a rapid and robust reduction in HAM-A scores. Some psychomotor impairment was observed at doses 2–8 times higher than the recommended single dose (25–50 mg). Thus, while not exhibiting the large dose separation predicted from preclinical tests, alpidem did not impair memory at therapeutic doses (and was reported to antagonize the amnestic actions of a BZ), nor did chronic treatment produce a withdrawal reaction after abrupt termination. These latter properties have also been observed in preclinical studies for many of the putative anxioselective agents described here [47], but alpidem is the only compound for which these effects have also been documented in the clinic. Based on these clinical studies, the side effect profile of alpidem differs significantly from BZs. Several cases of severe hepatitis were reported post-marketing, resulting in one death and several patients requiring liver transplantation [48]. Alpidem was withdrawn from the market in 1995.
Despite all four molecules exhibiting anxioselective profiles in the laboratory, a remarkably different picture emerged in the clinic. The common neurochemical thread among these molecules, identified prior to the availability of recombinant GABAA receptors, was a partial agonist profile evinced in native receptors using neurochemical and electrophysiological techniques [30, 32, 34, 38, 49–51]. Because the brain preparations used to characterize these compounds as partial agonists contain multiple receptor isoforms, it is instructive to compare the profiles of bretazenil (highly sedating) to alpidem (that appears to produce far less sedation at anxiolytic doses) in recombinant GABAA receptors of defined composition. Bretazenil exhibits very high (< 1 nM) affinities but no selectivity among recombinant GABAA receptors containing α1,2, 3,and 5 subunits. It is however, a partial agonist at these GABAA receptors, with efficacies between 20–44% compared to a standard BZ [52, 53]. By contrast, alpidem is >500-fold more selective for GABAA receptors bearing α1 compared to α5 subunits [54]. Moreover, despite compelling neurochemical evidence that alpidem is a partial agonist in multiple assays using native receptors [50, 51], it behaves as a full (i.e, BZ-like) agonist in electrophysiological assays using recombinant receptors bearing either α1 [55, 56] or α2 subunits [57]. If the profile of ocinaplon (which gave no indication of BZ-like side effects in the clinic) is now considered, there is little evidence of subtype selectivity, with efficacies (relative to diazepam) ranging from 0.85 in α1 β2 γ2 receptors to 0.25 at α5 β2 γ2 containing receptors [30]. Taken together, these data would seem consistent with the hypothesis that high efficacy at α1 containing receptors would not necessarily translate into sedation. Nonetheless, this hypothesis is inconsistent with the profile of zolpidem which, like alpidem, exhibits both α1 selectivity and high efficacy in recombinant GABAA receptors, yet in addition to possessing anxiolytic activity is sedating, and has been marketed primarily as a sedative. This enigma was recognized early on [58, 59] and until the recent publication of clinical findings with compounds like TPA 023 and MRK 409 [20, 29] was largely ignored, overshadowed by studies in genetically engineered mice [28, 60]. These latter studies led to the development of another wave of putative anxioselectives moving into the clinic predicated on efficacy at α2 (and to a lesser extent α3) containing receptors with low or no efficacy at α1 containing receptors.
Genetic engineering and shifting theories of anxioselectivity
In the late 1990s, studies with genetically engineered “diazepam-insensitive” mice bearing a point mutation at a key histidine residues on the α1,2,3, and 5 subunits (a knock-in strategy) altered contemporary thinking about the GABAA receptors responsible for the anxiolytic actions of BZs. The conclusions drawn from these studies could be viewed as surprising in the face of an existing body of published clinical data. Thus, independent studies using mice generated on different backgrounds indicated that an α1(H101R) mutation abolished the ability of diazepam to reduce motor activity [60, 61], which the authors used as a surrogate measure for sedation. This mutation also abolished the amnestic effects of diazepam [61], whereas the anxiolytic-like activities were retained [60,61]. McKernan et al. [60] also demonstrated that L–838,417, a partial agonist (with similar affinities) at α 2,3, and 5 containing receptors and null efficacy (i.e, an antagonist) at α1 containing receptors, retained anxiolytic-like activity in the elevated plus maze but did not reduce motor activity. The authors concluded that GABAA receptors bearing an α1 subunit are not required for an anxiolytic action, but are crucial for the sedative properties of a BZ. Löw et al. [62] extended these findings, demonstrating that the anxiolytic-like actions of diazepam in both the elevated plus maze and light/dark choice tests were abolished in α2 (H101R) knock-in mice, while the effects of diazepam on both motor activity and rotarod behavior (used as surrogates of sedation and ataxia) were unaffected. Using this knock-in strategy, Löw et al. [63] also concluded that α3 containing GABAA receptors do not contribute to the anxiolytic actions of diazepam.
However, follow-on studies revealed a more complex picture. Studies with an α3 preferring compound (TP 003) in α2 knock-in mice demonstrated that an intact α2 subunit was not necessary for the anxiolytic-like activity of this compound [63]. Electroencephalographic (EEG) studies in α 1 knock-in mice [64] demonstrated that the effects of diazepam on sleep EEG (arguably a better surrogate for the sedating potential of a compound than motor activity) were unaffected in these mice, but rather dependent on the presence of the α2 subunit [65]. Other studies using knock-in mice [66] indicated that the α2 subunit, and to a lesser extent the α3 subunit, contributed to the myorelaxant actions of diazepam. Despite the emergence of inconsistencies in the “new benzodiazepine pharmacology” *28], several companies embraced the conceptual framework that activation of α1 subunit containing receptors leads to sedation, whereas selective anxiolysis could be achieved by activating α2 subunit containing receptors. Because of the difficulties synthesizing compounds that are highly selective for α2 containing receptors, an alternative strategy was adopted: developing compounds with low or null efficacy at receptors bearing an α1 subunit, and partial agonist properties at α 2,3, and 5 bearing receptors. This strategy may have been partly based on the attractive preclinical profile of L-838,417, which exhibited anxiolytic-like properties in multiple models, with no evidence of motor impairment [60]. Multiple compounds with this neurochemical profile entered the clinic, yielding either disappointing or at best equivocal outcomes. Several of these compounds were developed by Merck, and the data generated with these molecules are especially instructive because of the ability to make head-to-head comparisons among molecules, including PET imaging studies (to measure receptor occupancy) in rodents, non-human primates, and humans.
MRK 409 (also known as MK 0343) is a triazolopyridazine structurally related to L-838,417 (Figure 1). MRK 409 also binds with very high affinity (Ki <1 nM), albeit nonselectively, to DS GABAA receptors. MRK 409 is a low efficacy agonist at receptors expressing α1 subunits (0.18 relative to chlordiazepoxide), and also exhibits partial agonist properties (with efficacies of 0.23, 0.45, and 0.18) at α2,3, and 5 bearing receptors, respectively [20]. MRK 409 produced dose-dependent anxiolytic-like effects in multiple preclinical models (both rodents and nonhuman primates) with oral MEDs ranging from 0.1–3 mg/kg. These doses correspond to receptor occupancies of 35%–65%, estimated by PET studies using 11C flumazenil as a radiotracer. By contrast, little or no sedation was noted at much higher doses corresponding to receptor occupancies estimated at 79–99%. Perhaps most remarkable were the findings in squirrel monkeys, in which no indication of sedation was observed at doses 100-fold higher than the MED for anxiolytic-like effects [20]. However, in healthy male volunteers, MRK 409 (0.75 mg) reduced both saccadic peak velocity [67] (a sensitive indicator of the sedative effects of drugs) and alertness scores assessed with a VAS. At a dose of 1mg (the maximum tolerated dose, MTD), there were reports of tiredness, drowsiness and dizziness, and at a dose of 2 mg, five of six subjects reported a marked sedation. At the MTD, receptor occupancy determined by PET was at the limit of sensitivity (i.e., <10% occupancy). While the marked contrast between preclinical and clinical findings may be viewed as surprising, the in vitro profile of MRK 409 at recombinant GABAA receptors is reminiscent of bretazenil, which also exhibited an anxioselective profile in preclinical models and produced a profound sedation in healthy volunteers.
The sedative effects of MRK 409 have been attributed to its partial agonist properties at α1 receptors [20], but clinical data with two other compounds developed in this program are inconsistent with this view. TPA023 (Figure 1) is a monofluorinated analog of L-838,417 [60], with null efficacy at α1 and low efficacies at α2,3, and 5 bearing receptors (0.11, 0.21, 0.05 compared to chlordiazepoxide, respectively). In view of the hypothesis that anxiolysis requires activation of α2 bearing receptors, the very low efficacy of TPA 023 might be considered a liability. However, BZs produce anxiolytic-like actions at low receptor occupancies (15–20%) [40,68]. TPA 023 exhibited anxiolytic-like activity in both rodents and primates with oral MED values ranging from 0.3–3 mg/kg, with no sedative effects at doses 10–30-fold higher (in PET studies using 11C flumazenil, these doses correspond to >99% receptor occupancy) [29]. However, in healthy male volunteers, doses of 0.5–1.5 mg of TPA 023 reduced saccadic peak velocity [29], indicative of a sedative action. In safety and tolerability studies, dizziness, drowsiness, and motor incoordination were observed at 2 mg (the defined MTD) that appears to produce between 35–65% receptor occupancy [29]. This compound was progressed to multiple ascending dose studies using an extended release formulation, with drowsiness and motor incoordination as the limiting side effects [29]. Despite these BZ-like side effects, TPA 023 was evaluated in several small GAD trials before the program was terminated due to preclinical toxicity findings [29]. By combining data from separate studies using flexible dosing of between 1.5–4.5 and 3–8 mg of the extended release formulation, it was possible to demonstrate a significant (3.5 point) separation from placebo in total HAM-A score by one week of dosing. This effect was sustained for three weeks but no longer apparent in week 4 of the study [29].
The third compound from the Merck development program examined in the clinic, TPA 023B, is structurally distinct from TPA 023 and MRK 409 (Fig. 1). TPA 023b binds with high affinity (0. 7–2 nM) across recombinant DS GABAA receptors and exhibited an “ideal” agonist profile: essentially null efficacy (0.03 relative to chlordiazepoxide) at α1 receptors with moderate efficacies at α 2,3, and 5 bearing receptors (0.38, 0.50, and 0.37 relative to chlordiazepoxide, respectively). Like its predecessors, TPA 023B was unequivocally anxioselective in preclinical models. Anxiolytic-like effects were reported in both primates and rodents [69] with no sedation observed at doses up to 30-fold higher, corresponding to receptor occupancies of >98% [69]. In Phase I studies, fatigue/tiredness and somnolence/drowsiness were noted in four of eight subjects at a 1 mg dose. Dose limiting adverse events at the MTD (2 mg) were fatigue and drowsiness, corresponding to a ~55% receptor occupancy determined by PET imaging [69].
It has been stated that compounds like TPA 023 and 023B are relatively less sedating than BZs because a higher receptor occupancy is required to produce sedation (50–60% versus receptor occupancies of <25% for BZs) [29]. This distinction appears to have little clinical significance with respect to the development of an anxioselective. Thus, although TPA 023 gave some indication of a positive signal in GAD [29], drowsiness and muscle incoordination were limiting side effects. The BZ-like side effects observed with these compounds, despite the demonstration of null efficacies at recombinant α1 bearing receptors, provides strong evidence that the clinical manifestation of sedation requires activation of other GABAA receptor subtypes. Consistent with this hypothesis, MRK 409 was sedating at lower receptor occupancies (at or below the limits of detection) than typically observed with BZs [69] despite an intrinsic efficacy at α1 containing receptors <20% that of chlordiazepoxide [20, 69].
Is there a path forward?
Thirty-five years have elapsed since the seminal discovery of BZ receptors catalyzed the development of GABAA receptor-based anxioselectives, and many hundreds of millions of dollars have been expended on a quest for the ‘Holy Grail.’ The economic incentives for successfully developing an anxioselective enabled multiple “shots on goal”. Based upon the commercialization (albeit briefly) of alpidem and two multicenter Phase II trials with ocinaplon, the goal of developing GABAA receptor-based anxiolytics with an improved side effect profile has, in principle, been achieved. Nonetheless, we are arguably no better informed about the GABAA receptors responsible for either the clinical profiles of these compounds or the sedation produced by compounds like MRK 409, bretazenil, and TPA 023. It is striking from a translational perspective that tiredness, drowsiness/sleepiness, light headedness, and dizziness are prominent adverse events of molecules possessing either null or low efficacy at α1 bearing GABAA receptors. Measuring motor activity in species (rats and mice) living horizontally may model sedation (a calming, quieting effect), but may not predict the potential to promote sleep (a hypnotic). This distinction, consistent with more rigorous EEG studies in α1 and α2 knock-in mice [64,65], has little clinical relevance for the development of an anxioselective. Perhaps more puzzling is the reported lack of sedative effects of compounds like bretazenil, TPA 023, and MRK 409 in primates. These studies have generally relied on reductions in operant responding (lever pressing) to predict a sedative effect [30], and thus may not be very sensitive. Although EEG studies have not been published with these compounds in non-human primates, overt signs of sedation should be obvious to a trained observer [30].
One potential explanation for the failure to accurately predict the pharmacological properties of these compounds across species may relate to species differences in GABAA receptor composition and distribution. Thus, while α1β2γ2 containing GABAA receptors are the most abundant isoform in rodent brain, a significant proportion (>25%) of α1 bearing receptors are heterogeneous (containing either α2 or α 3 subunits) [70]. Moreover, both α2 and α3 subunits exist as predominantly heteromeric GABAA receptors in rodent brain [70], but neither the proportion nor distribution of these heterogeneous receptors in other species, including humans, is known. Species differences in either the proportion or distribution of these heterogeneous receptors could dramatically alter the behavioral actions of a compound. Further, the properties of a molecule described (e.g., its efficacy) in recombinant GABAA receptors that are homogeneous with respect to the α subunit may not reflect the pharmacology of native receptors enriched in these heterogeneous receptors. The response to a drug in mice with a mutated, diazepam-insensitive α subunit (e.g., an α2 (H101R) subunit), where a majority of these receptors are heterogeneous [70], could explain some of the discrepant findings between studies in these mice and pharmacological studies using subtype-selective agents. Some glaring examples of these discrepant findings include the anxiolytic-like actions of an α3 preferring compound (TP 003) in both wild-type and knock-in mice carrying a mutated α2 subunit [63], and the ability of the selective α1 antagonist β-CCT (3-t-butoxy-β-carboline) [71] to block the anxiolytic-like (but not the muscle relaxant or ataxic) effects of BZs [72,73].
Concluding remarks
Despite the obvious therapeutic advantages of a GABAA receptor based anxioselective, the search for the Holy Grail has been abandoned. When viewed in the context of a risk-averse environment that has led many pharmaceutical companies to abandon the development of drugs for psychiatric disorders, the lack of a coherent hypothesis to explain the clinical profiles of molecules ranging from ocinaplon to bretazenil makes it unlikely that additional resources will be dedicated to developing GABAA receptor based anxioselectives. At a minimum, the development of an animal model which can predict the sedative properties of (e.g.) bretazenil and MRK 409 resolves the proximal cause of these failures in the clinic. Such a model, together with the availability of multiple compounds with defined clinical profiles and a powerful molecular toolbox could well revitalize the search for GABAA receptor based anxioselectives.
Acknowledgments
This paper is dedicated to my friend and colleague, Dr. Arnold S. Lippa. His energy and vision provided many of the tools that enabled this quest.
Footnotes
Compounds that bind to this allosteric modulatory domain on GABAA receptors [7] include 1,4-BZs and more than a dozen structurally distinct species. The molecular and structural requirements for high affinity ligand binding at recombinant GABAA receptors are well understood [8,9].
1,4-BZ are also excellent sedative-hypnotics, muscle relaxants, amnestics (useful as preoperative medications), and anticonvulsants. The demonstration of receptor heterogeneity led to the hypothesis that these pharmacological actions are separable; the in vivo profile of CL 218,872 provided the proof of principle that such a separation could be achieved [16].
A number of SSRIs and SNRIs were approved for the treatment of GAD beginning in the late 1990s; many of these compounds are now generic.
Disclaimer: The views expressed in this article do not necessarily reflect the views of the National Institute on Drug Abuse, the National Institutes of Health, or the Department of Health and Human Services.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stahl S. Don’t ask, don’t tell, but benzodiazepines are still the leading treatments for anxiety disorder. The Journal of clinical psychiatry. 2002;63:756–757. doi: 10.4088/jcp.v63n0901. [DOI] [PubMed] [Google Scholar]
- 2.Nash JR, Nutt D. Pharmacotherapy of anxiety. In: Sibley DR, et al., editors. Handbook of Contemporary Neuropharmacology. John Wiley & Sons; 2007. pp. 59–91. [Google Scholar]
- 3.Wittchen HU, Hoyer J. General anxiety disorder: nature and course. J Clinical Psychiatry. 2011;62(Suppl 11):15–21. [PubMed] [Google Scholar]
- 4.Pollack MH. Paroxetine in the treatment of generalized anxiety disorder: results of a placebo-controlled, flexible-dosage trial. The Journal of clinical psychiatry. 2001;62:350–357. doi: 10.4088/jcp.v62n0508. [DOI] [PubMed] [Google Scholar]
- 5.Pande A, et al. Pregabalin in generalized anxiety disorder: a placebo-controlled trial. The American journal of psychiatry. 2003;160:533–540. doi: 10.1176/appi.ajp.160.3.533. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW, et al. Chlordiazepoxide selectively augments GABA action in spinal cord cell cultures. Nature. 1977;269:342–344. doi: 10.1038/269342a0. [DOI] [PubMed] [Google Scholar]
- 7.Barnard EA, et al. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acid(A) receptors: Classification on the basis of subunit structure and receptor function. Pharmacological reviews. 1998;50:291–313. [PubMed] [Google Scholar]
- 8.Lüddens H, Korpi ER. Benzodiazepines. In: Sibley DR, et al., editors. Handbook of Contemporary Neuropharmacology. Vol. 2. John Wiley & Sons; 2007. pp. 93–131. [Google Scholar]
- 9.Richter L, et al. Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat Chem Biol. 2012;8:455–464. doi: 10.1038/nchembio.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohler H, Okada T. Benzodiazepine receptor: demonstration in the central nervous system. Science. 1977;198:849–851. doi: 10.1126/science.918669. [DOI] [PubMed] [Google Scholar]
- 11.Squires RF, Braestrup C. Benzodiazepine receptors in rat brain. Nature. 1977;266:732–734. doi: 10.1038/266732a0. [DOI] [PubMed] [Google Scholar]
- 12.Lippa AS, et al. Synthetic Non-Benzodiazepine Ligand for Benzodiazepine Receptors - Probe for Investigating Neuronal Substrates of Anxiety. Pharmacology Biochemistry and Behavior. 1979;11:99–106. doi: 10.1016/0091-3057(79)90304-6. [DOI] [PubMed] [Google Scholar]
- 13.Lippa AS, et al. Benzodiazepine Receptors - Cellular and Behavioral-Characteristics. Pharmacology Biochemistry and Behavior. 1979;10:831–843. doi: 10.1016/0091-3057(79)90342-3. [DOI] [PubMed] [Google Scholar]
- 14.Lippa AS, et al. The role of GABA in mediating the anticonvulsant properties of benzodiazepines. Brain Research Bulletin. 1980;5(Supplement 2):861–865. [Google Scholar]
- 15.Vogel JR, et al. A simple and reliable conflict procedure for testing anti-anxiety agents. Psychopharmacology. 1971;21:1–7. doi: 10.1007/BF00403989. [DOI] [PubMed] [Google Scholar]
- 16.Lippa AS, et al. Molecular substrates of anxiety: Clues from the heterogeneity of benzodiazepine receptors. Life sciences. 1982;31:1409–1417. doi: 10.1016/0024-3205(82)90001-7. [DOI] [PubMed] [Google Scholar]
- 17.Tallman JF, et al. Receptors for the Age of Anxiety - Pharmacology of the Benzodiazepines. Science. 1980;207:274–281. doi: 10.1126/science.6101294. [DOI] [PubMed] [Google Scholar]
- 18.Abbott A. Novartis to shut down brain research facility. Nature. 2011;480:161–162. doi: 10.1038/480161a. [DOI] [PubMed] [Google Scholar]
- 19.Czobor P, et al. A Multicenter, Placebo-Controlled, Double-Blind, Randomized Study of Efficacy and Safety of Ocinaplon (DOV 273,547) in Generalized Anxiety Disorder. CNS Neuroscience & Therapeutics. 2010;16:63–75. doi: 10.1111/j.1755-5949.2009.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atack JR, et al. MRK-409 (MK-0343), a GABAA receptor subtype-selective partial agonist, is a non-sedating anxiolytic in preclinical species but causes sedation in humans. Journal of Psychopharmacology. 2011;25:314–328. doi: 10.1177/0269881109354927. [DOI] [PubMed] [Google Scholar]
- 21.Schofield PR, et al. Sequence and functional expression of the GABAA receptor shows a ligand-gated receptor super-family. Nature. 1987;328:221–227. doi: 10.1038/328221a0. [DOI] [PubMed] [Google Scholar]
- 22.Shivers BD, et al. Two novel GABAA receptor subunits exist in distinct neuronal subpopulations. Neuron. 1989;3:327–337. doi: 10.1016/0896-6273(89)90257-2. [DOI] [PubMed] [Google Scholar]
- 23.Minier F, Sigel E. Positioning of the α subunit isoforms confers a functional signature to γ aminobutyric acid type A receptors. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7769–7774. doi: 10.1073/pnas.0400220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berezhnoy D, et al. Pharmacology of the GABAA receptor. In: Sibley DR, et al., editors. Handbook of Contemporary Neuropharmacology. John Wiley & Sons; 2007. pp. 465–568. [Google Scholar]
- 25.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends in Neurosciences. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 26.Wisden W, et al. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. The Journal of Neuroscience. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wieland HA, et al. A single histidine in GABAA receptors is essential for benzodiazepine agonist binding. Journal of Biological Chemistry. 1992;267:1426–1429. [PubMed] [Google Scholar]
- 28.Möhler H, et al. A new benzodiazepine pharmacology. J Pharmacol Exper Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 29.Atack JR. Subtype-Selective GABAA Receptor Modulation Yields a Novel Pharmacological Profile: The Design and Development of TPA023. In: Enna SJ, Williams M, editors. Advances in Pharmacology Contemporary Aspects of Biomedical Research Drug Discovery. Vol. 57. Academic Press; 2009. pp. 137–185. [DOI] [PubMed] [Google Scholar]
- 30.Lippa A, et al. Selective anxiolysis produced by ocinaplon, a GABAA receptor modulator. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:7380–7385. doi: 10.1073/pnas.0502579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lippa A, et al. Pre-clinical neuropsychopharmacological testing procedures for anxiolytic drugs. In: Fielding S, Lal H, editors. Anxiolytics. Futura Publishing Company; 1979. pp. 41–82. [Google Scholar]
- 32.Haefely W, et al. Novel anxiolytics that act as partial agonists at benzodiazepine receptors. Trends in Pharmacological Sciences. 1990;11:452–456. doi: 10.1016/0165-6147(90)90126-s. [DOI] [PubMed] [Google Scholar]
- 33.Van Steveninck A, et al. Pharmacokinetic and pharmacodynamic interactions of bretazenil and diazepam with alcohol. Br J Clin Pharmacol. 1996;41:565–573. doi: 10.1046/j.1365-2125.1996.38514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stephens DN, et al. Abecarnil, a metabolically stable, anxioselective beta-carboline acting at benzodiazepine receptors. Journal of Pharmacology and Experimental Therapeutics. 1990;253:334–343. [PubMed] [Google Scholar]
- 35.Duka T, et al. Human studies on abecarnil a new beta-carboline anxiolytic: safety, tolerability, and preliminary pharmacological profile. Br J Clin Pharmacol. 1993;35:386–394. doi: 10.1111/j.1365-2125.1993.tb04155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aufdembrinke B. Abecarnil, a new beta-carboline, in the treatment of anxiety disorders. Brit J Psychiat. 1998;173:55–63. [PubMed] [Google Scholar]
- 37.Pollack MH, et al. Abecarnil for the treatment of generalized anxiety disorder: a placebo-controlled comparison of two dosage ranges of abecarnil and buspirone. J Clinical Psychiatry. 1997;58(suppl 11):19–23. [PubMed] [Google Scholar]
- 38.Chilman-Blair K, et al. Ocinaplon. Drugs of the Future. 2003;28:115–120. [Google Scholar]
- 39.Zivkovic B, et al. Pharmacological and Behavioral Profile of Alpidem as an Anxiolytic. Pharmacopsychiatry. 1990;23:108–113. doi: 10.1055/s-2007-1014545. [DOI] [PubMed] [Google Scholar]
- 40.Jones GH, et al. Comparison of several benzodiazepine receptor ligands in two models of anxiolytic activity in the mouse: an analysis based on fractional receptor occupancies. Psychopharmacology. 1994;114:191–199. doi: 10.1007/BF02244836. [DOI] [PubMed] [Google Scholar]
- 41.Hascoet M, Bourin M. Anticonflict Effect of Alpidem as Compared with the Benzodiazepine Alprazolam in Rats. Pharmacology Biochemistry and Behavior. 1997;56:317–324. doi: 10.1016/s0091-3057(96)00293-6. [DOI] [PubMed] [Google Scholar]
- 42.Dimsdale M, et al. Alpidem. Drugs of the Future. 1988;13:106–109. [Google Scholar]
- 43.Musch B, et al. Clinical studies with the new anxiolytic alpidem in anxious patients: an overview of the European experiences. Pharmacol, Biochem, & Behav. 2012;29:803–806. doi: 10.1016/0091-3057(88)90211-0. [DOI] [PubMed] [Google Scholar]
- 44.Rosenzweig P, et al. Alpidem: lack of sedative effect on psychomotor performance at therapeutic doses. Hum Psychopharmacol. 1993;8:409–415. [Google Scholar]
- 45.Morton S, Lader M. Studies with Alpidem in Normal Volunteers and Anxious Patients. Pharmacopsychiatry. 1990;23:120–123. doi: 10.1055/s-2007-1014547. [DOI] [PubMed] [Google Scholar]
- 46.Patat A, et al. Assessment of the interaction between a partial agonist and a full agonist of benzodiazepine receptors, based on psychomotor performance and memory, in healthy volunteers. Journal of Psychopharmacology. 1995;9:91–101. doi: 10.1177/026988119500900203. [DOI] [PubMed] [Google Scholar]
- 47.Perrault G, et al. Repeated treatment with alpidem, a new anxiolytic, does not induce tolerance or physical dependence. Neuropharmacol. 1993;9:855–863. doi: 10.1016/0028-3908(93)90140-x. [DOI] [PubMed] [Google Scholar]
- 48.Baty V, et al. Hepatitis induced by alpidem (Ananxyl). Four cases, one of them fatal. Gastroenterol Clin Biol. 1994;18:1129–1131. [PubMed] [Google Scholar]
- 49.Pribilla I, et al. Abecarnil is a full agonist at some, and a partial agonist at other recombinant GABAA receptor subtypes. Psychopharmacol Ser. 1993;11:50–61. doi: 10.1007/978-3-642-78451-4_5. [DOI] [PubMed] [Google Scholar]
- 50.Zivkovic B, et al. Alpidem, an omega-1 receptor-selective partial agonist: a new approach to anxiety treatment. European Neuropsychopharmacology. 1991;1:202–205. [Google Scholar]
- 51.Langer SZ, et al. Zolpidem and Alpidem: Two Imidazopyridines with selectivity for Ω1- and Ω3-receptor subtypes. In: Biggio G, Costa E, editors. GABA and Benzodiazepine Receptor Subtypes. Raven Press; 1990. pp. 61–72. [PubMed] [Google Scholar]
- 52.Graham D, et al. Pharmacological profile of benzodiazepine site ligands with recombinant GABAA receptor subtypes. European Neuropsychopharmacology. 1996;6:119–125. doi: 10.1016/0924-977x(95)00072-w. [DOI] [PubMed] [Google Scholar]
- 53.Smith AJ, et al. Effect of α Subunit on Allosteric Modulation of Ion Channel Function in Stably Expressed Human Recombinant γ Aminobutyric AcidA Receptors Determined Using36Cl Ion Flux. Molecular Pharmacology. 2001;59:1108–1118. doi: 10.1124/mol.59.5.1108. [DOI] [PubMed] [Google Scholar]
- 54.Faure-Halley C, et al. Expression and properties of recombinant α1β2γ2 and α5β2γ2 forms of the rat GABAA receptor. Eur J Pharmacol - Mol Pharmacol Sect. 1993;246:283–287. doi: 10.1016/0922-4106(93)90043-9. [DOI] [PubMed] [Google Scholar]
- 55.Ducic I, et al. Triazolam is more efficacious than diazepam in a broad spectrum of recombinant GABA(A) receptors. European Journal of Pharmacology - Molecular Pharmacology Section. 1993;244:29–35. doi: 10.1016/0922-4106(93)90056-f. [DOI] [PubMed] [Google Scholar]
- 56.Im HK, et al. Potentiation of γ-aminobutyric acid-induced chloride currents by various benzodiazepine site agonists with the α1γ2, β2γ2, and α1 β 2 γ 2 subtypes of cloned γ-aminobutyric acid type A receptors. Molecular Pharmacology. 1993;44:866–870. [PubMed] [Google Scholar]
- 57.Wafford KA. Functional comparison of the role of gamma subunits in recombinant human γ-aminobutyric acidA/benzodiazepine receptors. Molecular Pharmacology. 1993;44:437–442. [PubMed] [Google Scholar]
- 58.Skolnick P. Is receptor heterogeneity relevant to the anxiolytic actions of benzodiazepine receptor ligands? In: Briley M, File S, editors. New Concepts in Anxiety. The Macmillan Press Ltd; 1991. pp. 190–202. [Google Scholar]
- 59.Sanger DJ. Recent developments in the behavioral pharmacology of benzodiazepine (Ω) receptors: Evidence for the functional significance of receptor subtypes. Neuroscience & biobehavioral reviews. 1994;18:355–372. doi: 10.1016/0149-7634(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 60.McKernan RM. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor α1 subtype. Nature neuroscience. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 61.Rudolph U. Benzodiazepine actions mediated by specific γ-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 62.Löw K, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- 63.Dias R, et al. Evidence for a Significant Role of α3-Containing GABAA Receptors in Mediating the Anxiolytic Effects of Benzodiazepines. The Journal of Neuroscience. 2005;25:10682–10688. doi: 10.1523/JNEUROSCI.1166-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tobler I, et al. Diazepam-induced changes in sleep: Role of the α1 GABAA receptor subtype. Proceedings of the National Academy of Sciences. 2001;98:6464–6469. doi: 10.1073/pnas.111055398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kopp C, et al. Modulation of rhythmic brain activity by diazepam: GABAA receptor subtype and state specificity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3674–3679. doi: 10.1073/pnas.0306975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crestani F, et al. Molecular Targets for the Myorelaxant Action of Diazepam. Molecular Pharmacology. 2001;59:442–445. doi: 10.1124/mol.59.3.442. [DOI] [PubMed] [Google Scholar]
- 67.de Haas SL, et al. Pharmacodynamic and pharmacokinetic effects of MK-0343, a GABAA α2,3 subtype selective agonist, compared to lorazepam and placebo in healthy male volunteers. Journal of Psychopharmacology. 2008;22:24–32. doi: 10.1177/0269881107082108. [DOI] [PubMed] [Google Scholar]
- 68.Lippa AS, et al. Relationship between benzodiazepine receptors and experimental anxiety in rats. Pharmacology Biochemistry and Behavior. 1978;9:853–856. doi: 10.1016/0091-3057(78)90368-4. [DOI] [PubMed] [Google Scholar]
- 69.Atack JR, et al. Preclinical and clinical pharmacology of TPA023B, a GABAA receptor α2/α3 subtype-selective partial agonist. Journal of Psychopharmacology. 2011;25:329–344. doi: 10.1177/0269881109354928. [DOI] [PubMed] [Google Scholar]
- 70.Benke D, et al. Analysis of the Presence and Abundance of GABAA Receptors Containing Two Different Types of α Subunits in Murine Brain Using Point-mutated α Subunits. Journal of Biological Chemistry. 2004;279:43654–43660. doi: 10.1074/jbc.M407154200. [DOI] [PubMed] [Google Scholar]
- 71.June HL, Eiler WJ. Dopaminergic and GABAergic regulation of alcohol-motivated behaviors: novel neuroanatomical substrates. In: Sibley D, et al., editors. Handbook of Contemporary Neuropharmacology. Vol. 2. John Wiley & Sons; 2007. pp. 465–533. [Google Scholar]
- 72.Shannon H, et al. β-Carboline-3-carboxylate-t-butyl ester; a selective BZ1 benzodiazepine receptor antagonist. Life sciences. 1984;35:2227–2236. doi: 10.1016/0024-3205(84)90464-8. [DOI] [PubMed] [Google Scholar]
- 73.Belzung C, et al. β-CCT, a selective BZ-Ω1 receptor antagonist, blocks the anti-anxiety but not the amnestic action of chlordiazepoxide in mice. Behav Pharmacol. 2000;11:125–131. doi: 10.1097/00008877-200004000-00004. [DOI] [PubMed] [Google Scholar]