

Summary
Microtubules are a vital part of the cytoskeleton of eukaryotic cells and are involved in various cellular processes. The cytoskeleton of Trypanosoma brucei is characterized by an array of subpellicular microtubules and is essential for maintenance of cell shape and polarity, but little is known about the regulation of the assembly and organization of the subpellicular microtubule corset. Here, we report that the orphan kinesin TbKIN-D regulates the organization of subpellicular microtubules and is required for maintaining cell morphology. TbKIN-D possesses in vitro ATPase activity, associates with cytoskeletal microtubules and is distributed throughout the cytoskeleton at all cell cycle stages. RNAi of TbKIN-D disrupts the organization of the subpellicular microtubule corset and distorts cell morphology, resulting in round cells with an elongated posterior filled with newly assembled microtubules. Depletion of TbKIN-D also abolishes the segregation of organelles and cytoskeletal structures, suggesting that cellular morphogenesis is essential for proper organelle segregation. Moreover, TbKIN-D deficiency impairs the attachment of the new flagellum without compromising the formation of the flagellum attachment zone. Finally, we identified TbKIN-C, a kinetoplastid-specific kinesin known to regulate subpellicular microtubules and cell morphogenesis in T. brucei, as a partner of TbKIN-D. Further, we demonstrate that interaction between TbKIN-C and TbKIN-D requires the coiled-coil motifs in the C-termini of both proteins. Altogether, our results suggest that TbKIN-D cooperates with TbKIN-C to maintain cell morphology by regulating the organization of the subpellicular microtubule corset.
Key words: Trypanosoma brucei, kinesin, microtubule, morphogenesis
Introduction
Microtubules constitute a vital part of the cytoskeleton of eukaryotic cells and are involved in various cellular processes, such as transport of cargos, chromosome segregation and cell morphogenesis (Nogales, 2001; Heald and Nogales, 2002). The cytoskeleton of Trypanosoma brucei, an early branched microbial eukaryote and the causative agent of human sleeping sickness, is defined by an array of subpellicular microtubules located underneath the plasma membrane (Gull, 1999). These subpellicular microtubules are cross-linked to each other and to the plasma membrane, and possess an intrinsic polarity, with the dynamic ends located at the posterior of the cell where cell growth takes place during early stages of the cell cycle (Sherwin and Gull, 1989b; Robinson et al., 1995). The microtubule cytoskeleton is essential for maintenance of cell shape and polarity and for distribution of organelles during the cell cycle in T. brucei, but little is known about the regulation of the assembly and organization of subpellicular microtubules.
Microtubules are a major component of the flagellum, which is composed of a canonical 9+2 microtubule axoneme and is essential for motility and cell morphogenesis in trypanosomes (Kohl et al., 2003; Broadhead et al., 2006; Ralston et al., 2009). The flagellum exits from the flagellar pocket and is attached to the cell body via a unique cytoskeletal structure, the flagellum attachment zone (FAZ) (Sherwin and Gull, 1989b). The FAZ contains a single filament and a specialized set of four microtubules associated with the smooth endoplasmic reticulum (Kohl et al., 1999) and is required for basal body segregation and cytokinesis (Absalon et al., 2007; Vaughan et al., 2008). The basal body is situated at the posterior of the cell and is linked to the mitochondrial DNA network, known as the kinetoplast (Gull, 1999). Replication and segregation of organelles and cytoskeletal structures such as the nucleus, kinetoplast, flagellum, Golgi and basal body are well coordinated with the growth of the new flagellum and the segregation of the new and old flagella during the cell cycle in trypanosomes (Woodward and Gull, 1990; Kohl and Gull, 1998; Vaughan, 2010).
Kinesins are microtubule-dependent motor proteins that participate in intracellular transport and regulate microtubule dynamics, which are essential for various cellular functions and cell morphogenesis (Hirokawa et al., 1998). The trypanosome genome encodes more than 40 kinesin-like proteins, among which 13 are kinetoplastid-specific kinesins and 15 are orphan kinesins (Wickstead and Gull, 2006). Strikingly, many well conserved mitotic kinesins, such as kinesin-5, kinesin-6 and kinesin-7, are missing from the trypanosome genome (Wickstead and Gull, 2006). However, the absence of these mitotic kinesins could be compensated by trypanosome-specific kinesins and orphan kinesins. Previously, we identified two orphan kinesins, TbKIN-A and TbKIN-B, that are essential for spindle assembly in trypanosomes (Li et al., 2008b). Inspired by the discovery of TbKIN-A and TbKIN-B as essential mitotic kinesins, we further explored the potential roles of other orphan kinesins in cellular morphogenesis, cell proliferation and cell cycle regulation in trypanosomes.
In this paper, we report the functional characterization of TbKIN-D, an orphan kinesin (Wickstead and Gull, 2006), in procyclic trypanosomes. We find that RNAi of TbKIN-D resulted in distorted cell morphology and disorganization of subpellicular microtubules. We also demonstrate that TbKIN-C, a trypanosome-specific kinesin previously shown to be essential for maintaining cell morphology in T. brucei (Hu et al., 2012), associates with TbKIN-D in vivo. Our data suggest a cooperative role of the two kinesin-like proteins in cell morphogenesis through regulating subpellicular microtubules.
Results
TbKIN-D encodes a divergent kinesin that associates with cytoskeletal microtubules
To search for essential orphan kinesins in T. brucei, we performed RNAi to knockdown each of the 15 orphan kinesins in procyclic trypanosomes. RNAi of one orphan kinesin (Tb927.11.10760) resulted in severe growth inhibition (see below). We named this kinesin TbKIN-D after the three kinesins, TbKIN-A, TbKIN-B and TbKIN-C, identified in our previous studies (Li et al., 2008b; Hu et al., 2012). TbKIN-D possesses an N-terminal motor domain and three coiled-coil motifs in the C-terminus (Fig. 1A). Although the microtubule-binding motif in the motor domain of TbKIN-D is conserved, the putative nucleotide-binding motif lacks the well-conserved lysine residue in the P-loop and two additional conserved motifs, SSRSH (switch I) and DLAGSE (switch II) (Sablin et al., 1996) (Fig. 1A). To test whether TbKIN-D possesses ATPase activity, we performed in vitro ATPase activity assay with purified motor domain of TbKIN-D, and found that it was capable of hydrolyzing ATP, as measured by phosphate release (Fig. 1B). To rule out the possibility that the detected activity was contributed by contamination from bacterial proteins, we mutated one of the two well-conserved glycine residues (G116; Fig. 1A, arrowhead) in the nucleotide-binding motif to alanine, and found that G116A mutation disrupted ATP hydrolysis activity (Fig. 1B). These results suggest that TbKIN-D possesses in vitro ATPase activity, despite lacking the lysine residue and the two switch motifs.
Fig. 1.
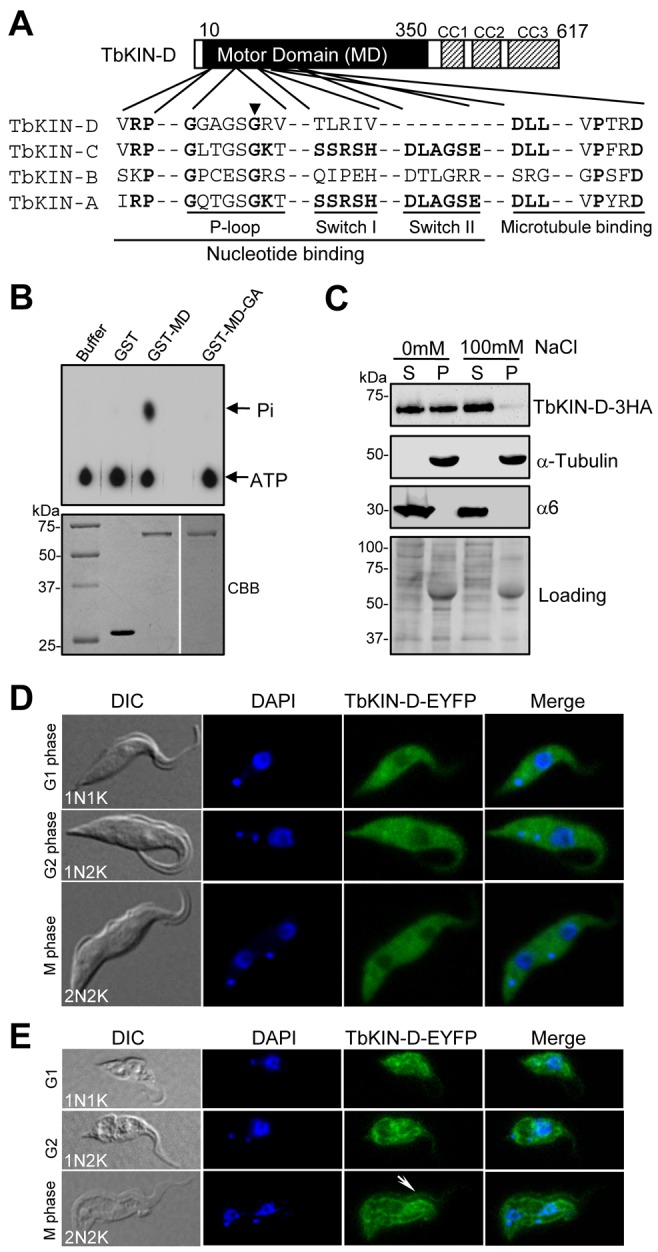
TbKIN-D possesses in vitro ATPase activity and associates with cytoskeletal microtubules in vivo. (A) Schematic representation of the structure of TbKIN-D. Three coiled-coil motifs (CC1–CC3) are indicated. Alignment of the conserved motifs in the motor domain of TbKIN-D with that of TbKIN-A, TbKIN-B, and TbKIN-C is presented. The black arrowhead points to the conserved glycine residue that is mutated to alanine in TbKIN-D (G116A) for the in vitro ATPase assay. (B) In vitro ATPase activity assay of the motor domain of TbKIN-D. The motor domain of TbKIN-D (MD) and its G116A mutant (MD-GA) were purified as GST fusion proteins from E. coli (lower panel) and used for in vitro ATPase assays (upper panel). Purified GST protein was included as the control. (C) Association of TbKIN-D with cytoskeletal microtubules. TbKIN-D–3HA in the soluble and insoluble fractions of trypanosome cells was detected with anti-HA antibody. The same blot was also probed with anti-α-tubulin antibody and anti-α6 antibody, which detects the α6 subunit of the 26S proteasome (Li et al., 2002), as controls for the insoluble and soluble fractions, respectively. The same blot was also stained with Coomassie Blue as the loading control. S, supernatant; P, pellet. (D,E) Subcellular localization of EYFP-tagged TbKIN-D during different cell-cycle stages in intact cells (D) and in detergent-treated cytoskeleton of the cell (E). The white arrow points to the flagellum to which TbKIN-D–EYFP is also localized.
To investigate whether TbKIN-D associates with cytoskeletal microtubules in vivo, soluble (cytosolic) and insoluble (cytoskeletal) fractions of trypanosome cells expressing a triple hemagglutinin (HA)-tagged TbKIN-D were prepared for western blotting. TbKIN-D was detected in both soluble and insoluble fractions (Fig. 1C). TbKIN-D in the insoluble fraction appeared to associate with microtubules as detected by anti-α-tubulin antibody, and the soluble fraction was detected by anti-α6 antibody that reacts with the α6 subunit of the 26S proteasome (Li et al., 2002) (Fig. 1C). Moreover, in the presence of high salt concentration, TbKIN-D in the cytoskeleton fraction was solubilized (Fig. 1C), indicating that association of TbKIN-D with cytoskeletal microtubules is dependent on ion strength. Together, these results suggest that some TbKIN-D proteins associate with cytoskeletal microtubules and some are present in the cytoplasm in the soluble form.
We next examined the subcellular distribution of TbKIN-D during the cell cycle by fluorescence microscopic analysis of EYFP-tagged TbKIN-D. In intact cells, TbKIN-D–EYFP was distributed throughout the cell body at all cell cycle stages, but it appeared to be excluded from the nucleus (Fig. 1D). When trypanosome cytoskeleton was prepared, TbKIN-D–EYFP remained to associate with the cytoskeleton at all cell cycle stages (Fig. 1E). Additionally, TbKIN-D–EYFP appeared to be also localized to the flagellum (Fig. 1E, arrow), although in some cells the localization to flagellum was not readily detectable (see the 1N1K and 1N2K cells in Fig. 1E). Since the trypanosome cytoskeleton is characterized by the subpellicular microtubule corset, the distribution of TbKIN-D throughout the cytoskeleton suggests that TbKIN-D may associate with subpellicular microtubules.
RNAi of TbKIN-D in T. brucei resulted in growth inhibition and cell death
To understand the role of TbKIN-D, we carried out RNAi in procyclic trypanosomes. TbKIN-D mRNA was significantly depleted after RNAi for 2 days (Fig. 2A), and TbKIN-D protein was gradually depleted from the cells after RNAi (Fig. 2B), which resulted in severe growth inhibition and cell death after 4 days (Fig. 2C), indicating that TbKIN-D is essential for cell proliferation and viability.
Fig. 2.

RNAi of TbKIN-D in the procyclic form of T. brucei inhibits cell proliferation. (A) TbKIN-D mRNA level in control and RNAi-treated cells, detected by northern blotting. (B) TbKIN-D protein level in control and RNAi-treated cells, detected by western blotting with anti-HA antibody. (C) RNAi of TbKIN-D resulted in growth inhibition and cell death. (D) Flow cytometry analysis of TbKIN-D RNAi cells. (E). Tabulation of cells with different numbers of nuclei (N) and kinetoplasts (K) upon TbKIN-D knockdown. (F) Percentage of TbKIN-D RNAi cells with a detached flagellum. (G) Morphology of TbKIN-D RNAi cells. Black arrows indicate the detached flagellum in TbKIN-D RNAi cells. N, nucleus; K, kinetoplast.
Flow cytometry was performed to examine whether the inhibited cell growth was attributed to any defects in the cell cycle. TbKIN-D RNAi resulted in a gradual decrease of cells with 2C DNA content, followed by an increase of cells with 4C and 8C DNA content after RNAi for 3 days (Fig. 2D), suggesting defects in cytokinesis. To further characterize the effect of TbKIN-D RNAi on cell cycle progression, cells with different numbers of nuclei and kinetoplasts were counted. After RNAi for 2–3 days, the proportion of cells with one nucleus and one kinetoplast (1N1K) and 1N2K was reduced from ∼70% to ∼10% (Fig. 2E). Strikingly, 2N1K cells made up ∼40% of the total population after RNAi for 3 days (Fig. 2E,G). The two nuclei in 2N1K and 2N2K cells appeared to be closer to each other than those in control cells (Fig. 2), suggesting that TbKIN-D may be also involved in mitosis. Moreover, cells with multiple nuclei and one kinetoplast (XN1K) and XN2K and XNXK also emerged (Fig. 2G), which, after RNAi for 4 days, constituted about 30%, 25% and 15% of the total population, respectively (Fig. 2E). The 2N1K and XN1K cells appeared to contain an enlarged kinetoplast, whereas the two kinetoplasts in the 2N2K and XN2K cells and the multiple kinetoplasts in XNXK cells were closer to each other than in the control 2N2K cells (Fig. 2G), suggesting that kinetoplast segregation was inhibited.
TbKIN-D RNAi does not inhibit the synthesis of the flagellum or FAZ
TbKIN-D RNAi caused flagellum detachment in more than 80% of the 2N2K, 2N1K, XN1K and XN2K cells and in all the XNXK cells (Fig. 2F,G, black arrows). Immunofluorescence assays with the L8C4 antibody, which recognizes the paraflagellar rod in trypanosomes (Kohl et al., 1999), showed that all the 2N2K, 2N1K, XN1K and XN2K cells contained two full-length flagella with one of them detached, and all the XNXK cells possessed multiple full-length flagella with majority of them detached (Fig. 3A). These results suggest that TbKIN-D deficiency does not affect the synthesis of new flagellum.
Fig. 3.

Effect of TbKIN-D RNAi on replication and segregation of the flagellum, flagellum attachment zone, basal body, flagellar pocket and Golgi. (A) Effect of TbKIN-D RNAi on the growth of the new flagellum. Cells were fixed in cold methanol, stained with L8C4 antibody for flagella, and counterstained with DAPI for DNA. Arrows point to the detached flagella. (B) Effect of TbKIN-D RNAi on the growth of the new FAZ. Cells were fixed in cold methanol, stained with L3B2 antibody to detect the FAZ and counterstained with DAPI (DNA). Arrows point to detached flagella that have a full-length FAZ. (C) Effect of TbKIN-D RNAi on basal body replication and segregation. Basal bodies (arrows) were stained with YL 1/2 antibody. The arrowhead indicates the accumulation of tyrosinated α-tubulin in the elongated posterior of the cell (see Fig. 4 below). (D) Effect of TbKIN-D RNAi on replication and segregation of the flagellar pocket (arrows) as monitored by anti-CRAM immunofluorescence. (E). Effect of TbKIN-D RNAi on Golgi replication and segregation. The Golgi (arrows) was labeled with anti-GRASP antibody. Scale bars: 5 µm.
Intriguingly, a closer examination of the detached flagellum in TbKIN-D RNAi cells showed that the detached flagellum appeared to associate with a small amount of cytoplasm (Fig. 2G, black arrows). Since the flagellum in a trypanosome cell is attached to the cell body via the FAZ, the presence of cytoplasm with the detached flagellum suggests that the FAZ was likely also detached. To test this possibility as well as to examine whether there was any defect in the FAZ in TbKIN-D RNAi cells, cells were stained with the L3B2 antibody, which specifically labels the FAZ in T. brucei (Kohl et al., 1999). The detached flagellum contained a full-length FAZ (Fig. 3B), suggesting that TbKIN-D RNAi does not impair the formation of the FAZ.
RNAi of TbKIN-D impairs organelle segregation
The accumulation of 2N1K and XN1K cells indicates that kinetoplast segregation was inhibited by TbKIN-D RNAi (Fig. 2G). Since kinetoplast segregation is mediated by basal body separation (Robinson and Gull, 1991), we examined whether basal body replication and/or segregation were compromised in TbKIN-D RNAi cells by immunostaining with the YL 1/2 antibody, which labels basal bodies and tyrosinated α-tubulin in trypanosomes (Woods et al., 1989). While the two basal bodies in control 2N2K cells were well segregated, the two basal bodies in 2N1K cells were very close to each other, and the multiple basal bodies in XN1K cells formed a cluster around the enlarged kinetoplast (Fig. 3C, arrows), indicating that segregation, but not replication, of basal bodies was abrogated after TbKIN-D RNAi. Therefore, defects in basal body segregation likely contributed to the inhibited kinetoplast segregation.
To investigate whether replication and/or segregation of other organelles and cytoskeletal structures were also affected by TbKIN-D RNAi, we monitored the flagellar pocket and Golgi by immunofluorescence microscopy. Flagellar pocket was stained with anti-CRAM antibody (Liu et al., 2000), and Golgi was detected with anti-GRASP antibody (He et al., 2004). We found that the 2N2K cells deficient in TbKIN-D contained two flagellar pockets and two Golgi apparatus that were separated ∼3–4 µm apart, but in control 2N2K cells the two organelles were separated around 6–7 µm (Fig. 3D,E). Additionally, the XN cells always possessed multiple flagellar pockets and multiple Golgi apparatus that were clustered around the single enlarged kinetoplast or kinetoplast cluster (Fig. 3D,E). These observations suggest that segregation of flagellar pockets and Golgi was also inhibited.
Depletion of TbKIN-D disrupts cell morphology, resulting in elongation of the posterior of the cell, which is filled with newly assembled microtubules
The most prominent effect of TbKIN-D RNAi is the distortion of cell shape, resulting in a fat cell with a slim and elongated posterior (Fig. 2G and Fig. 3). To quantitatively examine the effect of TbKIN-D RNAi on cell morphology, we measured the length of the posterior and the width of the cell body. The length of the posterior was defined as the distance between the kinetoplast and the posterior tip of the cell (Fig. 4A, white arrows), whereas the cell width was defined as the width of the middle portion of the cell between the two nuclei (Fig. 4A, red arrows). Only the cells during the first 2 days of RNAi were measured because we believe that the morphology change during the early stages of RNAi is likely attributed to the direct effect of TbKIN-D depletion. The posterior length of control cells was measured to be between 5 and 6 µm, with an average length of 5.3 µm (Fig. 4B). However, the average length of the posterior of TbKIN-D RNAi cells was calculated to be 10.8 µm, a two-fold increase compared to control cells. In some cells the posterior length exceeded 20 µm (Fig. 4B). The average width of control 2N cells was about 5 µm, but the average width of TbKIN-D RNAi cells was calculated to be 6.5 µm (Fig. 4C), a 30% increase in cell width after TbKIN-D RNAi.
Fig. 4.
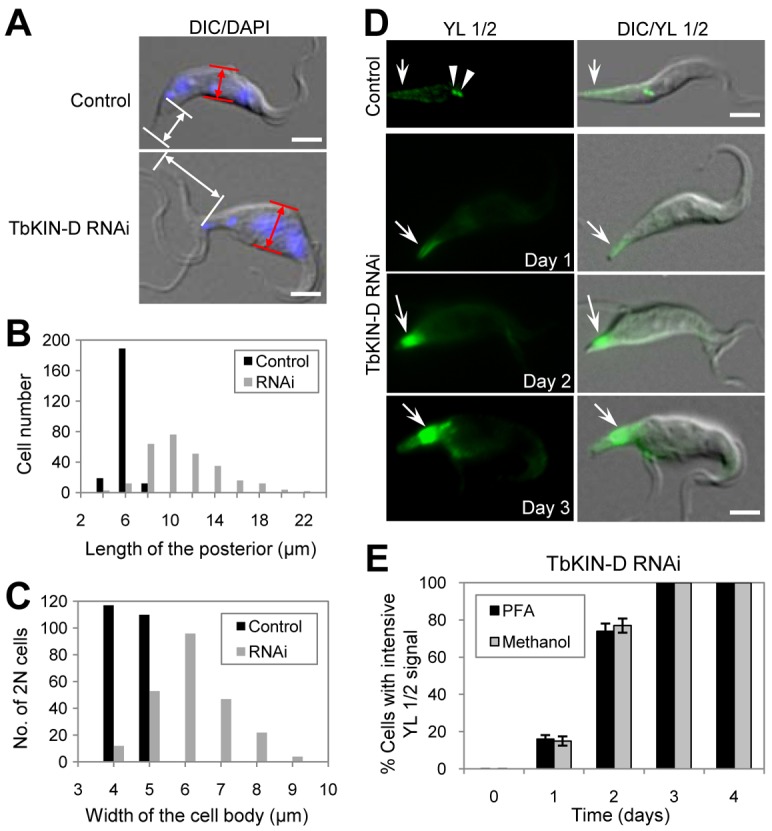
TbKIN-D RNAi caused accumulation of tyrosinated α-tubulin in the posterior portion of the cell. (A) TbKIN-D RNAi resulted in elongation of the posterior of the cell and rounding-up in the middle portion of the cell. White arrows show the length of the posterior of the cell, which is defined as the distance between the kinetoplast and the posterior tip of the cell, and red arrows indicate the width of the middle portion of the cell between the two nuclei. (B) The length of the posterior region was measured using ImageJ software, and plotted. 200 cells of each type were measured. (C) The width of the middle portion of 2N cells (∼200) was measured using ImageJ software, and plotted. (D) Cellular distribution of tyrosinated α-tubulin in TbKIN-D RNAi cells. Cells were fixed with either paraformaldehyde (PFA) or cold methanol and stained with YL 1/2 antibody, which stains tyrosinated α-tubulin. Arrows point to YL 1/2 signal, and arrowheads indicate the basal bodies in the control cells. Scale bars: 5 µm. (E) Percentage of TbKIN-D RNAi cells with strong YL 1/2 fluorescence signal in the posterior portion of the cell.
The average two-fold increase in posterior length of TbKIN-D RNAi cells promoted us to examine whether it was attributed to the extension of subpellicular microtubules towards the posterior. It has been well established that the subpellicular microtubule corset extends towards the posterior by assembling newly synthesized tubulins to the plus end of the microtubule corset during S and G2 phases in trypanosomes (Sherwin et al., 1987; Robinson et al., 1995). This can be detected by immunostaining with the YL 1/2 antibody (Kilmartin et al., 1982), which labels tyrosinated α-tubulins that are newly assembled at the plus ends of the subpellicular microtubules in trypanosomes (Sherwin et al., 1987). We found that while the posterior end of some control cells was lightly stained by YL 1/2 (Fig. 4D, arrow), as reported previously (Sherwin et al., 1987; Sherwin and Gull, 1989b; Robinson et al., 1995), the YL 1/2 fluorescence signal at the posterior of TbKIN-D RNAi cells was ∼10-fold of that in control cells (Fig. 4D, arrows). Strong YL 1/2 fluorescence signal was detected at the posterior tip of the cell after RNAi for 1 day, but at later stages of RNAi the signal was primarily detected in the region near basal bodies because YL 1/2-stained basal bodies were not separated from the strong YL 1/2 fluorescence signal (Fig. 4D, arrows). Similar YL 1/2 fluorescence pattern was observed when cells were fixed with either paraformaldehyde or methanol (Fig. 4E), indicating that the observed YL 1/2 fluorescence signal was not an artifact due to cell fixation. Strikingly, the number of TbKIN-D RNAi cells with strong YL 1/2 fluorescence signal increased to over 60% of the total population after RNAi for 2 days, and after RNAi for 3 days all TbKIN-D-deficient cells contained intensive YL 1/2 fluorescence signal (Fig. 4E). These results suggest the accumulation of newly assembled microtubules at the posterior portion of the cell after TbKIN-D RNAi, which likely contributed to the observed posterior elongation. These observations also suggest that RNAi of TbKIN-D likely interferes with the dynamics of the microtubules at the posterior of the cell.
Effect of TbKIN-D RNAi on the subpellicular microtubule corset
The trypanosome cytoskeleton is characterized by an array of subpellicular microtubules that are cross-linked to the plasma membrane and extend along the longitudinal axis from the anterior towards the posterior of the cell (Gull, 1999). Since TbKIN-D associates with cytoskeletal microtubules (Fig. 1), and RNAi of TbKIN-D distorts cell morphology and increases the abundance of newly assembled microtubules at the posterior of the cell (Fig. 4), we asked whether silencing of TbKIN-D affected the assembly and organization of microtubule corset. We examined the subpellicular microtubules as well as the flagellar axoneme by transmission electron microscopy. The flagellar axoneme in trypanosomes has the characteristic 9+2 organization of microtubules, and there appeared to be little change in the axoneme structure in TbKIN-D RNAi cells (Compare Fig. 5A and E). The subpellicular microtubules in control cells were arrayed underneath the plasma membrane with a regular inter-microtubule spacing (Fig. 5A). Strikingly, in TbKIN-D RNAi cells some subpellicular microtubules were placed away from the plasma membrane (Fig. 5B, black arrowheads), and additional microtubules were detected underneath the subpellicular microtubule corset (Fig. 5B, white arrowheads). These additional microtubules were either randomly distributed (Fig. 5B,E) or appeared to form another array underneath the regular subpellicular microtubule corset, albeit the spacing between these microtubules was not as even as that between the subpellicular microtubules (Fig. 5C). In other cases, the additional microtubules were distributed throughout the cell body, particularly at the posterior portion of the cell where there is no flagellum (Fig. 5D), consistent with the accumulation of tyrosinated microtubules at these locations (Fig. 4). Moreover, additional microtubules were also detected in the small portion of cytoplasm detached with the flagellum or at the anterior portion of the cell (Fig. 5F). These observations suggest disorganization of subpellicular microtubules and accumulation of internal microtubules in TbKIN-D RNAi cells.
Fig. 5.
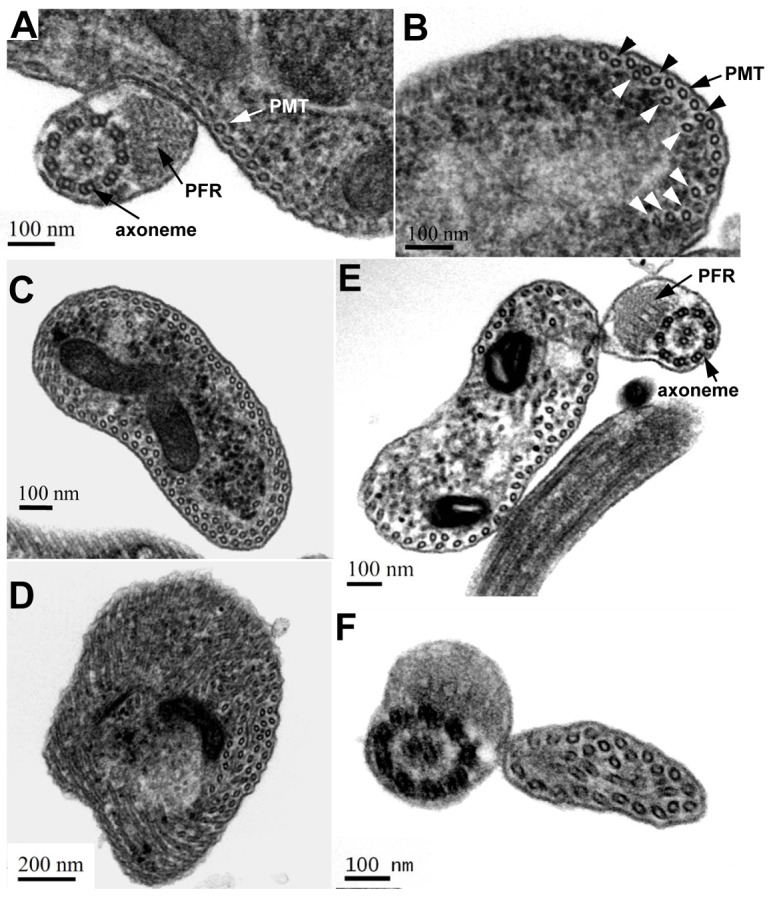
Effect of TbKIN-D RNAi on the organization of the subpellicular microtubule corset. (A) Thin section of a control cell. The flagellar axoneme, the paraflagellar rod (PFR) and the subpellicular microtubule corset (PMT) are indicated. (B) Thin section of a TbKIN-D RNAi cell. Black arrowheads point to the disorganized subpellicular microtubules, and white arrowheads indicate internal microtubules that are underneath the subpellicular microtubule corset. (C,D) Thin sections of the posterior portion of TbKIN-D RNAi cells (no flagellum is present) showing internal microtubules in addition to the subpellicular microtubule corset that is underneath the membrane. (E,F) Thin sections of TbKIN-D RNAi cells with a flagellum, which could be either the anterior portion of the cell or the small portion of cytoplasm along with the flagellum that is detached from the main cell body. Additional internal microtubules are detected throughout the cells, in addition to the subpellicular microtubule corset.
To examine the overall structure of the subpellicular microtubule corset, transmission electron microscopic analysis of detergent-extracted, negatively stained whole-mount cytoskeleton was performed. While the subpellicular microtubules in control cells were evenly spaced and organized (Fig. 6A,B), the subpellicular microtubules in TbKIN-D RNAi cells appeared to be unevenly spaced (Fig. 6C–H), which were detected at the posterior portion of the cell and in the portion of cytoskeleton detached along with the new flagellum (Fig. 6D,E,H). Strikingly, the spacing between the microtubules in the middle, rounded-up portion of the cell appeared to be significantly increased, whereas the microtubules in the slim and elongated posterior portion of the cell were tightly bundled together, making it difficult to distinguish between individual microtubules even at high magnifications (compare Fig. 6G,J and K,M). It is likely that accumulation of internal microtubules underneath the subpellicular microtubule corset (Fig. 5C,D) contributed to the observed tight microtubule bundle at the posterior portion of the cell (Fig. 6K). In some cases, there appeared to be a constricted region near the flagellar basal body (Fig. 6N, arrowhead). Finally, the cytoplasm that was detached with the flagellum was clearly visible (Fig. 6C,L,N, arrows), which further confirms the observations made under light microscope (Fig. 2G).
Fig. 6.

Whole-mount negative staining to examine the effect of TbKIN-D RNAi on the organization of the subpellicular microtubule corset. (A,B) A negatively stained control 1N cell visualized under the electron microscope. (B) An enlarged view of the boxed area in A, showing the even subpellicular microtubule corset. (C–E) A TbKIN-D RNAi cell with a detached flagellum (arrow). (D,E) Enlarged view of the boxed areas in C, showing the disorganized microtubules at the posterior of the cell, and a portion of detached cytoplasm, along with the flagellum, from the main cell body, respectively. (F–H) A fat TbKIN-D RNAi cell with an elongated posterior. (G,H) Enlarged view of the boxed areas in F, showing the disorganized microtubules at the rounded-up portion, and the elongated posterior, respectively. (I–K) A TbKIN-D RNAi cell with a very long and slender posterior. (J,K) Enlarged view of the boxed areas in I, showing disorganized microtubules in the middle portion of the cell, and tightly bundled microtubules at the slender posterior, respectively. (L,M) A multinucleated TbKIN-D RNAi cell with a fat cell shape, a slender, elongated posterior and a detached flagellum (arrow). (M) Enlarged view of the boxed area in L, showing the tight microtubule bundles at the posterior end of the cell. (N) A TbKIN-D RNAi cell with a detached flagellum (arrow) and a constricted region near the flagellar basal body (arrowhead).
TbKIN-D interacts with TbKIN-C, a kinetoplastid-specific kinesin
To identify TbKIN-D partners, we carried out tandem affinity purification. Endogenous TbKIN-D was tagged at its C-terminus with the PTP (protein C–tobacco etch virus protease–protein A) epitope (Schimanski et al., 2005). TbKIN-D–PTP was then purified from trypanosomes consecutively through IgG affinity chromatography and anti-protein C epitope immunoaffinity chromatography. The purification was highly efficient (Fig. 7A), and a major protein was co-precipitated with TbKIN-D–PTP (Fig. 7B). Liquid chromatography–tandem mass spectrometry (LC/MS/MS) analysis indicated that this protein was TbKIN-C, a kinetoplastid-specific kinesin (Hu et al., 2012). To further confirm the in vivo interaction between TbKIN-D and TbKIN-C, co-immunoprecipitation was performed using cells co-expressing endogenously PTP-tagged TbKIN-D and triple HA-tagged TbKIN-C. We found that immunoprecipitation of TbKIN-D–PTP was able to pull down TbKIN-C–3HA from the cell lysate (Fig. 7C), suggesting that the two proteins do interact with each other in trypanosomes. Since TbKIN-D and TbKIN-C contain three coiled-coil motifs at their C-termini (Fig. 1A) (Hu et al., 2012), we tested whether the coiled-coil motif is required for interaction between them. Yeast two-hybrid assays indicated that TbKIN-D and TbKIN-C were capable of interacting with each other, but neither protein was able to interact with itself (Fig. 7D). Further, deletion of the third coiled-coil motif (CC3) from TbKIN-D and TbKIN-C completely abolished the interaction (Fig. 7D), suggesting that the third coiled-coil motif is involved in interaction between the two proteins.
Fig. 7.

Tandem affinity purification identifies TbKIN-C as a partner of TbKIN-D. (A) Procyclic cells expressing PTP-tagged TbKIN-D were subject to the two-step chromatography affinity purification. Western blot with anti-protein-C antibody was performed to monitor the purification of TbKIN-D-PTP fusion protein. (B) Identification of TbKIN-C as the partner of TbKIN-D by SDS-PAGE followed by mass spectrometry. (C) Co-immunoprecipitation to verify the in vivo interaction between TbKIN-C and TbKIN-D in trypanosomes. TbKIN-D was tagged with a PTP epitope and TbKIN-C was tagged with a triple HA epitope in the same cell line. TbKIN-D-PTP was precipitated by incubating with IgG–Sepharose, and the immunoprecipitate was immunoblotted with anti-HA and anti-protein-C antibody to detect 3HA-tagged TbKIN-C and PTP-tagged TbKIN-D, respectively. (D) Interaction of TbKIN-C and TbKIN-D in yeast, and the requirement of the coiled-coil motifs for the interaction between the two proteins.
Discussion
In this paper, we report the functional characterization of TbKIN-D in the procyclic form of T. brucei. TbKIN-D possesses a highly divergent motor domain in which the conserved lysine residue in the P-loop is replaced by arginine and two well-conserved motifs, the switch I (SSRSH) and switch II (DLAGSE), are missing (Fig. 1A). The two switch motifs form a salt-bridge that closes the nucleotide-binding cleft, enabling the motor to hydrolyze ATP (Sablin et al., 1996; Parke et al., 2010). Nevertheless, our results showed that despite lacking these conserved residues, TbKIN-D was capable of hydrolyzing ATP (Fig. 1B), suggesting that the absence of the lysine residue and the two switch motifs may be compensated by arginine and other residues, respectively. Replacement of the lysine residue by arginine and the absence of switch I and II motifs were also observed in TbKIN-B, another orphan kinesin that plays essential roles in spindle assembly and chromosome segregation in trypanosomes (Li et al., 2008b; Li et al., 2008a). Intriguingly, among the 15 orphan kinesins in trypanosomes, only TbKIN-D and TbKIN-B lack the lysine residue and the two switch motifs (Fig. 1A), both of which are essential for trypanosome viability (Fig. 2) (Li et al., 2008b). Moreover, the conserved threonine/serine residue immediately downstream of the lysine residue found in many kinesins is replaced with a valine residue in TbKIN-D (Fig. 1A). The Thr/Ser residue in the kinesin motor domain appears to control motor activity because a Thr/Asn or Thr/Ile mutation is able to convert the protein to a so-called ‘rigor’ kinesin that binds microtubules very tightly even in the presence of ATP (Nakata and Hirokawa, 1995). In this regard, it would be interesting to investigate whether TbKIN-D possesses similar properties like the rigor kinesin.
TbKIN-D is required for maintaining cell morphology in procyclic trypanosomes (Figs 2, 4 and 6). It has been well established that the basic cell shape of a trypanosome cell is maintained by the subpellicular microtubule corset underlying the plasma membrane and extending towards the posterior of the cell (Gull, 1999). Additionally, it is known that the size and morphology of a trypanosome cell is also defined by the position and length of the flagellum (Kohl et al., 2003). However, RNAi of TbKIN-D apparently does not impair the synthesis of the new, full-length flagella that contain normal axoneme structures (Figs 3 and 5), indicating that the morphology change of TbKIN-D RNAi cells is not due to any defects in the flagellum. Instead, TbKIN-D deficiency significantly interferes with cytoskeletal microtubules, resulting in excessive accumulation of internal microtubules underneath the subpellicular microtubule corset and disorganization of the subpellicular microtubule corset (Figs 4–6). These observations suggest that the distorted cell shape caused by TbKIN-D RNAi is very likely attributed to the drastic change in the abundance and organization of cytoskeletal microtubules.
One unusual phenotype caused by TbKIN-D RNAi is the detachment of flagellum along with a small amount of cytoplasm (Figs 2 and 6). RNAi of TbKIN-C caused similar phenotype (Hu et al., 2012). It remains unknown how the new flagellum and the small portion of cytoplasm were split from the main cell body after TbKIN-D or TbKIN-C RNAi. Presumably, the cell could have undergone an asymmetrical cytokinesis due to mis-positioning of the cleavage plane between the new and old flagella. However, this notion is not supported by the observation that the chromosomal passenger complex, an essential regulator of cytokinesis initiation in trypanosomes (Li et al., 2008b), failed to be targeted to the cytokinesis initiation site after TbKIN-C RNAi (Hu et al., 2012). Alternatively, since TbKIN-D RNAi cells lost their regular shape before the new flagellum and the small portion of cytoplasm were detached, it is possible that the cell was unable to maintain the new flagellum in the right position due to distorted morphology.
Several lines of evidence suggest that TbKIN-D functions together with TbKIN-C to regulate cytoskeletal microtubules and cellular morphogenesis. First, TbKIN-D interacts with TbKIN-C in vivo in trypanosomes (Fig. 7). Second, the morphology of TbKIN-D-deficient cells is almost identical to that of TbKIN-C RNAi cells; both cells are round-shaped with an elongated posterior filled with tyrosinated microtubules (Fig. 4). Finally, RNAi of TbKIN-D disrupts the organization of the subpellicular microtubule corset (Fig. 5), a striking phenotype that was also observed in TbKIN-C RNAi cells. Given that most kinesins perform their cellular functions as dimer via coiled-coil-mediated dimerization (Wordeman, 2010), our data suggest that TbKIN-D and TbKIN-C likely form a heterodimer through interactions between the C-terminal coiled-coil motifs (Fig. 7).
RNAi of TbKIN-D appeared to exert three effects on cytoskeletal microtubules: extension of the subpellicular microtubule corset towards the posterior of the cell, which resulted in elongation of the posterior (Fig. 6), disorganization of the subpellicular microtubule corset (Fig. 5), and de novo assembly of internal microtubules underneath the subpellicular microtubule corset (Fig. 5). Although we do not know the exact function of TbKIN-D in trypanosomes, the elongation of posterior suggests that TbKIN-D, together with TbKIN-C, may regulate microtubule dynamics by either inhibiting microtubule assembly or promoting microtubule disassembly at the plus end or both. Kinesins are known to not only move cargos along the microtubules but also regulate microtubule dynamics (Endow et al., 2010). The best-described function in the latter capacity is microtubule depolymerization, and it is known that kinesin-8, kinesin-13 and kinesin-14 all have depolymerization activity (Endow et al., 2010). Trypanosomes express an expanded kinesin-13 family consisting of seven members, among which TbKif13-1 regulates spindle disassembly and TbKif13-2 controls flagellum length (Chan and Ersfeld, 2010; Chan et al., 2010; Wickstead et al., 2010). The trypanosome genome also encodes two kinesin-14 proteins, but lacks kinesin-8 homolog (Wickstead and Gull, 2006). Since RNAi of TbKIN-D, as well as TbKIN-C (Hu et al., 2012), appears to primarily promote the assembly of cytoskeletal microtubules (Fig. 5), it will be interesting to investigate whether TbKIN-D and TbKIN-C possess depolymerization activity. Alternatively, the two kinesin-like proteins may be involved in positioning or organizing microtubule nucleation centers or in inserting nascent microtubules into the subpellicular microtubule corset. Future work will be directed to understand the function of the two proteins in regulating the assembly and organization of the corset.
Materials and Methods
Trypanosome cell culture and RNA interference
Procyclic 427 strain was cultured at 27°C in SDM-79 medium supplemented with 10% fetal bovine serum (Atlanta Biologicals, Inc.), whereas procyclic 29–13 cell line (Wirtz et al., 1999) was cultivated in SDM-79 medium containing 10% fetal bovine serum, 15 µg/ml G418 and 50 µg/ml hygromycin. Cells were diluted routinely when the density reaches 5× 106/ml.
RNAi was carried out as described previously (Dang and Li, 2011). A 400–500-bp fragment from the coding region of each of the 14 orphan kinesins was cloned into the pZJM vector (Wang et al., 2000). The resulting plasmids were linearized and electroporated into the 29-13 cell line. After 24 hours, cells were selected under 2.5 µg/ml phleomycin in addition to G418 and hygromycin. Successful transfectants were selected under 2.5 µg/ml phleomycin and cloned by limiting dilution. To induce RNAi, the monoclonal cell line was incubated with 1.0 µg/ml tetracycline, and cell growth was monitored daily by counting the number of cells under a light microscope with a hemacytometer.
Northern blotting
Total RNA was purified from T. brucei cells with the TRIzol reagent (Invitrogen). Northern blotting was performed as previously described (Li et al., 2003). Briefly, 30 µg total RNA was transferred onto a nitrocellulose membrane in 20× SSC solution. Northern hybridization was carried out overnight at 42°C in 50% formamide, 6× SSC, 0.5% SDS, 5× Denhardt's solution with 0.1 mg/ml salmon sperm DNA. The membrane was washed three times in 2× SSC, 0.1% SDS, 1× SSC, 0.1% SDS, and 0.5× SSC, 0.1% SDS for 30 min, respectively.
Purification of GST fusion protein and in vitro ATPase assays
Wild-type TbKIN-D and the mutant TbKIN-D bearing G116A mutation in its ATP-binding motif were each cloned into pGEX-4T-3 vector for expression of recombinant proteins in E. coli. The motor domain (MD) of TbKIN-D and the G116A mutant were also cloned into pGEX-4T-3 vector. Recombinant proteins were expressed in E. coli BL21 strain and purified through a column of glutathione Sepharose 4B beads. Purified proteins were dialyzed against 50mM Tris-HCl, pH 8.0 and 50 mM NaCl. Only GST–MDTbKIN-D and GST–MDTbKIN-D-G116A were soluble and used for in vitro ATPase activity assay.
In vitro ATP hydrolysis catalyzed by GST–MDTbKIN-D and its G116A mutant was assayed by measuring the release of radiolabelled Pi from [γ-32P]ATP according to our published procedures (Dang and Li, 2011; Hu et al., 2012). GST–MDTbKIN-D or its G116A mutant was incubated with 20 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 30 mM KCl, 4% (w/v) sucrose, 80 µg/ml BSA, 1 mM ATP, 0.2 µCi [γ-32P]ATP at 37°C for 2 hrs, and the reaction was stopped by chilling on ice. 1.0 µl of the reaction solution was spotted onto a polyethyleneimine (PEI) cellulose thin-layer plate, and ascending chromatography was performed in 0.5 M LiCl and 1.0 M formic acid at room temperature for 20 min. The plate was air dried and exposed to X-ray films.
In situ epitope tagging of endogenous TbKIN-D in the procyclic form
A 500-bp DNA fragment corresponding to the C-terminal portion of TbKIN-D was cloned into the pC-EYFP-Neo vector, and the resulting constructs were electroporated into the 427 cell line according to our published procedures (Dang and Li, 2011; Hu et al., 2012). The same DNA fragment was also cloned into the pC-3HA-Bla vector, and the resulting construct was transfected into pZJM-TbKIN-D RNAi cell line for monitoring TbKIN-D protein level after TbKIN-D RNAi. Stable transfectants were selected under 40 µg/ml G418 or 10 µg/ml blasticidin, and monoclonal cell lines were obtained by limiting dilution. Correct in situ tagging of one of the two TbKIN-D alleles was confirmed by PCR and subsequent sequencing of the PCR fragment (see supplementary material Fig. S1 for details).
Preparation of cytoskeletal proteins
Preparation of the cytoskeleton of trypanosomes was performed according to published procedures (Robinson et al., 1991). Cells expressing endogenously 3HA-tagged TbKIN-D were incubated in PEME buffer (0.1 M PIPES, pH 6.9, 2 mM EGTA, 1 mM magnesium sulfate, 0.1 mM EDTA) containing 1% Nonidet P-40 at room temperature for 5 min. To solubilize any cytoskeleton-associated proteins, 100 mM NaCl was added to the PEME buffer. Cells were then spun down, and soluble (S) fraction and insoluble pellet (P) fraction were separated. The pellet fraction was further washed three times with PEME buffer. Both fractions were boiled in 1× SDS-PAGE sampling buffer for 5 min and analyzed by western blotting with anti-HA to detect TbKIN-D-3HA, anti-α-tubulin for insoluble (cytoskeletal) fraction control, and anti-α6 antibody against the α6 subunit of the 26S proteasome for soluble (cytosolic) fraction control (Li et al., 2002), respectively.
Flow cytometry
Flow cytometry analysis of propidium-iodide-stained trypanosome cells was carried out as previously described (Li et al., 2006). Briefly, T. brucei cells were fixed in ethanol, washed once with PBS, and re-suspended in PBS containing DNase-free RNase (10 µg/ml) and propidium iodide (20 µg/ml). The DNA content of propidium-iodide-stained cells was analyzed with a fluorescence-activated cell sorting scan (FACScan) analytical flow cytometer (BD Biosciences).
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde or in cold methanol and were adhered to poly-L-lysine-treated coverslips. Cells were then incubated with primary antibodies at room temperature for 1 hr. The following antibodies were used: YL 1/2 mAb for tyrosinated α-tubulin (1:400 dilution) (Kilmartin et al., 1982); α-CRAM pAb for flagellar pocket (1:400 dilution) (Liu et al., 2000); α-GRASP pAb for Golgi (1:300 dilution) (He et al., 2004); L8C4 mAb for the flagellum (1:50 dilution) (Kohl et al., 1999); and L3B2 for the FAZ (1:50 dilution) (Kohl et al., 1999). After three washes, cells were incubated with FITC-conjugated or Cy3-conjugated anti-mouse IgG or anti-rabbit IgG at room temperature for 1 hr. The slides were mounted in VectaShield mounting medium (Vector Labs) containing DAPI and examined using an inverted microscope (Model IX71, Olympus) equipped with a cooled CCD camera (model Orca-ER, Hamamatsu) and a PlanApo N 60× 1.42 NA DIC objective. Images were acquired and processed using the Slidebook5 software (Intelligent Imaging Innovations, Inc).
Preparation of whole-mount cytoskeletons and thin sections of trypanosomes, and transmission electron microscopy
Preparation of whole-mount cytoskeleton was performed according to published procedures (Sherwin and Gull, 1989a). Briefly, cells were washed twice with PBS, settled onto freshly charged carbon and Formvar-coated grids and incubated with PEME buffer containing 1% Nonidet P-40 for 5 min. Cells were then fixed with glutaradehyde for 20 sec and negatively stained with aurothioglucose in distilled water.
Trypanosome cells were fixed in glutaraldehyde, treated with Millonig's buffer for 10 min and then incubated with 2% OsO4 at 4°C for 60 min. Cells were dehydrated with ethanol (50% ethanol for 5 min, 70% ethanol for 10 min, 95% ethanol for 10 min and finally treated three times with 100% ethanol for 10 min) and then embedded in resin (treated twice with 100% propylene oxide for 15 min, 50% LX-112 resin/50% propylene oxide for 2 hrs and finally with 100% LX-112 resin for 2 hrs). The fixed cells were embedded in BEEM capsules before polymerizing overnight at 70°C. Subsequently, the 120 nm thin sections were cut using a Leica Ultracut-R microtome and a diamond knife (Daitome-US), placed on 150 mesh copper grids (EMS) and stained with 2% uranyl acetate for 15 min. The thin sections were rinsed with water and incubated with Renold's lead citrate for 5 min. Grids were imaged using a JEOL 1400 TEM at 60 kV and captured with a Gatan CCD camera.
PTP tagging of TbKIN-D and tandem affinity purification
A 900-bp DNA fragment corresponding to the C-terminal portion of TbKIN-D was cloned into the pC-PTP-Neo vector (Schimanski et al., 2005), and the resulting construct was linearized with Nsi I and electroporated into the 427 cell line for in situ tagging of one of the two TbKIN-D alleles. Successful transfectants were selected under 40 µg/ml G418 and cloned by limiting dilution. Correct in situ tagging of one of the two alleles of TbKIN-D gene was confirmed by PCR and subsequent sequencing. Expression of TbKIN-D–PTP fusion protein was verified by western blotting with anti-protein-C antibody (Roche).
Tandem affinity purification was carried out according to our previous procedures (Li et al., 2008b). Briefly, 500 ml procyclic cells expressing TbKIN-D–PTP was lysed by sonication, and the crude lysate was incubated with 200 µl IgG Sepharose beads at 4°C for 2 hrs. After overnight incubation with TEV protease (Promega), the eluate was incubated with 200 µl anti-protein C beads at 4°C for 2 hrs. Proteins were then eluted with EGTA/EDTA elution buffer. Proteins were further precipitated with StrataClean resin (Stratagene), separated on SDS-PAGE and stained with Coomassie Blue. Protein bands were excised from the gel and analyzed by LC/MS/MS for protein identification.
Co-immunoprecipitation and western blotting
Procyclic cells expressing TbKIN-D–PTP were transfected with the pC-TbKIN-C-3HA-Bla vector to express TbKIN-C–3HA from one of its endogenous loci. Successful transfectants were selected under 10 µg/ml blasticidin. For immunoprecipitation, 20 ml of the double transfectants were lysed in trypanosome immunoprecipitation (IP) buffer (25 mM Tris-HCl, pH 7.6, 100 mM NaCl, 1 mM DTT, 1% Nonidet P-40 and protease inhibitor cocktail), and incubated with 20 µl IgG Sepharose beads at 4°C for 2 hrs. After six washes with IP buffer, beads were re-suspended in SDS-PAGE sampling buffer and boiled for 5 min. The supernatant was separated on SDS-PAGE and probed with anti-HA antibody and anti-protein-C antibody, respectively.
Procyclic cells expressing 3HA-tagged TbKIN-D were re-suspended in SDS-PAGE sampling buffer, boiled for 5 min, and cleared by centrifugation. The cell lysate was fractionated in SDS-PAGE and immunoblotted with anti-HA antibody as described previously (Li and Wang, 2008). Total proteins on the membrane were stained with Coomassie Blue as the loading control.
Yeast two-hybrid analysis
Full-length coding sequences of TbKIN-D and TbKIN-C and their truncation mutants were each cloned into pGADT7 vector to express prey proteins or pGBKT7 vector to express bait proteins. Yeast strains AH109 (mating type a) and Y187 (mating type α) were transformed with prey or bait plasmids, respectively. Strains carrying different combinations of bait and prey plasmids were generated by mating the haploids in YPDA medium and were selected on SD-Leu-Trp plates Each combination strain was then spotted in three ten-fold serial dilutions onto SD-Leu-Trp and SD-Leu-Trp-His plates.
In-gel tryptic digestion and on-line capillary LC-MS and LC-MS-MS analysis
In-gel digestions of proteins were carried out according to our published procedures (Li et al., 2008b). 160 ng of trypsin (Promega, Madison, WI) was used for each band, and digestion was carried out at 37°C for 4 hrs. Peptides were then extracted from gel pieces with 50 µl of 50% acetonitrile, 2% acetic acid, and the extraction solution was concentrated to ∼ 10 µl.
The trypsin digest (1.0 µl) was injected into a Dionex Ultimate 30000 RSLCnano UHPLC system (Dionex Coporation, Sunnyvale, CA), and separated by a 75 µm×25 cm PepMap RSLC column (100 Å, 2 µm) at a flow rate of ∼450 nl/min. The HPLC eluent was connected directly to the nanoelectrospray ionization source of an LTQ Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA). LC-MS data were acquired in an information-dependent acquisition mode, cycling between a MS scan (m/z 310-2,000) acquired in the Orbitrap, followed by a low-energy CID analysis on three most intense multiply charged precursors acquired in the linear ion trap. The centroided peak lists of the CID spectra were searched against a UniProt protein database (UniProtKB.2011.07.27), to which a randomized version had been concatenated using Batch-Tag, a program in the in-house version of the University of California San Francisco Protein Prospector version 5.9.2. A precursor mass tolerance of 15 ppm and a fragment mass tolerance of 0.5 Da were used for protein database search. Protein hits were reported with a Protein Prospector protein score ≥22, protein discriminant score ≥0.0, and a peptide expectation value ≤0.01 (Chalkley et al., 2005). This set of protein identification parameters threshold did not return any substantial false positive protein hit from the randomized half of the concatenated database.
Acknowledgements
We are grateful to George A. M. Cross, Rockefeller University, for providing the 29-13 cell line and to Paul Englund, Johns Hopkins School of Medicine, for providing the pZJM vector. We also thank Arthur Günzl, University of Connecticut Health Center, for the pC-PTP-Neo and pC-HA-Bla vectors and Keith Gull, University of Oxford, for the L8C4 and L3B2 antibodies. Anti-CRAM antibody and anti-GRASP antibody are kind gifts from Mary Lee, New York University, and Graham Warren, Max F. Perutz Laboratories, Austria, respectively. We thank Steven Kolodziej, University of Texas Medical School at Houston, for help with electron microscopy.
Supplementary Material
Footnotes
Funding
This work was supported by start-up funds from the University of Texas Medical School at Houston [grant number UTHSC-H-34001 to Z. L.] and, in part, by the National Institutes of Health [grant number AI090070 to Z. L.]; and the New Hampshire Agricultural Experiment Station and National Institute of Food and Agriculture/US Department of Agriculture [grant number H00567 to F.C.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.106534/-/DC1
References
- Absalon S., Kohl L., Branche C., Blisnick T., Toutirais G., Rusconi F., Cosson J., Bonhivers M., Robinson D., Bastin P. (2007). Basal body positioning is controlled by flagellum formation in Trypanosoma brucei. PLoS ONE 2, e437 10.1371/journal.pone.0000437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadhead R., Dawe H. R., Farr H., Griffiths S., Hart S. R., Portman N., Shaw M. K., Ginger M. L., Gaskell S. J., McKean P. G.et al. (2006). Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 440, 224–227 10.1038/nature04541 [DOI] [PubMed] [Google Scholar]
- Chalkley R. J., Baker P. R., Huang L., Hansen K. C., Allen N. P., Rexach M., Burlingame A. L. (2005). Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell. Proteomics 4, 1194–1204 10.1074/mcp.D500002-MCP200 [DOI] [PubMed] [Google Scholar]
- Chan K. Y., Ersfeld K. (2010). The role of the Kinesin-13 family protein TbKif13-2 in flagellar length control of Trypanosoma brucei. Mol. Biochem. Parasitol. 174, 137–140 10.1016/j.molbiopara.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. Y., Matthews K. R., Ersfeld K. (2010). Functional characterisation and drug target validation of a mitotic kinesin-13 in Trypanosoma brucei. PLoS Pathog. 6, e1001050 10.1371/journal.ppat.1001050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang H. Q., Li Z. (2011). The Cdc45·Mcm2-7·GINS protein complex in trypanosomes regulates DNA replication and interacts with two Orc1-like proteins in the origin recognition complex. J. Biol. Chem. 286, 32424–32435 10.1074/jbc.M111.240143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endow S. A., Kull F. J., Liu H. (2010). Kinesins at a glance. J. Cell Sci. 123, 3420–3424 10.1242/jcs.064113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gull K. (1999). The cytoskeleton of trypanosomatid parasites. Annu. Rev. Microbiol. 53, 629–655 10.1146/annurev.micro.53.1.629 [DOI] [PubMed] [Google Scholar]
- He C. Y., Ho H. H., Malsam J., Chalouni C., West C. M., Ullu E., Toomre D., Warren G. (2004). Golgi duplication in Trypanosoma brucei. J. Cell Biol. 165, 313–321 10.1083/jcb.200311076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald R., Nogales E. (2002). Microtubule dynamics. J. Cell Sci. 115, 3–4 [DOI] [PubMed] [Google Scholar]
- Hirokawa N., Noda Y., Okada Y. (1998). Kinesin and dynein superfamily proteins in organelle transport and cell division. Curr. Opin. Cell Biol. 10, 60–73 10.1016/S0955-0674(98)80087-2 [DOI] [PubMed] [Google Scholar]
- Hu L., Hu H., Li Z. (2012). A kinetoplastid-specific kinesin is required for cytokinesis and for maintenance of cell morphology in Trypanosoma brucei. Mol. Microbiol. 83, 565–578 10.1111/j.1365-2958.2011.07951.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin J. V., Wright B., Milstein C. (1982). Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. J. Cell Biol. 93, 576–582 10.1083/jcb.93.3.576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl L., Gull K. (1998). Molecular architecture of the trypanosome cytoskeleton. Mol. Biochem. Parasitol. 93, 1–9 10.1016/S0166-6851(98)00014-0 [DOI] [PubMed] [Google Scholar]
- Kohl L., Sherwin T., Gull K. (1999). Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J. Eukaryot. Microbiol. 46, 105–109 10.1111/j.1550-7408.1999.tb04592.x [DOI] [PubMed] [Google Scholar]
- Kohl L., Robinson D., Bastin P. (2003). Novel roles for the flagellum in cell morphogenesis and cytokinesis of trypanosomes. EMBO J. 22, 5336–5346 10.1093/emboj/cdg518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Wang C. C. (2008). KMP-11, a basal body and flagellar protein, is required for cell division in Trypanosoma brucei. Eukaryot. Cell 7, 1941–1950 10.1128/EC.00249-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Zou C. B., Yao Y., Hoyt M. A., McDonough S., Mackey Z. B., Coffino P., Wang C. C. (2002). An easily dissociated 26 S proteasome catalyzes an essential ubiquitin-mediated protein degradation pathway in Trypanosoma brucei. J. Biol. Chem. 277, 15486–15498 10.1074/jbc.M109029200 [DOI] [PubMed] [Google Scholar]
- Li Y., Li Z., Wang C. C. (2003). Differentiation of Trypanosoma brucei may be stage non-specific and does not require progression of cell cycle. Mol. Microbiol. 49, 251–265 10.1046/j.1365-2958.2003.03575.x [DOI] [PubMed] [Google Scholar]
- Li Z., Tu X., Wang C. C. (2006). Okadaic acid overcomes the blocked cell cycle caused by depleting Cdc2-related kinases in Trypanosoma brucei. Exp. Cell Res. 312, 3504–3516 10.1016/j.yexcr.2006.07.022 [DOI] [PubMed] [Google Scholar]
- Li Z., Umeyama T., Wang C. C. (2008a). The chromosomal passenger complex and a mitotic kinesin interact with the Tousled-like kinase in trypanosomes to regulate mitosis and cytokinesis. PLoS ONE 3, e3814 10.1371/journal.pone.0003814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Lee J. H., Chu F., Burlingame A. L., Günzl A., Wang C. C. (2008b). Identification of a novel chromosomal passenger complex and its unique localization during cytokinesis in Trypanosoma brucei. PLoS ONE 3, e2354 10.1371/journal.pone.0002354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Qiao X., Du D., Lee M. G. (2000). Receptor-mediated endocytosis in the procyclic form of Trypanosoma brucei. J. Biol. Chem. 275, 12032–12040 10.1074/jbc.275.16.12032 [DOI] [PubMed] [Google Scholar]
- Nakata T., Hirokawa N. (1995). Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J. Cell Biol. 131, 1039–1053 10.1083/jcb.131.4.1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E. (2001). Structural insight into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 30, 397–420 10.1146/annurev.biophys.30.1.397 [DOI] [PubMed] [Google Scholar]
- Parke C. L., Wojcik E. J., Kim S., Worthylake D. K. (2010). ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J. Biol. Chem. 285, 5859–5867 10.1074/jbc.M109.071233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston K. S., Kabututu Z. P., Melehani J. H., Oberholzer M., Hill K. L. (2009). The Trypanosoma brucei flagellum: moving parasites in new directions. Annu. Rev. Microbiol. 63, 335–362 10.1146/annurev.micro.091208.073353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. R., Gull K. (1991). Basal body movements as a mechanism for mitochondrial genome segregation in the trypanosome cell cycle. Nature 352, 731–733 10.1038/352731a0 [DOI] [PubMed] [Google Scholar]
- Robinson D., Beattie P., Sherwin T., Gull K. (1991). Microtubules, tubulin, and microtubule-associated proteins of trypanosomes. Methods Enzymol. 196, 285–299 10.1016/0076-6879(91)96027-O [DOI] [PubMed] [Google Scholar]
- Robinson D. R., Sherwin T., Ploubidou A., Byard E. H., Gull K. (1995). Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J. Cell Biol. 128, 1163–1172 10.1083/jcb.128.6.1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablin E. P., Kull F. J., Cooke R., Vale R. D., Fletterick R. J. (1996). Crystal structure of the motor domain of the kinesin-related motor ncd. Nature 380, 555–559 10.1038/380555a0 [DOI] [PubMed] [Google Scholar]
- Schimanski B., Nguyen T. N., Günzl A. (2005). Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot. Cell 4, 1942–1950 10.1128/EC.4.11.1942-1950.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin T., Gull K. (1989a). The cell division cycle of Trypanosoma brucei brucei: timing of event markers and cytoskeletal modulations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 323, 573–588 10.1098/rstb.1989.0037 [DOI] [PubMed] [Google Scholar]
- Sherwin T., Gull K. (1989b). Visualization of detyrosination along single microtubules reveals novel mechanisms of assembly during cytoskeletal duplication in trypanosomes. Cell 57, 211–221 10.1016/0092-8674(89)90959-8 [DOI] [PubMed] [Google Scholar]
- Sherwin T., Schneider A., Sasse R., Seebeck T., Gull K. (1987). Distinct localization and cell cycle dependence of COOH terminally tyrosinolated alpha-tubulin in the microtubules of Trypanosoma brucei brucei. J. Cell Biol. 104, 439–446 10.1083/jcb.104.3.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan S. (2010). Assembly of the flagellum and its role in cell morphogenesis in Trypanosoma brucei. Curr. Opin. Microbiol. 13, 453–458 10.1016/j.mib.2010.05.006 [DOI] [PubMed] [Google Scholar]
- Vaughan S., Kohl L., Ngai I., Wheeler R. J., Gull K. (2008). A repetitive protein essential for the flagellum attachment zone filament structure and function in Trypanosoma brucei. Protist 159, 127–136 10.1016/j.protis.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Wang Z., Morris J. C., Drew M. E., Englund P. T. (2000). Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275, 40174–40179 10.1074/jbc.M008405200 [DOI] [PubMed] [Google Scholar]
- Wickstead B., Gull K. (2006). A “holistic” kinesin phylogeny reveals new kinesin families and predicts protein functions. Mol. Biol. Cell 17, 1734–1743 10.1091/mbc.E05-11-1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B., Carrington J. T., Gluenz E., Gull K. (2010). The expanded Kinesin-13 repertoire of trypanosomes contains only one mitotic Kinesin indicating multiple extra-nuclear roles. PLoS ONE 5, e15020 10.1371/journal.pone.0015020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz E., Leal S., Ochatt C., Cross G. A. (1999). A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89–101 10.1016/S0166-6851(99)00002-X [DOI] [PubMed] [Google Scholar]
- Woods A., Sherwin T., Sasse R., MacRae T. H., Baines A. J., Gull K. (1989). Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J. Cell Sci. 93, 491–500 [DOI] [PubMed] [Google Scholar]
- Woodward R., Gull K. (1990). Timing of nuclear and kinetoplast DNA replication and early morphological events in the cell cycle of Trypanosoma brucei. J. Cell Sci. 95, 49–57 [DOI] [PubMed] [Google Scholar]
- Wordeman L. (2010). How kinesin motor proteins drive mitotic spindle function: Lessons from molecular assays. Semin. Cell Dev. Biol. 21, 260–268 10.1016/j.semcdb.2010.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.