

Abstract
5-Hydroxymethylcytosine (5hmc) is a newly discovered DNA base present at detectable levels in most mammalian cell types and tissues. It is generated by Tet-enzyme–mediated oxidation of 5-methylcytosine (5mc). 5hmc is important both because of its potential role in regulating gene expression and because it may be an intermediate in DNA demethylation. Here we describe a technique termed GLIB (glucosylation, periodate oxidation and biotinylation), which combines several enzymatic and chemical modification steps to attach biotin to 5hmc. Biotin-containing genomic DNA fragments are then enriched using streptavidin beads, eluted and sequenced. GLIB is capable of quantitatively tagging and precipitating fragments containing a single 5hmc molecule. sample preparation and GLIB can be conducted in 2–3 d.
INTRODUCTION
A full description of the generation and function of 5hmC, a comparison of different 5hmC precipitation techniques and a discussion of how precipitation techniques can be appraised are all contained in the introduction of the accompanying manuscript1. We limit ourselves here to a description of the GLIB method and a comparison of the GLIB and anti–cytosine 5-methylenesulfonate (CMS) methodologies.
GLIB relies on a combination of enzymatic and chemical treatment to selectively biotinylate 5hmC (ref. 2). In place of regular cytosine, T-even bacteriophages (T2, T4 and T6 phages) contain glycosylated 5hmC (ref. 3), generated by enzymes unrelated to Tet. T4 phages express an enzyme, β-glucosyltransferase (BGT), that can be expressed in bacteria and can reliably add a glucose to 5hmC in vitro4. The glycols (hydroxyls on adjacent carbons) present in glucose can be oxidized with sodium periodate to generate aldehydes5, which in turn can be functionalized with biotins in aqueous solvent under mild reaction conditions6 (Fig. 1). A similar strategy has been used to precipitate glycosylated proteins7. Because glucose contains hydroxyls at the 2-, 3- and 4- positions, and because adjacent aldehydes and hydroxyls can also be oxidized, three distinct reaction products are possible if the reaction does not proceed to completion (Fig. 1). However, all of them will contain two aldehydes that can be biotinylated and subsequently precipitated with streptavidin-conjugated beads.
Figure 1.

The GLIB method. Glucose is added to 5hmC using BGT, oxidized with sodium periodate to yield aldehydes and reacted with ARP, yielding two biotins at the site of every 5hmC.
GLIB has certain advantages and disadvantages relative to anti-CMS and other methodologies. GLIB allows near-quantitative precipitation of DNA fragments containing even a single 5hmC, which is not possible with the anti-5hmC or even anti-CMS techniques (Fig. 2a). The GLIB technique also does not precipitate CA and CT repeats, an artifact observed when using 5hmC-specific antibodies8. At least in embryonic stem cells (ESCs), the GLIB and anti-CMS methods yield very similar results, as discussed in the accompanying manuscript1.
Figure 2.

Effect of sonication on background precipitation in genomic DNA samples. (a) Extent of precipitation of variably hydroxymethylated PCR amplicons using the technique indicated. The red dots indicate the percentage of amplicons in a population expected to contain at least one 5hmC. This therefore corresponds to the theoretical degree of precipitation expected for a ‘perfect’ pull-down method that pulls down all hydroxymethylated amplicons and nothing else. The blue dots indicate the observed percentage of precipitation. Note that with the GLIB technique the observed pull-down is close to the theoretical maximum, whereas the anti-CMS technique has a lower degree of background precipitation. (b) The extent of pull-down observed from GLIB using endogenous sources containing low (293), intermediate (mouse ESCs) or high (293 cells expressing TET1) levels of 5hmC. Error bars show means ± s.d. of 3–6 independent experiments. Note that DNA fragmented with the restriction enzyme MseI has a lower degree of background pull-down than DNA fragmented with a Covaris S2 AFA instrument. pAb, polyclonal antibody. Reproduced from ref. 2 with permission.
Biotinylation of 5-formylcytosine with the GLIB method is predicted to be inefficient. Although 5-formylcytosine contains an aldehyde that can react with the biotin-containing aldehyde reactive probe (ARP), it is less active than a normal aldehyde because it is conjugated to an aromatic ring and requires special reaction conditions9. 5-Carboxylcytosine is not predicted to react via GLIB. Both bases are very rare relative to 5hmC and unlikely to confound analysis10.
A drawback of GLIB is that it produces markedly higher background precipitation of DNA than anti-CMS. Approximately 1% of cytosine- or 5mC-containing oligos are precipitated by the GLIB technique (Fig. 2a). A similar precipitation percentage is observed for DNA that contains virtually no 5hmC, such as HEK293 cell DNA. Fragmentation with a Covaris Adaptive Focused Acoustics (AFA) instrument increases the background pull-down to ~3% precipitation, even if aldehydes in the sonicated DNA are quenched with methoxyamine before GLIB treatment (Fig. 2b). Fragmentation with a Diagenode Bioruptor produces similar levels of background (data not shown). This high background only occurs if DNA is treated with sodium periodate before biotinylation, indicating that it is a product of minor side reactions of periodate with the DNA. Fortunately, the GLIB technique contains a built-in negative control: by omitting BGT, it is possible to generate a parallel sample that recapitulates the sources of background in the GLIB technique. Both samples are sequenced and regions of 5hmC can be identified as areas that show a high density of reads in the + BGT but not the − BGT sample. The background pull-down in GLIB requires that more reads be generated to distinguish signal from background, but is not otherwise problematic.
Although lower background can be attained by fragmenting DNA with a restriction enzyme such as MseI (Fig. 2b), the non-random fragmentation caused by such enzymes creates serious systematic bias in the data and is strongly discouraged for sequencing applications.
Another drawback of GLIB is that because the GLIB adduct has a very slight inhibitory effect on PCR (delay of ~0.1 cycles per converted 5hmC residue)2, heavily hydroxymethylated regions may be underrepresented when using sequencing platforms that rely on PCR amplification. As a result, we have so far always used a Helicos instrument to sequence GLIB-treated DNA. The protocol described below is written for sequencing with a Helicos instrument. Users who want to apply GLIB to a Solexa or SOLiD instrument will need to perform size selection and adapter ligation between Steps 5 and 6 of the PROCEDURE (see steps 4–19 of the Procedure in ref. 1). To avoid PCR bias altogether, we are testing a version of the ARP that contains a disulfide and thus can be cleaved by thiols to yield a smaller adduct.
There has as yet been no rigorous comparison of the GLIB method and the ‘click’ chemistry–based 5hmC precipitation method engineered by Song et al.11. Because of the strength of the biotin-streptavidin interaction, both are likely to be very effective for precipitation of 5hmC.
Experimental design
Genomic DNA is isolated from cells or tissues and sheared using a Covaris AFA instrument. The GLIB protocol is then implemented as described in the PROCEDURE below. To verify the efficiency of 5hmC-immunoprecipitation (IP), it is strongly recommended that hydroxymethylated and control DNA oligonucleotides be added to (i.e., spiked into) the genomic DNA (see Box 1 and Fig. 3 for a description of the generation of these oligos and for the oligonucleotide sequences). A–BGT control should be performed to serve as a negative control so that enriched regions can be identified.
Box 1. Generation of spike-in control DNA fragments  ~6 h.
~6 h.
As controls, GLIB precipitations should include a substrate containing a single 5hmC residue, as well as a substrate without 5hmC of the same approximate size. An example of how such fragments can be generated is given below. The pUC19NH fragment is a control spike-in DNA fragment that contains a single 5hmC, incorporated by Klenow fill-in of 5′-A-G-5hmC-T-3′ opposite the 5′ overhang. The pUC19AF fragment is a control spike-in DNA fragment that contains no 5hmC. These should be added to the pool of genomic DNA that will be subjected to GLIB treatment. The quantity added will depend on the desired molar proportion of the spike-in DNA fragment compared with the pool of genomic DNA (e.g., 0.1 pg of pUC19 fragments per microgram of human genomic DNA would be roughly equimolar for single-copy sequences in the human genome). Recovery of the pUC19NH fragment relative to the pUC19AF fragment after GLIB pull-down should be quantified. Sequences of each fragment are provided in Figure 3.
1. Digest 6 μg of pUC19 plasmid with 20 U each of NdeI and HindIII for 2 h at 37 °C (pUC19NH) in a 50-μl reaction volume. Set up a separate reaction to digest 6 μg of pUC19 plasmid with 10 U each of AhdI and FspI for 2 h at 37 °C (pUC19AF) in a 50-μl reaction volume.
2. Fill in the overhangs on pUC19NH only using the following conditions, and then incubate for 30 min at 37 °C:
| Reagent | Volume (μl) | Final |
|---|---|---|
| pUC19NH digest from step 1 |
50 | 6 μg |
| Klenow enzyme (5 U μl−1) | 2 | 10 U |
| NEB buffer 2, 10× | 1 | 1× |
| dhmCTP (2 mM) | 1 | 33 μM |
| dTTP (2 mM) | 1 | 33 μM |
| dATP (2 mM) | 1 | 33 μM |
| dGTP (2 mM) | 1 | 33 μM |
| ddH2O | 3 μl |
3. Run the pUC19NH and pUC19AF reactions on a 2.5% (wt/vol) agarose gel, and cut out the respective 267- and 222-bp fragments. Purify with a Qiagen gel extraction kit according to the manufacturer’s instructions, eluting in 50 μl of buffer EB (included in the kit).
4. Measure DNA concentration using NanoDrop.
 DNA can be stored at −20 °C for months.
DNA can be stored at −20 °C for months.
Figure 3.

Sequence of hmC-containing oligos.
Fresh reagents must be used where indicated below. It is recommended that new users perform quick validation with synthetic substrates to confirm efficient precipitation before undertaking expensive sequencing of genomes. This optimization is described in Box 2 (see also Table 1); oligonucleotide sequences are included in Figure 3.
Box 2. Optimization of GLIB conditions  32 h.
32 h.
Preparing DNA oligonucleotides containing different amounts of 5hmc 28 h
28 h
1. Generate a 201-bp DNA oligonucleotide using PCR (DNA sequence and primer information is listed in Fig. 3). 5hmC-containing oligonucleotides are generated by replacing dCTP with hmdCTP. Make oligonucleotides containing different ratios of 5hmC and C by using different ratios of dCTP to hmdCTP as listed in table 1. Note that other ~200-bp amplicons with an ~25% cytosine content can also be used for this application. Set up and run the PCR as tabulated below.
| Reagent | Volume (μl) | Final |
|---|---|---|
| Template, 5 ng μl−1 | 2 | 10 ng |
| Primer mixture, 10 μM | 2 | 0.2 μM |
| dNTP (10 mM, containing varying ratios of dCTP and dhmCTP) |
5 | 1 mM |
| Reaction buffer, 10× | 5 | 1× |
| Taq polymerase (5 U μl−1) | 1 | 0.1 U |
| ddH2O | Up to 50 |
| Cycle | Denature | Anneal | Extend | Hold |
|---|---|---|---|---|
| 1 | 95 °C, 2 min | — | — | — |
| 2–36 | 95 °C, 20 s | 65 °C, 30 s | 72 °C, 2 min | — |
| 37 | — | — | 72 °C, 10 min | — |
| 38 | — | — | — | 4 °C, ∞ |
2. Run the PCR product on a 2% (wt/vol) agarose gel (1h, 90 V) and purify with a Qiagen gel extraction kit according to the manufacturer’s instructions.
3. Perform glucosylation, periodate treatment and biotinylation according to Steps 6–16 of the main PROCEDURE.
End-labeling GLIB-modified DNA oligonucleotides with 32P  2 h
2 h
4. End-label 500 ng of modified DNA using T4 PNK enzyme (NEB) in 50-μl volume as tabulated below; incubate at 37 °C for 1 h.
| Reagent | Volume (μl) | Final |
|---|---|---|
| DNA, 250 ng | X | 250 ng |
| T4 PNK (10 U μl−1) | 1 | 0.4 U |
| T4 PNK buffer, 10× | 2.5 | 1× |
| ATP [γ-32P] | 1 | |
| ddH2O | Fill to 25 |
 ATP [γ-32P] is a radioactive material. Radiation should be handled carefully and only after training, according to rules set by the facility at which the experiments occur.
ATP [γ-32P] is a radioactive material. Radiation should be handled carefully and only after training, according to rules set by the facility at which the experiments occur.
5. Remove unincorporated ATP [γ-32P] by processing samples through a G50 Illustra MicroSpin column (GE Healthcare) according to the manufacturer’s instructions.
 DNA can be stored at −20 °C for some time, but as γ-32P has a half-life of 2 weeks, sensitivity will drop as the DNA is stored. Do not store longer than 2 weeks.
DNA can be stored at −20 °C for some time, but as γ-32P has a half-life of 2 weeks, sensitivity will drop as the DNA is stored. Do not store longer than 2 weeks.
Optimizing GLIB pull-down conditions  2 h
2 h
6. Use 20 μl of streptavidin M280 Dynabeads slurry (Invitrogen) for test pull-downs. Wash beads three times with 200 μl of 1× binding and wash buffer (5 mM Tris (pH 7.5), 0.5 mM EDTA and 1 M NaCl), and collect the beads after each resuspension using a magnetic rack.
7. Resuspend the beads in 20 μl of 2× binding and wash buffer. Add 20 μl of a mixture of radiolabeled DNA and water.
8. Rotate the beads for 15 min at room temperature on a nutator.
9. Collect the beads on the magnet and save the supernatant.
10. Wash three times with 200 μl of 1× binding and wash buffer. Save the washes.
11. Resuspend beads in 100 μl of 1× binding and wash buffer and transfer the entire solution to a scintillation vial.
12. Transfer the supernatant from pull-down to a second scintillation vial, and one-fifth of all five washes (pooled) in a third scintillation vial.
13. Measure the radiation in each vial using a scintillation counter.
 Measured counts per minute tends to drop as beads settle in the scintillation vial, and thus vials must be inverted repeatedly before counts in order to ensure accuracy.
Measured counts per minute tends to drop as beads settle in the scintillation vial, and thus vials must be inverted repeatedly before counts in order to ensure accuracy.
14. Calculate percentage pull-down by the formula (measured IP)/(measured IP + measured supernatant + 5× measured wash). Pull-down can also be estimated by calculating measured IP/input, but this is less precise because even small pipetting errors can cause variations in the initial input amount.
15. Compare the observed pull-down with the theoretical pull-down in table 1.
![]()
Table 1.
Calculated 5hmC level and IP efficiency per amplicon.
| Average no. of 5hmCs per amplicon |
81 | 20 | 4 | 2 | 1 | 0.5 | 0.25 | 0 |
|---|---|---|---|---|---|---|---|---|
| 5hmdCTP (%) | 100 | 24.69 | 4.94 | 2.47 | 1.23 | 0.62 | 0.31 | 0 |
| dCTP (%) | 0 | 75.31 | 95.06 | 97.53 | 98.77 | 99.38 | 99.69 | 100 |
| Theoretical IP efficiency (%) |
100 | 100 | 86.8 | 61.9 | 63.3 | 39.6 | 22.2 | 0 |
An overview of the working procedure is shown in Figure 4. At least two biological replicates are recommended in order to obtain reliable results.
Figure 4.

Workflow of the GLIB method.
MATERIALS
REAGENTS
DNA, at least 20 μg, RNA-free
pUC19 (New England Biolabs, cat. no. N3041S)
EDTA (Sigma-Aldrich, cat. no. E9884)
Sodium chloride (NaCl; Sigma-Aldrich, cat. no. S3014)
GenElute-LPA (Sigma-Aldrich, cat. no. 56575)
Methoxyamine-HCl (Sigma-Aldrich, cat. no. 226904)
HEPES (Mediatech CellGro, cat. no. 25-060-Cl)
Magnesium chloride (MgCl2) (Sigma-Aldrich, cat. no. M8266)
Uridine diphosphoglucose (UDPG; New England Biolabs, cat. no. M0357S)
Sodium periodate (NaIO4; Sigma-Aldrich, cat. no. 311448)
 Sodium periodate solution must be prepared fresh.
Sodium periodate solution must be prepared fresh.Sodium phosphate, monobasic (Fisher Scientific, cat. no. S397-500)
Sodium phosphate, dibasic (Fisher Scientific, cat. no. S375-500)
Sodium sulfite (Sigma-Aldrich, cat. no. 239321)
 Sodium sulfite solution must be prepared fresh.
Sodium sulfite solution must be prepared fresh.Aldehyde-reactive probe (Invitrogen, cat. no. A-10550)
Dimethyl sulfoxide (DMSO; American Bioanalytical, cat. no. AB03091-00050)
 DMSO is absorbed by skin and is somewhat toxic. Wear protective clothing and gloves when handling.
DMSO is absorbed by skin and is somewhat toxic. Wear protective clothing and gloves when handling.Formamide (Sigma-Aldrich, cat. no. F9037-100 ml)
 Formamide is toxic upon inhalation or on contact with skin or eyes. Wear protective clothing and gloves and handle under a fume hood.
Formamide is toxic upon inhalation or on contact with skin or eyes. Wear protective clothing and gloves and handle under a fume hood.Sodium acetate (Sigma-Aldrich, cat. no. S2889-250G)
QIAquick PCR purification kit (Qiagen, cat. no. 28106)
QIAquick gel extraction kit (Qiagen, cat. no. 28704)
Illustra MicroSpin G-50 columns (GE Healthcare, cat. no. 27-5330-01)
Dynabeads M280-Streptavidin (Invitrogen, cat. no. 11206D)
QuantIt OliGreen kit (Invitrogen, cat. no. O11492)
PBS, 10× (Meditech CellGro, cat. no. 46-013-CM)
Deoxynucleotide solution set (New England Biolabs, cat. no. N0446S)
Hydroxymethyl dCTP (Bioline, cat. no. BIO-39046)
pUC19 DNA (New England Biolabs, cat. no. N3041S)
T4 phage β-glucosyltransferase (T4-BGT; New England Biolabs, cat. no. M0357S)
AhdI (New England Biolabs, cat. no. R0584S)
FspI (New England Biolabs, cat. no. R0135S)
HindIII (New England Biolabs, cat. no. R0104S)
NdeI (New England Biolabs, cat. no. R0111S)
DNA polymerase, Klenow fragment (New England Biolabs, cat. no. M0210S)
Ethanol (Sigma-Aldrich, cat. no. E7023)
Tris (Sigma-Aldrich, cat. no. 154563)
Agarose (Invitrogen, cat. no. 16500500)
EQUIPMENT
SpectraMax M2 multimode microplate reader (Molecular Devices)
AFA S2 (Covaris)
DynaMag-2 (Invitrogen, cat. no. 123-21D)
NanoDrop 1000 (Thermo Scientific)
Thermocycler (Applied Biosystems, 2720)
Nutator (Southwest Science, SB3D2300)
Desktop centrifuge (Eppendorf, 5424)
REAGENT SETUP
 All of the reagents listed below can be stored indefinitely at room temperature (20 °C) but must be discarded if a visible precipitate is formed.
All of the reagents listed below can be stored indefinitely at room temperature (20 °C) but must be discarded if a visible precipitate is formed.
Binding and wash buffer, 2× Binding and wash buffer contains 10 mM Tris (pH 8.0), 1 mM EDTA and 2 M NaCl.
TE buffer TE contains 10 mM Tris (pH 8.0) and 1 mM EDTA.
TAE buffer TAE contains 40 mM Tris (pH 8.1), 20 mM acetate and 2 mM EDTA.
Sodium phosphate buffer, 1 M, pH 7.0 Sodium phosphate buffer contains 57.7 ml of 1 M Na2HPO4 and 42.3 ml of 1 M NaH2PO4.
PROCEDURE
Dna preparation  3–4 h
3–4 h
1| Pre-chill the Covaris AFA S2 instrument to 4 °C and degas for 30 min.
2| Dilute 10 μg of genomic DNA into 130 μl of TE buffer and transfer to a Covaris microtube (6 × 16 mm). The DNA input amount can be increased or decreased. If the amount of DNA is increased, shear no more than 10 μg of DNA at a time.
 We anticipate that, in the negative control sample or a sample that contains little 5hmC, 99.75% of input will be lost during processing steps or will not be precipitated during pull-down. To ensure a final yield of at least 25 ng of DNA for sequencing, we recommend an input of at least 10 μg.
We anticipate that, in the negative control sample or a sample that contains little 5hmC, 99.75% of input will be lost during processing steps or will not be precipitated during pull-down. To ensure a final yield of at least 25 ng of DNA for sequencing, we recommend an input of at least 10 μg.
3| Shear genomic DNA in a microtube using the Covaris AFA according to the manufacturer’s instructions. The target fragment size depends on the sequencing platform and application. For the Helicos S2 sequencing platform, a target size of 300 bp is recommended2.
4| Clean up sheared DNA using the Qiagen MinElute PCR cleanup kit according to the manufacturer’s instructions and elute in 20 μl of elution buffer.
5| Run 100 ng of fragmented genomic DNA on a 2% (wt/vol) agarose gel (90 V, ~1 h) to confirm accurate sonication.  Size selection of DNA is not necessary for Helicos sequencing. If the GLIB technique is being applied for use with an Illumina instrument, size selection using AmpurXP beads or gel extraction and adapter ligation are necessary (see steps 4–19 of the Procedure in ref. 1).
Size selection of DNA is not necessary for Helicos sequencing. If the GLIB technique is being applied for use with an Illumina instrument, size selection using AmpurXP beads or gel extraction and adapter ligation are necessary (see steps 4–19 of the Procedure in ref. 1).
 DNA can be stored at −20 °C for months.
DNA can be stored at −20 °C for months.
GLIB protocol  32 h (with 1 week for sequencing)
32 h (with 1 week for sequencing)
6| (Optional) Add spike-in hydroxymethylated and cytosine-containing oligo at a concentration of 1 pg per 10 μg of DNA (see Box 1 and Fig. 3). This will ensure that it is present in approximately equimolar amounts with comparably sized, fragmented genomic DNA.
7| (Optional) Prereact sheared (and if appropriate for the sequencing platform, size-selected) genomic DNA with 5 mM methoxyamine-HCl in 1× PBS for 1 h at 37 °C. This will slightly reduce background, but this step can be omitted to save time and conserve the sample.
8| If the methoxyamine-HCl incubation in Step 7 was conducted, it is essential to purify the DNA on Qiagen PCR purification columns according to the manufacturer’s instructions. Use one PCR purification column per 10 μg of DNA.
9| Glucosylate 1 μg of DNA with 2 U of BGT in 50 μl of 50 mM HEPES (pH 8.0), 25 mM MgCl2 and 50 μM UDPG for 3 h at 30 °C (see Box 3 and Fig. 5 for definition of BGT units and optimization of glucosylation). For larger amounts of DNA, reaction volume and BGT quantity should be scaled up accordingly. If large quantities of DNA must be glucosylated (>20 μg), it is acceptable to use up to 4 μg of DNA and 8 U of BGT in 50-μl volume.
Figure 5.
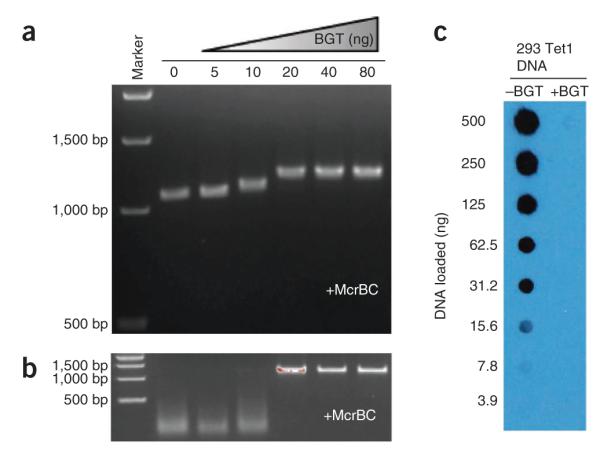
Anticipated results of experiments to verify successful glucosylation. (a) Treatment with increasing amounts of BGT causes slower migration of a 1,001-bp hydroxymethylated PCR product on an agarose gel. (b) Treatment with increasing amounts of BGT protects the hydroxymethylated PCR product from degradation by McrBC. (c) Treatment with BGT eliminates 5hmC signal in an anti-5hmC dot blot. The lack of signal in the dot blot indicates complete or near-complete glucosylation. Reproduced from ref. 2 with permission.
 It is important to run a negative control reaction in parallel, in which BGT is omitted during this step. Otherwise, the negative control sample is to be treated exactly in the same way as the experimental sample.
It is important to run a negative control reaction in parallel, in which BGT is omitted during this step. Otherwise, the negative control sample is to be treated exactly in the same way as the experimental sample.
10| Purify DNA on a Qiagen PCR purification column according to the manufacturer’s instructions, using 10 μg of DNA per column, and eluting with 36 μl of 10 mM Tris (pH 8.0).
 DNA can be stored at −20 °C for months.
DNA can be stored at −20 °C for months.
11| Incubate DNA with 23 mM NaIO4 in 100 mM sodium phosphate (pH 7.0) at 22 °C for 16 h. Use a 90-μl reaction volume for every 36 μl of eluent from the end of Step 10.
 Freshly prepare the sodium periodate stock solution.
Freshly prepare the sodium periodate stock solution.
12| Quench the unreacted periodate via incubation with sodium sulfite. Add 10 μl of 460 MM sodium sulfite per 90-μl reaction in Step 11 (final concentration 46 mM sodium sulfite).  Freshly prepare the sodium sulfite stock solution.
Freshly prepare the sodium sulfite stock solution.
13| Pre-exchange a G50 column with 1× PBS. This entails centrifuging for 1 min at 735g at room temperature to drain the liquid, adding 300 μl of 1× PBS, and centrifuging at 735g for 1 min again at room temperature to remove the 1× PBS. Add and drain fresh PBS two more times.
14| Apply up to 50 μl of sample to the column, use multiple columns for samples with volumes >50 μl, and then centrifuge for 2 min at 735g at room temperature.
 Some newer G50 columns require spins at 2,000g instead of 735g. Follow the manufacturer’s guidelines regarding column spin speed.
Some newer G50 columns require spins at 2,000g instead of 735g. Follow the manufacturer’s guidelines regarding column spin speed.
15| Dissolve ARP (Invitrogen) in DMSO to a final concentration of 100 mM. Dissolved ARP can be stored at −20 °C until required.
16| React DNA with a final concentration of 2 mM ARP for 1 h at 37 °C (the DNA should already be in 1× PBS), and then purify it with Qiagen PCR purification columns according to the manufacturer’s instructions, eluting with 10 mM Tris (pH 8) and 0.1 mM EDTA. As described earlier, do not load more than 10 μg of DNA per column.
 DNA can be stored at −20 °C for months.
DNA can be stored at −20 °C for months.
17| Wash 100 μl of the streptavidin M280 Dynabeads slurry (Invitrogen) three times with 1 ml of 1× binding and wash buffer (5 mM Tris (pH 7.5), 0.5 mM EDTA and 1 M NaCl) and collect the beads after each resuspension using a magnetic rack.
18| Resuspend the beads in 100 μl of 2× binding and wash buffer. Bring the volume of DNA from Step 16 to 100 μl by adding water, and add it to the beads. Rotate the beads for 15 min at room temperature on a nutator.
19| Wash the beads three times with 1 ml of 1× binding and wash buffer.
20| Elute DNA by incubating the beads with 100 μl of 95% (vol/vol) formamide and 10 mM EDTA at 90 °C for 5 min. Collect the beads on a magnet, save the eluent and immediately place it on ice.
21| Incubate the beads with another 100 μl of 95% formamide and 10 mM EDTA at 90 °C for 5 min to elute any residual DNA. Pool this eluent with the first sample.
22| Place the chilled eluent once again on the magnet to remove any residual beads—there will usually be a few—and save the eluent.
23| Mix the eluent with 200 μl of chilled water, 40 μl of 3 M sodium acetate (pH 8.0), 20 μg of linear polyacrylamide and 1 ml of chilled 100% (vol/vol) ethanol.
24| Precipitate the DNA by incubating this mixture for 1 h at −80 °C, and then centrifuging at 14,000g for 30 min at 4 °C.
25| Wash the pellet three times with 900 μl of room-temperature 70% (vol/vol) ethanol and remove residual ethanol by brief centrifugation and subsequent aspiration.
 Be very careful not to disturb the pellet, which may be hard to see. It is preferable to leave a little ethanol at the bottom of the tube during each wash step than to accidentally lose the pellet.
Be very careful not to disturb the pellet, which may be hard to see. It is preferable to leave a little ethanol at the bottom of the tube during each wash step than to accidentally lose the pellet.
26| Air-dry the DNA 5 min at room temperature. Do not allow the air-drying to proceed longer than 5 min.
27| Resuspend the purified DNA with a type and quantity of buffer appropriate for the downstream sequencing application (in the case of Helicos sequencing, 30 μl of 0.1× TE (pH 7.5) is appropriate). Vortex vigorously.
28| Quantify samples before Helicos sequencing; we use the QuantIt OliGreen kit. This kit is recommended because it performs well with single-stranded DNA, and the formamide-eluted DNA will be single-stranded. Create stocks of the QuantIt oligomer at concentrations of 1 μg ml−1 and 500, 200 and 100 ng ml−1. Perform quantification using the QuantIt OliGreen kit according to the manufacturer’s instructions, adapted for a 384-well plate. As a standard, use 10 μl of the standard mix and 10 μl of diluted OliGreen reagent to generate a standard curve of absorbance. Combine 1 μl of the precipitated material, 9 μl of water and 10 μl of the OliGreen reagent. Run all samples and standards in duplicate. When calculating the concentration of the precipitated material, make sure to factor in the tenfold dilution relative to the standards.
29| Carry out adapter ligation and sequencing of GLIB-treated DNA, which can be performed in-house at Helicos.
![]()
![]()
Troubleshooting advice can be found in table 2.
Table 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solutions |
|---|---|---|---|
| Box 2 | Low precipitation efficiency during optimization |
Bad reagents | Use fresh reagents, as directed in the protocol |
| Carryover of sodium periodate into biotinylation step |
Make sure to perform sodium sulfite quench in PROCEDURE Step 12. Also, in PROCEDURE Step 14 be sure to pipette into the middle of the G50 column, liquid dispensed at the edge of the column may not be properly buffer exchanged |
||
| High background precipitation during optimization |
Bad reagents | Use fresh reagents, as directed in the protocol | |
| Incorrect streptavidin beads |
Use only streptavidin beads intended for specific DNA precipitation such as the M280 beads in the protocol. DNA sticks nonspecifically to other beads |
||
| Damaged DNA | DNA with many lesions or abasic sites will undergo nonspecific biotinylation. Pretreat with methoxyamine in PROCEDURE Step 7 |
||
| 29 | Inadequate final yield | Lost DNA pellet | Be sure to include linear polyacrylamide in Step 23 to enhance pellet visibility. Be careful when removing ethanol after spins |
| Insufficient starting DNA |
Obtain at least 20 μg of starting DNA. Also, be sure that DNA concentration was measured correctly initially: improperly dissolved DNA will give erratic spec measurements and should be remeasured after sonication. A high optical density ratio at 260/280 nm (>1.9) is indicative of RNA contamination |
Steps 1–3, shearing DNA: 1–2 h (depending on the number of samples)
Steps 4 and 5, cleanup and verification of DNA fragment size: 2 h
Steps 6–10, glucosylate DNA: 4 h
Steps 11–16, add biotin to DNA: 1 d
Steps 17–28, precipitation and quantification of biotinylated DNA: 8 h
Step 29, adapter ligation and sequencing: 1 week
Box 1, generation of spike-in control DNA: ~6 h
Box 2, optimization of GLIB conditions: 32 h
Box 3, optimization of glucosylation: ~12 h
Box 3. Optimization of glucosylation  ~12 h.
~12 h.
To ensure that BGT efficiently glucosylates hydroxymethylated substrates, we developed a simple method relying on the observation that glucosylated DNA runs more slowly than unmodified DNA2 on an agarose gel, detailed below. This is not the most quantitative method, but it is a quick way to test many glucosylation conditions without resorting to sophisticated technology such as mass spectrometry.
Two other methods that can be used to confirm complete glucosylation were used in the initial development of GLIB (ref. 2). The procedures for those methods are not described at length here. The first is based on the fact that McrBC degrades hydroxymethylated DNA but not glucosylated DNA. Therefore, completely glucosylated DNA cannot be digested by McrBC. Second, glucosylation masks 5hmC and eliminates the signal in an anti-5hmC dot blot. The anticipated results of these verification experiments can be found in Figure 5.
Determination of BGt activity  12 h
12 h
1. Generate a 1,000-bp PCR amplicon using dhmCTP instead of dCTP. Any sequence can be used, although a greater shift is observable for amplicons with high GC content.
2. Glucosylate 1 μg of the PCR product using various amounts of BGT enzyme; incubate for 3 h at 30 °C in 50 mM HEPES (pH 8.0), 25 mM MgCl2 and 50 μM UDP-glucose.
 The glucosylation reaction is performed using T4-BGT from NEB. However, we have found the manufacturer’s suggested reaction conditions (1 h 37 °C NEB buffer 4 with 1× UDP-glucose) to be suboptimal. Roughly eight times the activity can be obtained using our conditions.
The glucosylation reaction is performed using T4-BGT from NEB. However, we have found the manufacturer’s suggested reaction conditions (1 h 37 °C NEB buffer 4 with 1× UDP-glucose) to be suboptimal. Roughly eight times the activity can be obtained using our conditions.
3. To desalt, collect the glucosylated DNA using a Qiagen PCR purification kit according to the manufacturer’s instructions and elute in 10 mM Tris (pH 8.0).
4. Run the product on a 1.5% (wt/vol) agarose gel (90 V, ~1 h). In reactions in which more BGT has been added, the band shifts higher, eventually reaching a maximal shift corresponding to complete or near-complete glucosylation. We define 1 U of BGT as the amount required to completely shift 1 μg of fully hydroxymethylated DNA in a 50-μl volume, and we use 2 U for each microgram of genomic DNA. This is a substantial excess, as genomic DNA will contain a much lower concentration of 5hmC than the PCR amplicon.
ANTICIPATED RESULTS
As much as 75% of DNA is lost during processing steps. We therefore recommend starting with 20 μg of DNA for each GLIB precipitation, half for the + BGT sample and half for the negative control. A 1% recovery is anticipated for the pull-down step in the negative control sample, and thus a yield of 25 ng of DNA is anticipated for 10 μg of negative control sample. For the + BGT sample, the yield will depend on the 5hmC content of the sample DNA, with a minimum of 25 ng. For example, a yield of 100 ng is anticipated for 5hmC-rich mouse ESC DNA.
The hydroxymethylated, but not the cytosine-containing spike-in oligo, should be heavily enriched in the + GLIB sample. Neither oligonucleotide should be enriched in the − BGT sample.
The pattern of enrichment for 5hmC will vary depending on the species and sources of the DNA. Between 10 and 50 million reads will probably be necessary to reach saturation and to define all 5hmC-enriched regions2.
ACKNOWLEDGMENTS
W.A.P. was supported by a predoctoral graduate research fellowship from the National Science Foundation. Y.H. is supported by a postdoctoral fellowship from the Leukemia and Lymphoma Society. This study was supported by US National Institutes of Health (NIH) grants AI44432 and HD065812, grant RM1-01729 from the California Institute for Regenerative Medicine and Translational Research grant 6187-12 from the Leukemia and Lymphoma Society (to A.R.) as well as NIH grant HL089150 and a pilot grant from the Harvard Clinical and Translational Science Center (NIH grant 1 UL1 RR 025758-02) (to S.A.).
Footnotes
AUTHOR CONTRIBUTIONS A.R. and S.A. conceptualized and directed the project. W.A.P. and Y.H. developed the GLIB method. H.R.H. contributed to optimizing the GLIB method.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Huang Y, Pastor WA, Zepeda-Martínez JA, Rao A. The anti-CMS method for genome-wide mapping of 5-hydroxymethylcytosine. Nat. Protoc. 2012;7:1897–1908. doi: 10.1038/nprot.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehman IR, Pratt EA. On the structure of the glucosylated hydroxymethylcytosine nucleotides of coliphages T2, T4, and T6. J. Biol. Chem. 1960;235:3254–3259. [PubMed] [Google Scholar]
- 4.Kornberg SR, Zimmerman SB, Kornberg A. Glucosylation of deoxyribonucleic acid by enzymes from bacteriophage-infected Escherichia coli. J. Biol. Chem. 1961;236:1487–1493. [PubMed] [Google Scholar]
- 5.Bobbitt JM. Periodate oxidation of carbohydrates. Adv. Carbohydr. Chem. 1956;48:1–41. doi: 10.1016/s0096-5332(08)60115-0. [DOI] [PubMed] [Google Scholar]
- 6.Ide H, et al. Synthesis and damage specificity of a novel probe for the detection of abasic sites in DNA. Biochemistry. 1993;32:8276–8283. doi: 10.1021/bi00083a031. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 8.Matarese F, Carrillo-de Santa Pau E, Stunnenberg HG. 5-Hydroxymethylcytosine: a new kid on the epigenetic block? Mol. Syst. Biol. 2011;7:562. doi: 10.1038/msb.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfaffeneder T, et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. Int. Ed. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 10.Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song CX, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]