

Synopsis
The neuromuscular medicine, and physiatry specialists are key health care providers who work cooperatively with a multidisciplinary team to provide coordinated care for persons with Neuromuscular diseases (NMDs). The director or coordinator of the team must be aware of the potential issues specific to NMDs and be able to access the interventions that are the foundations for proper care in NMD. These include health maintenance and proper monitoring of disease progression and complications to provide anticipatory, preventive care and optimum management. Ultimate goals include maximizing health and functional capacities, performing medical monitoring and surveillance to inhibit and prevent complications, and promoting access and full integration into the community in order to optimize quality of life.
This issue of the Physical Medicine and Rehabilitation Clinics of North America is intended to provide the reader with a comprehensive over-view of the diagnostic approach, clinical characteristics, and care and management of patients with neuromuscular disease (NMD), with emphasis on the most common hereditary and acquired neuromuscular disorders. Neuromuscular diseases (NMDs), a classification category that describes hereditary and acquired diseases of the peripheral neuromuscular system, includes those that affect anterior horn cells, peripheral nerves, neuromuscular junctions and muscle.
The estimated total prevalence of the most common neuromuscular diseases (NMDs) in the United States is 500,000 (see Appendix – Prevalence Table).1–7 Combined with all forms of acquired NMDs the prevalence exceeds 4 million which is an impressive figure when compared for example to the prevalence of spinal cord injury, estimated to be between 239,000 to 306,000.8–11 There is tremendous diversity of etiologies for both acquired and hereditary neuromuscular diseases. Some NMDs are acquired with diverse causes distinct from genetic etiologies such as autoimmune, infectious, metabolic, toxic, or paraneoplastic (e.g. amyotrophic lateral sclerosis (ALS), myasthenia gravis, lambert eaton syndrome, botulism, Guillain Barre syndrome, or diabetic peripheral neuropathy). There are over 500 distinct NMDs identified to date which have specific genes that have been linked causally to these conditions.12–14 Limb girdle muscular dystrophy, for example, has over 20 genetically distinct subtypes that have been identified to date. The genetic heterogeneity of hereditary neuromuscular diseases has created challenges for clinicians and researchers.
Although currently incurable, NMDs are not untreatable. The neuromuscular medicine, and physiatry specialists are key health care providers who work cooperatively with a multidisciplinary team to maximize health, maximize functional capacities (including mobility, transfer skills, upper limb function, and self-care skills), inhibit or prevent complications (such as disuses weakness, skeletal deformities, disuses weakness, airway clearance problems, respiratory failure, cardiac insufficiency and dysrhythmias, bone health problems, excessive weight gain or weight loss, metabolic syndrome), and promote access to full integration into the community with optimal quality of life.
The molecular basis of hereditary NMDs has been emerging and coming into sharper focus over the past three decades. Many promising therapeutic strategies have since been developed in animal models. Human trials of these strategies have started, leading to the hope of definitive treatments for many of these currently incurable diseases. Although specific treatments for NMD have not yet reached the clinic, the natural history of these diseases can be changed by the targeting of interventions to known manifestations and complications. Diagnosis can be swiftly reached; the family and patient can be well supported, and individuals who have NMD can reach their full potential in education and employment. In Duchenne muscular dystrophy for example, corticosteroid, respiratory, cardiac, orthopaedic, and rehabilitative interventions have led to improvements in function, quality of life, health, and longevity, with children who are diagnosed today having the possibility of a life expectancy into their fourth decade.15–28
Advocacy organizations report variable and inconsistent health care for individuals with NMD. Although anticipatory and preventive clinical management of NMDs is essential, recommendations exist in only a few areas. Addressing the many complications of NMDs in a comprehensive and consistent way is also crucial for planning multicenter trials, as well as for improving care worldwide. The development and implementation of standardized care recommendations have been emphasized by stakeholders in heterogeneous NMD communities including government agencies, clinicians, scientists, academicians, volunteer health agencies, and advocacy organizations. The purpose of this issue is to provide a framework for recognizing the primary manifestations and possible complications and for planning optimum treatment across different specialties with a coordinated multidisciplinary team.
Reported Needs of Persons with Neuromuscular Diseases
The greatest needs of the population of persons with NMDs were recently evaluated by our The NIDRR-funded Rehabilitation Research and Training Center (RRTC) in NMD at the University of California Davis Medical Center. We performed a comprehensive quality of life survey of over 1,000 individuals with neuromuscular diseases.29, 30 The most frequent problems impacting quality of life in NMD are shown in Table 1. Consumers with NMD reported that the most significant problems impacting their health, function and quality of life were secondary conditions including weakness, fatigue and poor endurance, weight management, sleep disturbances, muscle contractures, and breathing problems. These problems translate into functional issues that consumers with NMD note to be significant, such as difficulty walking, exercising, controlling weight, and doing activities of daily living.
Table 1.
Selected Problems Impacting Health-Related Quality of Life in NMD (n = 1169)
| How much difficulty do you have performing the following functions? | Moderate or Severe | Slight | Not a problem |
|---|---|---|---|
| Difficulty with muscle weakness. | 72.4% | 11.6% | 6.0% |
| Difficulty getting exercise. | 66.1% | 17.5% | 16.3% |
| Difficulty with fatigue. | 70.8% | 21.7% | 7.6% |
| Difficulty controlling weight. | 36.0% | 27.9% | 36.2% |
| Difficulty with sleeping. | 36.2% | 28.6% | 35.1% |
| Difficulty with muscle contractures. | 33.6% | 29.9% | 36.6% |
| How much does your health limit you in the following activities? | A lot | A little | Not at all |
|---|---|---|---|
| Vigorous activities such as running? | 93% | 3% | 4% |
| Walking more than a mile? | 84% | 9% | 7% |
| Walking several blocks? | 73% | 17% | 10% |
| Walking one block? | 54% | 27% | 18% |
The Interdisciplinary NMD team
The comprehensive management of all of the varied clinical problems associated with NMDs is a complex task. For this reason, the interdisciplinary approach is critical. It takes advantage of the expertise of many clinicians, rather than placing the burden on one. This interdisciplinary approach to caring for patients with NMD, and participation by committed providers that have NMD disease-specific expertise are key ingredients to the provision of optimal care (figure 1).31, 32 The patient and family / care providers should actively engage with the medical professional who coordinates clinical care. Depending on the patient’s circumstances, such as area/country of residence or insurance status, this role might be served by, but is not limited to, a neurologist or pediatric neurologist, physiatrist / rehabilitation specialist, neurogeneticist, pediatric orthopedist, pediatrician, or primary-care physician. In the U.S. the coordination of care is often done by the Neuromuscular Medicine specialist (ACGME approved fellowships in Neuromuscular Medicine now exist for subspecialty certification within the American Board of Psychiatry and Neurology and the American Board of Physical Medicine and rehabilitation). The director or coordinator of the team must be aware of the potential issues specific to NMDs and be able to access the interventions that are the foundations for proper care in NMD. These include health maintenance and proper monitoring of disease progression and complications to provide anticipatory, preventive care and optimum management.
Figure 1. Interdisciplinary NMD Clinical Care Coordination (.
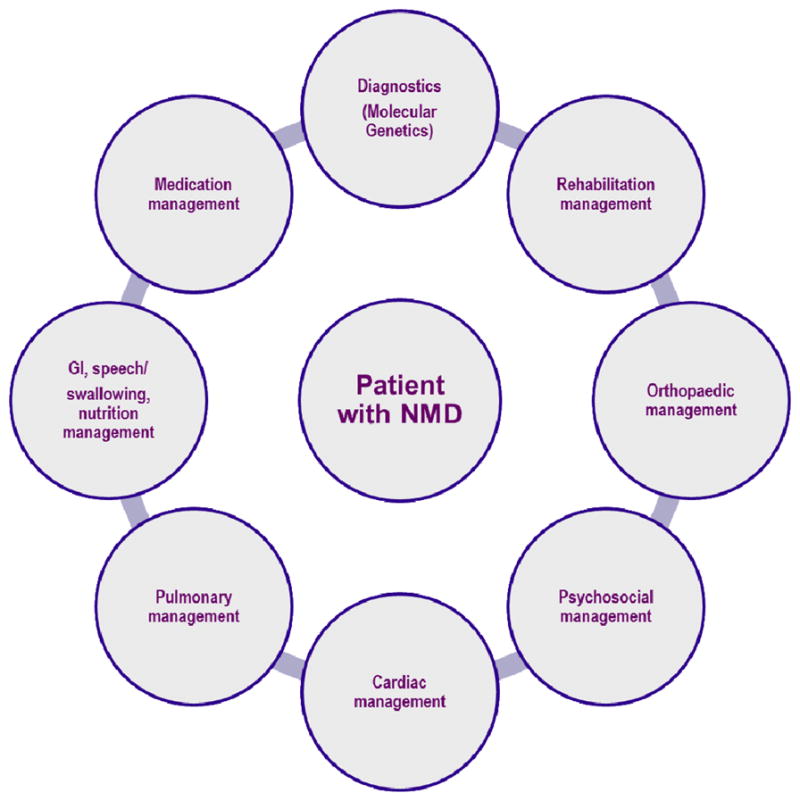
Data from Bushby, K., et al., Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurology, 2010. 9(1): p. 77–93.)
NMD Management is best carried out by a team consisting of physicians; physical, occupational and speech therapists; social workers; vocational counselors; and psychologists, among others. Ideally, owing to the significant mobility problems associated with most NMDs, the neuromuscular medicine specialist, physiatrist and all the key clinical personnel should be available at each visit. Tertiary care medical centers in larger urban areas usually can provide this type of service. This may be an independent clinic or may be sponsored by one or more of the consumer-driven organizations that sponsor research and clinical care for people with NMDs, including the Muscular Dystrophy Association (MDA), the Amyotrophic Lateral Sclerosis (ALS) Association, the Charcot-Marie-Tooth International and Association groups, and the Fascioscapulohumeral Society, among others. Although the physiatrist (Greek for physis for “nature” and iatrikos for “healing”) is well-suited to direct the rehabilitation team and to oversee a comprehensive, goal-oriented treatment plan, physiatrists are co-directors of less than 20% of the MDA clinics in the United States.33,34 Bach has previously described the many major advances physiatrists have contributed to the care of NMD patients and has pointed out the rather significant need for more physiatric involvement in the care of NMD patients.34
Toolkits of Assessments and Interventions for the Interdisciplinary NMD Team
There are varied toolkits of assessments and interventions applicable to NMD management (Table 2).31 Input from different specialties and the specific assessments, and interventions will change as the NMD progresses. Some measures such as strength assessment, functional grading, timed function measures, and pulmonary function measures are core measures performed at least annually for most NMDs. Others are performed regularly depending on the expected impairments for specific NMDs.
Table 2.
Multidisciplinary Domains, Assessments, and Interventions for the NMD Team
| Multidisciplinary Domains | Tools / Assessments | Interventions |
|---|---|---|
| Diagnostics (Neuromuscular medicine specialist, geneticist, pathologist) | Biomarkers (e.g. Creatine Kinase) Electrodiagnostics Molecular Genetics Muscle Biopsy |
Genetic Counseling Family Support |
| Medication Management (Neuromuscular medicine specialist, neurodevelopmental pediatrician, physiatrist) | Clinical Evaluation Strength Function Range of Motion |
Considerations Age of patient Stage of disease Risk factors for side-effects Available medications Choice of regimen Side-effect monitoring and and prophylaxis Dose alteration |
| Rehabilitation Management | ROM Strength Posture Function Alignment Gait |
Stretching Positioning Splinting Orthoses Submaximum exercise / activity Seating Standing devices Adaptive equipment Assistive technology Strollers/scooters Manual / power wheelchairs |
| Orthopedic Management | Assessment of ROM Spinal assessment Spinal radiograph Bone age (left wrist and hand radiograph) Bone densitometry |
Tendon surgery Osteotomies / arthrodesis Posterior spinal fusion |
| GI, speech/ swallowing, nutrition management | Upper and lower GI investigations Anthropometry |
Diet control and supplementation Gastrostomy Pharmacological management of gastric reflux and constipation |
| Pulmonary Management | Spirometry Static Airway pressures (MIP/MEP) Peak cough Flow Pulse oximetry Capnography / End tidal CO2 ABG Sleep Studies / Polysomnography |
Volume recruitment Noninvasive Ventilation (via mask and nasal interfaces) Invasive Ventilation (via tracheostomy) Airway Clearance (Mechanical insufflator/Exsufflator, Theravest) Immunizations |
| Cardiac management | ECG Echo Holter Cardiac MRI |
ACE inhibitors Angiotension Receptor Blockers β blockers Diuretcs Inotropes Other heart failure medication Antiarrhythmics |
| Psychosocial management | Coping Neurocognitive Speech and language Autism Social work |
Psychotherapy Pharmacological interventions Social support Educational support Palliative / Supportive care |
The World Health Organization International Classification of Functioning (ICF) Framework)
Although their degrees and severity can vary, the characteristics or complications of most NMDs include progressive weakness, limb contractures, spine deformity, and decreased pulmonary function; some patients suffer cardiac and intellectual impairment. In determining the severity of impairment and disability, a comprehensive evaluation of patients with NMDs should be done at routine intervals or as clinically indicated. The World Health Organization’s International Classification of Functioning, Disability and Health (ICF)35 provides a useful framework for evaluating the manifestations and complications of NMDs. The bioecological ICF framework35 incorporates domains covering body structure and function, individual activities and participation, and environmental factors that impact the overall physical and mental health of the individual in a societal context. In this framework body structures are anatomical parts of the body such as organs, limbs, and their counterparts and body functions are the physiological functions of body systems (including physiological functions). Impairments are problems in body structure or body function as a significant deviation from normal or loss. Activity is the execution of a task or action by an individual and activity limitations are difficulties an individual may have in executing activities. Participation is involvement in a life situation and participation restrictions are problems an individual may have in involvement in life situations. While the ICF framework covers environmental domains such as external barriers to participation, we typically do not include assessments of environmental domains of the ICF framework because of limited ability of therapeutic agents, surgeries or rehabilitation interventions evaluated in clinical trials to impact these domains.
Patient-reported outcomes (PROs) encompass self-perceived or caregiver-proxy perceived concepts of health and well-being defined broadly including such concepts as health-related quality of life, satisfaction, physical functioning, basic mobility and transfers, sports and physical functioning, mobility and ambulation, upper extremity functioning and ADLs, pain, fatigue, quality of sleep, emotional health, social health, depression, anxiety, stigma, etc.
Selected Assessments for NMD
There are diverse assessments that are performed in NMDs for clinical studies, natural history studies, and clinical trials. For example, table 3 summarizes the diverse nature of clinical endpoints and assessments that have been used clinically and in natural history studies involving persons with Duchenne muscular dystrophy (DMD) organized according to a modified ICF framework. Some core measures such as manual muscle testing for strength, ROM, timed function, pulmonary function, and selected cardiac measures are routinely performed annually or more frequently if clinically indicated.
Table 3.
Clinical Endpoints used in Duchenne Muscular Dystrophy Prospective Natural History Studies and Clinical Trails (ICF Framework35)
| Body Structure / Function | |
|
|
| Activities (Clinical Evaluator-Determined Scales) | |
|
|
| Activities (Functional Tests with Timed Dimension) | |
|
|
| Participation Measures | |
|
|
| Patient-Reported Outcome Measures (PROs) | |
|
|
Important clinical data that should be obtained initially on each patient include: gender, birth date, family history, date of disease (symptom) onset, disease duration, dominant limb, weight and height, cardiovascular and pulmonary symptoms and findings, presence of contractures and spine deformity, muscle strength, ambulatory status (including age at cessation of ambulation and years of wheelchair use), and any treatment interventions. When relevant, these data should be updated at each patient visit. In large clinics data can be recorded on a standardized form and entered into a computer database or electronic health record. Records of respiratory and cardiovascular symptoms and findings should be maintained at each clinic visit. The history should include questions relating to shortness of breath with ambulation, at rest, and during sleep; palpitations; dyspnea; and chest pain. A thorough systems review should be made of cardiopulmonary complications, including pneumonia, prolonged upper respiratory tract infections, respiratory compromise requiring assisted ventilation, and heart failure.
Strength Assessments
Precise measures of strength are important for evaluating clinical progression in NMD as well as assessing the efficacy of any interventions. Strength traditionally has been assessed with manual muscle testing (MMT) using the Medical Research Council (MRC) scale for muscle grading (Table 4), although this is not reliable in muscles that are only mildly affected.36–42 Care must be exercised for consistent inter-examiner measures of the antigravity muscles.
Table 4.
MRC Grade Degree of Strength
| 5. | Normal strength. |
| 5− | Barely detectable weakness. |
| 4S | Same as 4 but stronger than reference muscle. |
| 4 | Muscle is weak but moves the joint against a combination of gravity and some resistance. |
| 4W | Same as 4, but weaker than reference muscle. |
| 3+ | The muscle is capable of transient resistance but collapses abruptly. This degree of weakness is difficult to put into words, but it is a muscle that is able to move the joint against gravity and an additional small amount of resistance. It is not to be used for muscles capable of sustained resistance throughout their whole range of movement. |
| 3 | Muscle cannot move against resistance but moves the joint fully against gravity. With the exception of knee extensors, the joint must be moved through its full mechanical range against gravity. If a patient has contractures that limit movement of the joint, the mechanical range obviously will be to the point at which the contractures cause a significant resistance to the movement. |
| 3− | Muscle moves the joint against gravity but not through the full extent of the mechanical range of the joint. |
| 2 | Muscle moves the joint when gravity is eliminated. |
| 1 | A flicker of movement is seen or felt in the muscle. |
| 0 | No movement. |
Quantitative strength measurements are somewhat more labor intensive, but provide more sensitive and reproducible information.38, 40–42 These include static (isometric) and dynamic (isokinetic) measurements, most often done in selected muscle groups (usually bilateral knee, elbow, shoulder, and neck flexors and extensors) with a force transducer that displays force output through a digital force monitor. Static pinch strength and grip strength also may be measured using a force transducer, and these measurements followed serially at clinic visits. The highest score from three maximal trials usually is recorded. The hand-held dynamometer (HHD) is perhaps the most practical yet reliable way to obtain quantitative strength testing in the clinic. The HHD is a small device equipped with an internal load cell that operates as a force transducer. The HHD is capable of measuring force generated by a subject against the examiner who holds the device firmly against the subject and provides stabilization (counter-resistance). The maximum force is recorded. Although it does not replace formal quantitative strength testing, the HHD has reasonably good reliability in NMD for weaker muscle groups and is a good alternative to MMT.
Dynamic strength may be assessed using an isokinetic dynamometer at a fixed speed (e.g., 30 degrees per second) for both concentric (shortening) and eccentric (lengthening) contractions40. Flexors and extensors should be evaluated through a full range of motion. Parameters that can be measured include peak torque, total work, work per repetition, peak torque to body weight ration, joint angle at peak torque, range of motion, and fatigue index (decrement in work performance over the exercise bout).
Range of Motion Assessments
Passive Joint range of motion (PROM measurements) should be done with a standard goniometer following the protocol used by Brooke and colleagues,4 and Fowler and colleagues.40 Joints to be evaluated for contractures include elbow and wrist extension, hip adduction for iliotibial band tightness, hip and knee extension, and ankle dorsiflexion. The definition of clinically significant contractures varies according to the joint. In some joints, even as little as a 7° flexion contracture can result in the center of gravity (COG) falling to an unstable plane (e.g., anterior to the hip joint and posterior to the knee center of rotation). Care must be taken to measure the ROM of two-joint muscles in position of function (e.g., ankle ROM measured with the knee fully extended).
Active ROM (AROM) assesses the participant’s ability to recruit muscle strength to perform a muscle contraction through an available ROM. Passive ROM (PROM) is first performed by a clinical evaluator to assess the extensibility of muscle, tendons and ligaments passively through a ROM. This is followed by evaluation of AROM. The assessment of ROM must be performed taking into account what PROM is available due to contracture. AROM and PROM are necessary for appropriate biomechanics in activities such as walking and using the upper extremities for functional activities.
Timed Function Tests
Time to rise from the floor (supine to stand)
The time to rise from the floor from a supine position (in seconds) follows the protocol reported previously by CIDD36, UC Davis40, and the Cooperative International Neuromuscular Research Group (CINRG)42. The assessment is performed in all ambulatory study participants who can perform the test. For standing from supine the velocity is calculated as 1 divided by the time to complete the task. Subjects are given 30 to 60 seconds to complete the task. A subject who is unable to complete the task is given a score of 99 and a velocity which approaches zero.
Time to climb four stairs
The time to climb 4 standard stairs follows the protocol as described previously by CIDD36, UC Davis40, and CINRG42. The assessment is performed in children age 2 and older. For the total task of climbing 4 standard stairs, velocity was calculated as 1 divided by the time to complete the task. Subjects are given 30 to 60 seconds to complete the task. A subject who is unable to complete the task are given a score of 99 and a velocity which approaches zero.
Time to walk/run ten meters
The time to walk/run ten meters follows the protocol reported by CIDD36, and UC Davis for 30 feet40, 43, and CINRG for 10 meters42. The assessment is performed in children age 2 and older. Timed function test velocities were calculated as distance divided by completion time. Velocity for the 10 meter walk/run test is determined by dividing distance (10 meters) by the time to complete the task (in seconds). Subjects are given 30 to 60 seconds to complete the task. A subject who is unable to complete the task are given a score of 99 and a velocity which approaches zero. Other Timed function tests. These may include time to stand from a chair, time to propel a manual wheelchair 10 meters or 30 feet, time to put on a T-shirt, and time to cut out a 3″ × 3″ pre-marked square from a piece of paper with safety scissors.
Six-minute Walk Test (6MWT)
We have recently modified the ATS version of the six-minute walt test (6MWT) and validated the test as a clinical endpoint for DMD44–46. Subsequently, there has been widespread utilization of this measure and success of the measure as a primary endpoint in multicenter clinical trials. The modified 6MWT utilizes standard video instructions, a safety chaser to assist the subject up in the event of a fall, and constant rather than intermittent encouragement. Subjects walk around two cones placed 25 meters apart. For the CINRG DMD natural history study, the 6MWT is performed in all participants who can be expected to walk at least 75 meters. A subject who is unable to ambulate 10 meters on a 10 meter walk/run test is given a “0” value for the 6MWT. Using this protocol44, we determined the 6MWT to be reliable and valid in DMD at a single center. The primary variable derived from the 6MWT is the six-minute walk distance (6MWD) in meters. For clinical trials where subjects are tested serially, lower values of 6MWD can be obtained in more marginal ambulators. To account for maturational influences we have described the use of age- and height- based percent predicted values for 6MWT 46. For the DMD subjects we also measure the number of steps taken during the 6MWT with a tally counter or with a stepwatch activity monitor placed on the right ankle. This allows the calculation of average stride length and cadence. The 6MWT takes approximately 15 minutes to complete.
9-hole peg test (9-HPT)
The 9-HPT is commercially available, easy and quick to administer, portable test that is used to measure upper limb function and dexterity.47–49 The 9-HPT records the time to pick up 9 pegs from a container, put into the holes, and then returned to the container. The test has been validated in all age groups and has high inter-rater and test-retest validity. The 9-HPT shows concurrent and convergent validity, and the measure appears sensitive to change in adults with neuromuscular and musculoskeletal disorders 50, 51. Both adult and pediatric norms are available 48, 49. It has been chosen by the NIH Toolbox as a measure of dexterity because it is a very viable tool for longitudinal epidemiologic studies and intervention trials (www.nihtoolbox.org). The primary variable derived from the 9-HPT is completion time in seconds. Total time for administration of the 9-HPT is 10 minutes or less.
Disease-specific Functional Rating Scales
Disease specific functional rating systems exist for many NMDs. For example, the ALS Functional Rating scale (ALSFRS) is commonly obtained in ALS patients52. In DMD, Functional classifications utilize the upper extremity scale reported by Brooke et al 36 and the lower extremity scales used by Vignos et al.53,54.
ALS Functional rating Scale-revised (ALSFRS-R)
The ALSFRS-R assesses patients’ levels of self-sufficiency in areas of feeding, grooming, ambulation, communication, and respiratory52. The assessment determines the degree of impairment in ALS patients’ abilities to function independently in activities of daily living, locomotion, communication, and breathing. It consists of 12 items to evaluate bulbar function, motor function and respiratory function and each item is scored from 0 (unable) to 4 (normal). The ALSFRS has been validated both cross-sectionally and longitudinally against muscle strength testing, the Schwab and England ADL rating scale, the Clinical Global Impression of Change (CGIC) scale, and independent assessments of patient’s functional status. The ALSFRS-R is an attractive primary outcome measure in clinical trials of ALS because it is validated, easy to administer, minimizes dropout, reduces cost, and correlates with survival. Unlike the other standard outcome measures currently employed, the ALSFRS-R is also a measure of global function52.
Vignos Lower Extremity Functional Grade
The following functional grade was originally described by Vignos 53, 54: 1: walks and climbs stairs without assistance; 2: walks and climbs stairs with the aid of a railing; 3: walks and climbs stairs slowly with the aid of a railing (over 12 seconds for 4 standard stairs); 4: walks unassisted and rises from chair but cannot climb stairs; 5: walks unassisted but cannot rise from chair or climb stairs; 6: walks only with the assistance or walks independently with long leg braces; 7: walks in long leg braces but requires assistance for balance; 8: Stands in long leg braces but unable to walk even with assistance; 9: is in a wheelchair; 10: is confined to bed.
Brooke Upper Extremity Functional Grade
The following functional grade was originally described by Brooke et al. 36: 1: Starting with arms at the sides, the patient can abduct the arms in a full circleuntil they touch above the head; 2: Can raise arms above the head only by flexing the elbow; (i.e. shortening the circumference of the movement or using) or using accessory muscles; 3) Cannot raise hands above head but can raise an 8 ounce glass of water to mouth using both hands if necessary; 4: Can raise hands to mouth but cannot raise an 8-ounce glass of water to mouth; 5: Cannot raise hands to mouth but can use hands to hold pen or pick up pennies from the table; 6: Cannot raise hans to mouth and has no useful function of hands. As an optional measure if the patient has a Brooke Grade of 1 or 2 measured by the therapist, it is determined how many Kg of weight can be placed on a shelf above eye level, using one hand.
North Star Ambulatory Assessment (NSAA)
The NSAA is a clinician rated 17-item functional scale designed for ambulant boys with DMD who are able to stand 55–60. This evaluation tool assesses functional activities including standing, getting up from the floor, negotiating steps, hopping, and running. The assessment is based on a 3 point rating scale of 2= ability to perform the test normally, 1= Modified method or assistance to perform test, 0=unable to perform the test. Thus, total score can range from 0 (completely non-ambulant) to 34 - no impairment on these assessments. The North Star Ambulatory Assessment is currently also used in several other countries and international clinical trials61. NSAA has shown good reliability and validity in multi-center studies as well as good clinical validity through Rasch analysis 56, 58.
Motor Function Measure (MFM)
The MFM is a recently developed instrument to assess motor function in ambulant and non-ambulant patients with neuromuscular diseases (NMD) aged 6–62 years 62. The scale is comprised of 32 items, in three dimensions: D1: standing position and transfers (13 items), D2: axial and proximal motor function (12 items), and D3: distal motor function (7 items). Each test is scored by the therapist on a 4-point Likert scale. Scores for subscales and the composite total score range from 0= worse to 100%= better. In heterogeneous NMD patients, internal consistency, intra- and inter-rater reliability for the global scale and the subscales was excellent, and face validity, convergent validity and discriminant validity were good 62. Vuillerot et al. 63 showed that the MFM was able to measure changes in motor function over time in DMD and the total score predicted loss of ambulation. The MFM takes 30–40 minutes to complete.
Egen Klassification (EK2) Scale
The EK2 scale was developed by the Danish Muscular Dystrophy Association as a clinical tool to assess overall functional ability in non-ambulatory patients with DMD 64. This tool includes assessments comprised of functional ability measuring upper extremity grade, muscle strength measured with the manual muscle test, and forced vital capacity defined as a percentage of normal values (FVC%). The construct is based on the interaction of physical components such as muscle strength, range of motion, respiratory status, wheelchair dependence and age. The EK scale assesses ten functional categories (EK 1–10), each on a scale of 0=normal to 3= very impaired, contributing to an overall function score of 0 to 30. The EK scale has shown high inter- and intra-rater reliability (ICC-.98) and good construct validity 65.
Spine deformity Evaluations
For many NMD patients spine deformity should be evaluated at every clinic visit. Data obtained at the time of the first patient visit and thereafter should include the presence or absence of spine deformity, any interventions, and the patient’s age at time of observation. Radiographs should be reviewed and the results for kyphosis and scoliosis recorded along the guidelines recommended by Carman et al 66, Fon et al. 67, and Gupta et al. 68.
Pulmonary Evaluations
Depending on the specific NMD diagnosis, pulmonary evaluations by a pulmonologist may be indicated. Pulmonary function tests (PFTs) include forced vital capacity (FVC), forced expiratory volume in 1 second (FEV1), FEV1/FVC, maximal voluntary ventilation (MVV), residual volume (RV), peak expiratory flow rate (PEFR), and peak cough flow. Measurements are made using a spirometer (e.g. KoKo spirometer and digidoser, nSpire Health, Inc.) and the pulmonary function data may be interpreted using the Crapo et al.69 and Polgar et al.70 normative reference set for 6–7 year old participants or the Hankinson et al.71 normative reference set for ≥8 year old participants. Maximal inspiratory pressure (MIP) and maximal expiratory pressure (MEP) are helpful and are measured near RV and total lung capacity (TLC), respectively, following the technique described by Black and Hyatt 72 using a direct ready dial gauge force meter and ventilated T-tube assembly. Three technically satisfactory measurements should be obtained and the maximum reading recorded. Interpretation of MIP and MEP values can be based on Wilson et al.73 and Domenech-Clar et al.74 normative pediatric reference sets. Participants are evaluated in a seated position with support for the back and feet and they wear nose clips or have their noses held closed by hand during testing. If necessary, cardboard mouthpiece adapters can be used to enable participants to make a full lip seal. PFTs should be done at least yearly and more frequently if clinical indications exist. If clinically indicated, arterial blood gas studies, pulse oximetry, and/or capnography (CO2 monitoring) should be obtained. Formal sleep studies may be indicated depending on the NMD diagnosis and results of regular screening studies such as overnight pulse oximetry.
Cardiac Evaluations
Depending on the specific NMD diagnosis, cardiac evaluations by a cardiologists may be indicated. Regular assessments may include ECG, echocardiography, Holter monitoring, and Cardiac: MR Imaging. Standard 12-lead electrocardiograms (ECGs) should be obtained at 1-year intervals. In some diseases, particularly the myopathies with associated cardiomyopathy, echocardiograms are indicated.
Neuropsychological tests
Neuropsychologic measurements may be helpful in some of these diseases, particularly if there are educational and vocational problems. However, previous reports that used some of the standard measurement tools suggested that subtle physical impairments may have negatively affected the test results. Therefore, caution is advised in interpreting these tests. Tools such as the Category Test, Seashore Rhythm Test, and Speech-Perception Test should be used, if possible, because performance on these instruments is not dependent on motor function75–77.
Patient-Reported Outcomes (PROs)
Consumers, clinical researchers, the Food and Drug Administration, and industry have increasingly recognized the importance of patient-reported outcome (PRO) measures in the determination of clinically meaningful outcomes and validation of clinical and surrogate endpoints for therapeutic trials 78. There are regulatory requirements that registration studies must incorporate primary endpoints that objectively measure clinically meaningful “life-changing” events with significant impact on health and well-being. In addition, the FDA has strongly recommended inclusion of PRO measures such as health-related quality of life (HRQOL) assessments as an endpoints in all clinical trials 79, 80. Both global measures of HRQOL and disease-specific NMD measures have been used in NMD populations.
Ongoing Management / Anticipatory Guidance
Once the diagnosis is confirmed, the patient and family should be thoroughly educated about the expected outcome and what problems may be encountered. The Neuromuscular specialist and /or physiatrist should then assess the patient’s and family goals and develop a medical management and rehabilitative program that matches those goals. Palliative care focuses on living well with optimized quality of life despite life expectancy.
Major advances in the understanding of the molecular basis of many NMDs has greatly enhanced diagnostic accuracy and provides the basis for novel therapeutic interventions. There have also been major pharmacologic advances in the treatment of some NMDs, particularly ALS and DMD. The physiatrist may become involved in the prescription of disease-altering medications for the various NMDs, and therefore should familiarize him/herself with the appropriate pharmacologic agents available. In addition, if not directly involved in research, the physiatrist should nonetheless encourage enrollment in experimental protocols, which not only furthers science but provides some hope for the patient. Education and employment are very important with respect to self-esteem, quality of life, and integration into the community and should be emphasized in people with slowly progressive NMD. Patients should be referred to a support group. Support groups often are a great resource, not only for psychologic support but for problem-solving and recycling of equipment.
Given the many advances that have occurred in the management of people with NMD, many patients will survive through their childbearing years, possibly having children, and can expect to enjoy a good quality of life. The physiatrist can play a critical role during important life transitions and provide care which can maximize function and quality of life.
Summary
Although currently incurable, NMDs are not untreatable. The neuromuscular medicine, and physiatry specialists are key health care providers who work cooperatively with a multidisciplinary team to maximize health and functional capacities, inhibit or prevent complications, and provide access to resources which promote full integration into the community to optimize quality of life. Addressing the many complications of NMDs in a comprehensive and consistent way is also crucial for planning multicenter trials, as well as for improving care worldwide. The development and implementation of standardized care recommendations have been emphasized by government agencies, clinicians, scientists, academicians, volunteer health agencies, and advocacy organizations. The Neuromuscular specialists and physiatrists are well suited to working cooperatively to provide this type of multidisciplinary care and they can play a significant role in improving care for patients.
Key Points.
Both neuromuscular medicine specialists and physiatrists are key health care providers who work cooperatively with a multidisciplinary team to provide coordinated care for persons with Neuromuscular diseases (NMDs).
Ongoing input from different specialties is important as the specific assessments, and interventions which are necessary will change as the NMD progresses.
Addressing the many complications of NMDs in a comprehensive and consistent way is also crucial for planning and determining eligibility for clinical trials, as well as for improving care worldwide.
The development and implementation of standardized care recommendations for NMDs will lead to improved health, function, participation, and quality of life.
Acknowledgments
This work was supported by Grant# H133B0900001 from the National Institute of Disability and Rehabilitation Research. The authors take full responsibility for the contents of this paper, which do not represent the views of the National Institute of Disability and Rehabilitation Research or the United States Government.
Footnotes
Disclosures. The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Craig M. McDonald, Department of Physical Medicine and Rehabilitation, Professor of Pediatrics, University of California Davis School of Medicine, Sacramento, CA.
William M. Fowler, Jr, Department of Physical Medicine and Rehabilitation, University of California Davis School of Medicine, Sacramento, CA.
References
- 1.Emery AEH. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromusc Disord. 1991;1(1):19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 2.Fedele D, et al. A multicenter study on the prevelance of diabetic neuropathy in Italy. Diabetes Care. 1997;20(5):836–843. doi: 10.2337/diacare.20.5.836. [DOI] [PubMed] [Google Scholar]
- 3.De Domenico, et al. Amyotrophic Lateral Sclerosis: An epidemiological study in the Provine of Messina, Italy, 1976–1985. Neuroepidem. 1988;7:152–158. doi: 10.1159/000110149. [DOI] [PubMed] [Google Scholar]
- 4.Somnier FE. Myasthenia gravis. Danish Medical Bulletin. 1996;43(1):1–10. [PubMed] [Google Scholar]
- 5.Hughes MI, et al. The prevalence of inherited neuromuscular disease in Northern Ireland. Neuromusc Disord. 1996;6(1):69–73. doi: 10.1016/0960-8966(94)00017-4. [DOI] [PubMed] [Google Scholar]
- 6.Ahlstrom G, et al. Epidemiology of neuromuscular disease, including the postpolio sequelae in a Swedish Country. Neuroepidem. 1993;12(5):262–9. doi: 10.1159/000110327. [DOI] [PubMed] [Google Scholar]
- 7.Shoenberg BS. Epidemiologic approaches to peripheral neuropathy. In: Dyck PJ, editor. Peripheral Neuropathy. W.B. Saunders; Philadelphia: 1993. [Google Scholar]
- 8.DeVivo MJ, et al. Prevalence of spinal cord injury: a re-estimation employing life table techniques. Arch Neurol. 1980;37:707–8. doi: 10.1001/archneur.1980.00500600055011. [DOI] [PubMed] [Google Scholar]
- 9.Harvey C, Rothschild BB, Asmann AJ, Stripling T. New estimates of traumatic SCI prevalence: a survey-based approach. Paraplegia. 1990;28:537–44. doi: 10.1038/sc.1990.73. [DOI] [PubMed] [Google Scholar]
- 10.Lasfarques JE, et al. A Model for estimating spinal cord injury prevalence in the United States. Paraplegia. 1995;33:62–68. doi: 10.1038/sc.1995.16. [DOI] [PubMed] [Google Scholar]
- 11.Meal LaPlante. Disability in the United States: Prevalence and Causes. Washington, DC: U.S. Dept. of Education and the National Institute on Disability and Rehabilitation Research; 1992. [Google Scholar]
- 12.Kaplan JC. The 2009 version of the gene table of neuromuscular disorders. Neuromusc Disord. 2009;19:77–98. doi: 10.1016/j.nmd.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Mitochondrial encephalopathies: gene mutation. Neuromuscular Disorders. 2004;14:107–116. doi: 10.1016/s0960-8966(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 14.http://www.musclegenetable.org
- 15.Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008;1:CD003725. doi: 10.1002/14651858.CD003725.pub3. [DOI] [PubMed] [Google Scholar]
- 16.Moxley RT, 3rd, Ashwal S, Pandya S, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005;64:13–20. doi: 10.1212/01.WNL.0000148485.00049.B7. [DOI] [PubMed] [Google Scholar]
- 17.Jeppesen J, Green A, Steffensen BF, Rahbek J. The Duchenne muscular dystrophy population in Denmark, 1977–2001: prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul Disord. 2003;13:804–12. doi: 10.1016/s0960-8966(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 18.Yasuma F, Konagaya M, Sakai M, Kuru S, Kawamura T. A new lease on life for patients with Duchenne muscular dystrophy in Japan. Am J Med. 2004;117:363. doi: 10.1016/j.amjmed.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 19.Eagle M, Bourke J, Bullock R, et al. Managing Duchenne muscular dystrophy—the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord. 2007;17:470–75. doi: 10.1016/j.nmd.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 20.Eagle M. Report on the muscular dystrophy campaign workshop: exercise in neuromuscular diseases Newcastle, January 2002. Neuromuscul Disord. 2002;12:975–83. doi: 10.1016/s0960-8966(02)00136-0. [DOI] [PubMed] [Google Scholar]
- 21.Markham LW, Kinnett K, Wong BL, Woodrow Benson D, Cripe LH. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul Disord. 2008;18:365–70. doi: 10.1016/j.nmd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 22.American Academy of Pediatrics Section on Cardiology and Cardiac Surgery. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005;116:1569–73. doi: 10.1542/peds.2005-2448. [DOI] [PubMed] [Google Scholar]
- 23.Bushby K, Muntoni F, Bourke JP. 107th ENMC International Workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy; 7th–9th June 2002; Naarden, the Netherlands Neuromuscul Disord. 2003. pp. 166–72. [DOI] [PubMed] [Google Scholar]
- 24.Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Bécane HM. Eff ect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855–57. doi: 10.1016/j.jacc.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 25.Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley RT CIDD Group. Duchenne muscular dystrophy: patterns of clinical progression and effects of supportive therapy. Neurology. 1989;39:475–81. doi: 10.1212/wnl.39.4.475. [DOI] [PubMed] [Google Scholar]
- 26.Vignos PJ, Wagner MB, Karlinchak B, Katirji B. Evaluation of a program for long-term treatment of Duchenne muscular dystrophy. Experience at the University Hospitals of Cleveland. J Bone Joint Surg Am. 1996;78:1844–52. doi: 10.2106/00004623-199612000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Rahbek J, Werge B, Madsen A, Marquardt J, Steffensen BF, Jeppesen J. Adult life with Duchenne muscular dystrophy: observations among an emerging and unforeseen patient population. Pediatr Rehabil. 2005;8:17–28. doi: 10.1080/13638490400010191. [DOI] [PubMed] [Google Scholar]
- 28.Tatara K, Shinno S. Management of mechanical ventilation and prognosis in duchenne muscular dystrophy. IRYO Jap J Natl Med Serv. 2008;62:566–71. [Google Scholar]
- 29.Abresch RT, Seyden NK, Wineinger MA. Quality of life. Issues for persons with neuromuscular diseases. Phys Med Rehabil Clin N Am. 1998;9:233–248. [PubMed] [Google Scholar]
- 30.McDonald CM. Physical activity, health impair-ments, and disability in neuromuscular disease. Am J Phys Med Rehabil. 2002;81:S108–S120. doi: 10.1097/00002060-200211001-00012. [DOI] [PubMed] [Google Scholar]
- 31.Bushby K, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurology. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 32.Bushby K, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurology. 2010;9(2):177–89. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 33.Results of Muscular Dystrophy Association Clinic Directors Survey, presented at the 2012 MDA Clinical Conference; Las Vegas NV. March 6, 2012. [Google Scholar]
- 34.Bach JR. The historical role of the physiatrist in the management of Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1996;75(3):239–241. doi: 10.1097/00002060-199605000-00020. [DOI] [PubMed] [Google Scholar]
- 35.Agenda item 13.9. Resolution of the World Health Assembly: International classification of function, disability and health. Fifty-fourth World Health Assembly; World Health Organization. 2001. [Google Scholar]
- 36.Brooke MH, Griggs RC, Mendell JR, Fenichel GM, Shumate JB, Pellegrino RJ. Clinical trial in Duchenne dystrophy. I. The design of the protocol. Muscle Nerve. 1981;4(3):186–197. doi: 10.1002/mus.880040304. [DOI] [PubMed] [Google Scholar]
- 37.Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP, Province MA. Clinical investigation in Duchenne dystrophy: 2. Determination of the “power” of therapeutic trials based on the natural history. Muscle Nerve. 1983;6(2):91–103. doi: 10.1002/mus.880060204. [DOI] [PubMed] [Google Scholar]
- 38.Aitkens SG, Lord J, Bernauer E, et al. Relationship of manual muscle testing to objective strength measurements. Muscle Nerve. 1989;12:173–177. doi: 10.1002/mus.880120302. [DOI] [PubMed] [Google Scholar]
- 39.Kilmer DD, Abresch RT, Fowler WM., Jr Serial manual muscle testing in Duchenne muscular dystrophy. Arch Phys Med Rehabil. 1993;74:1168–1171. [PubMed] [Google Scholar]
- 40.Fowler WM, Jr, Abresch RT, Aitkens S, Carter GT, Johnson ER, Kilmer DD, McCrory MA, Wright NC. Profiles of neuromuscular diseases. Design of the protocol. Am J Phys Med Rehabil. 1995;74(5 Suppl):S62–69. doi: 10.1097/00002060-199509001-00002. [DOI] [PubMed] [Google Scholar]
- 41.Escolar DM, Henricson EK, Mayhew J, Florence J, Leshner R, Patel KM, Clemens PR. Clinical evaluator reliability for quantitative and manual muscle testing measures of strength in children. Muscle Nerve. 2001;24(6):787–793. doi: 10.1002/mus.1070. [DOI] [PubMed] [Google Scholar]
- 42.Mayhew JE, Florence JM, Mayhew TP, Henricson EK, Leshner RT, McCarter RJ, Escolar DM. Reliable surrogate outcome measures in multicenter clinical trials of Duchenne muscular dystrophy. Muscle Nerve. 2007;35(1):36–42. 11. doi: 10.1002/mus.20654. [DOI] [PubMed] [Google Scholar]
- 43.McDonald CM, Abresch RT, Carter GT, Fowler WM, Jr, Johnson ER, Kilmer DD, Sigford BJ. Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1995;74(5 Suppl):S70–92. doi: 10.1097/00002060-199509001-00003. [DOI] [PubMed] [Google Scholar]
- 44.McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL, Atkinson L, Reha A, Hirawat S, Miller LL. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. 2009;41(4):500–510. doi: 10.1002/mus.21544. [DOI] [PubMed] [Google Scholar]
- 45.McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Atkinson L, Elfring GL, Reha A, Miller LL. The 6-minute walk test in Duchenne/Becker muscular dystrophy: Longitudinal Observations. Muscle Nerve. 2010;42(6):966–974. doi: 10.1002/mus.21808. [DOI] [PubMed] [Google Scholar]
- 46.Henricson E, Abresch R, Han JJ, Nicorici A, Goude Keller E, Elfring G, Reha A, Barth J, McDonald CM. Percent-Predicted 6-Minute Walk Distance in Duchenne Muscular Dystrophy to Account for Maturational Influences. PLoS Curr. 2012 Jan 25; doi: 10.1371/currents.RRN1297. [revised 2012 Feb 2];4:RRN1297. [DOI] [PMC free article] [PubMed] [Google Scholar]; Backman CL, CS, Gibson D, Parsons J. Assessment of hand function: the relationship between pegboard dexterity and applied dexterity. Can J Occup Ther. 59:208–213. [Google Scholar]
- 47.Smith YA, Hong E, Presson C. Normative and validation studies of the Nine-hole Peg Test with children. Percept Mot Skills. 2000;90(3 Pt 1):823–43. doi: 10.2466/pms.2000.90.3.823. [DOI] [PubMed] [Google Scholar]
- 48.Michimata A, et al. The manual function test: norms for 20- to 90-year-olds and effects of age, gender, and hand dominance on dexterity. Tohoku J Exp Med. 2008;214(3):257–67. doi: 10.1620/tjem.214.257. [DOI] [PubMed] [Google Scholar]
- 49.Poole JL, et al. Measuring dexterity in children using the Nine-hole Peg Test. J Hand Ther. 2005;18(3):348–51. doi: 10.1197/j.jht.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Svensson E, Hager-Ross C. Hand function in Charcot Marie Tooth: test retest reliability of some measurements. Clin Rehabil. 2006;20(10):896–908. doi: 10.1177/0269215506072184. [DOI] [PubMed] [Google Scholar]
- 51.Eklund E, Svensson E, Hager-Ross C. Hand function and disability of the arm, shoulder and hand in Charcot-Marie-Tooth disease. Disabil Rehabil. 2009;31(23):1955–62. doi: 10.1080/09638280902874170. [DOI] [PubMed] [Google Scholar]
- 52.Gordon PH, Miller RG, Moore DH. ALSFRS-R. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004 Sep;5(Suppl 1):90–3. doi: 10.1080/17434470410019906. Review. [DOI] [PubMed] [Google Scholar]
- 53.Vignos PJ, Jr, Spencer GE, Jr, Archibald KC. Management of progressive muscular dystrophy of childhood. JAMA. 1963;184:89–96. doi: 10.1001/jama.1963.03700150043007. [DOI] [PubMed] [Google Scholar]
- 54.Vignos PJ. Management of musculoskeletal complications in neuromuscular disease: Limb contractures and the role of stretching, braces and surgery. In: Fowler WM, editor. Advances in the Rehabilitation of Neuromuscular Diseases. Philadelphia: Hanley & Belfus; 1988. pp. 509–536. [Google Scholar]
- 55.Scott E, Mawson SJ. Measurement in Duchenne muscular dystrophy: considerations in the development of a neuromuscular assessment tool. Dev Med Child Neurol. 2006 Jun;48(6):540–4. doi: 10.1017/S0012162206001137. Review. [DOI] [PubMed] [Google Scholar]
- 56.Mazzone ES, Messina S, Vasco G, Main M, Eagle M, D’Amico A, Doglio L, Politano L, Cavallaro F, Frosini S, Bello L, Magri F, Corlatti A, Zucchini E, Brancalion B, Rossi F, Ferretti M, Motta MG, Cecio MR, Berardinelli A, Alfieri P, Mongini T, Pini A, Astrea G, Battini R, Comi G, Pegoraro E, Morandi L, Pane M, Angelini C, Bruno C, Villanova M, Vita G, Donati MA, Bertini E, Mercuri E. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscul Disord. 2009 Jul;19(7):458–61. doi: 10.1016/j.nmd.2009.06.368. Epub 2009 Jun 23. [DOI] [PubMed] [Google Scholar]
- 57.Mazzone E, Martinelli D, Berardinelli A, Messina S, D’Amico A, Vasco G, Main M, Doglio L, Politano L, Cavallaro F, Frosini S, Bello L, Carlesi A, Bonetti AM, Zucchini E, De Sanctis R, Scutifero M, Bianco F, Rossi F, Motta MC, Sacco A, Donati MA, Mongini T, Pini A, Battini R, Pegoraro E, Pane M, Pasquini E, Bruno C, Vita G, de Waure C, Bertini E, Mercuri E. North Star Ambulatory Assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2010;20(11):712–716. doi: 10.1016/j.nmd.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 58.Mayhew A, Cano S, Scott E, Eagle M, Bushby K, Muntoni F. Moving towards meaningful measurement: Rasch analysis of the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Dev Med Child Neurol. 2011;53(6):535–542. doi: 10.1111/j.1469-8749.2011.03939.x. [DOI] [PubMed] [Google Scholar]
- 59.Mazzone E, Vasco G, Sormani MP, Torrente Y, Berardinelli A, Messina S, D’Amico A, Doglio L, Politano L, Cavallaro F, Frosini S, Bello L, Bonfiglio S, Zucchini E, De Sanctis R, Scutifero M, Bianco F, Rossi F, Motta MC, Sacco A, Donati MA, Mongini T, Pini A, Battini R, Pegoraro E, Pane M, Gasperini S, Previtali S, Napolitano S, Martinelli D, Bruno C, Vita G, Comi G, Bertini E, Mercuri E. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology. 2011 Jul 19;77(3):250–6. doi: 10.1212/WNL.0b013e318225ab2e. Epub 2011 Jul 6. [DOI] [PubMed] [Google Scholar]
- 60.Scott E, Eagle M, Mayhew A, Freeman J, Main M, Sheehan J, Manzur A, Muntoni F. The North Star Clinical Network for Paediatric Neuromuscular Disease. Development of a Functional Assessment Scale for Ambulatory Boys with Duchenne Muscular Dystrophy. Physiother Res Int. 2011 Sep 23; doi: 10.1002/pri.520. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 61.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, Abbs S, Garralda ME, Bourke J, Wells DJ, Dickson G, Wood MJ, Wilton SD, Straub V, Kole R, Shrewsbury SB, Sewry C, Morgan JE, Bushby K, Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011 Aug 13;378(9791):595–605. doi: 10.1016/S0140-6736(11)60756-3. Epub 2011 Jul 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berard C, et al. A motor function measure for neuromuscular diseases. Construction and validation study. Neuromuscul Disord. 2005;15(7):463–70. doi: 10.1016/j.nmd.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 63.Vuillerot C, et al. Monitoring changes and predicting loss of ambulation in Duchenne muscular dystrophy with the Motor Function Measure. Dev Med Child Neurol. 2010;52(1):60–5. doi: 10.1111/j.1469-8749.2009.03316.x. [DOI] [PubMed] [Google Scholar]
- 64.Steffensen B, Hyde S, Lyager S, Mattsson E. Validity of the EK scale: a functional assessment of non-ambulatory individuals with Duchenne muscular dystrophy or spinal muscular atrophy. Physiother Res Int. 2001;6(3):119–34. doi: 10.1002/pri.221. [DOI] [PubMed] [Google Scholar]
- 65.Brunherotti MA, Sobreira C, Rodrigues-Júnior AL, de Assis MR, Terra Filho J, Baddini Martinez JA. Correlations of Egen Klassifikation and Barthel Index scores with pulmonary function parameters in Duchenne muscular dystrophy. Heart Lung. 2007 Mar-Apr;36(2):132–9. doi: 10.1016/j.hrtlng.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 66.Carman DL, Browne RH, Birch JG. Measurement of scoliosis and kyphosis radiographs. J Bone Joint Surg. 1990;72:328–333. [PubMed] [Google Scholar]
- 67.Fon GT, Pitt MJ, Thies AC., Jr Thoracic kyphosis: Range in normal subjects. Am J Roentgenol AJR. 1980;134(5):979–983. doi: 10.2214/ajr.134.5.979. [DOI] [PubMed] [Google Scholar]
- 68.Gupta MC, Wijesekera S, Sossan A, Martin L, Vogel LC, Boakes JC, Lerman JA, McDonald CM, Betz RR. Reliability of radiographic parameters in neuromuscular scoliosis. Spine. 2007;32(6):691–5. doi: 10.1097/01.brs.0000257524.23074.ed. [DOI] [PubMed] [Google Scholar]
- 69.Crapo RO, Morris AH, Gardner RM. Reference spirometric values using techniques and equipment that meet ATS recommendations. Am Rev Respir Dis. 1981;123(6):659–664. doi: 10.1164/arrd.1981.123.6.659. [DOI] [PubMed] [Google Scholar]
- 70.Polgar G. Pulmonary function tests in children. J Pediatr. 1979;95(1):168–170. [PubMed] [Google Scholar]
- 71.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159(1):179–187. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 72.Black LF, Hyatt RE. Maximal respiratory pressures: Normal values and relationship to age and sex. Am Rev Respir Dis. 1969;99:696–702. doi: 10.1164/arrd.1969.99.5.696. [DOI] [PubMed] [Google Scholar]
- 73.Wilson SH, Cooke NT, Edwards RH, Spiro SG. Predicted normal values for maximal respiratory pressures in caucasian adults and children. Thorax. 1984;39(7):535–538. doi: 10.1136/thx.39.7.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Domenech-Clar R, Lopez-Andreu JA, Compte-Torrero L, De Diego-Damia A, Macian-Gisbert V, Perpina-Tordera M, Roques-Serradilla JM. Maximal static respiratory pressures in children and adolescents. Pediatr Pulmonol. 2003;35(2):126–132. doi: 10.1002/ppul.10217. [DOI] [PubMed] [Google Scholar]
- 75.Fowler WM, Abresch RT, Aitkens SA, et al. Impairment and disability profiles of neuromuscular diseases: Design of the protocol. Am J Phys Med Rehabil. 1995;74(2):S62–69. doi: 10.1097/00002060-199509001-00002. [DOI] [PubMed] [Google Scholar]
- 76.Fowler WM. Rehabilitation management of muscular dystrophy and related disorders. II. Comprehensive care. Arch Phys Med Rehabil. 1982;63(7):322–328. [PubMed] [Google Scholar]
- 77.Fowler WM, Goodgold J. Rehabilitation management of neuromuscular diseases. In: Goodgold J, editor. Rehabilitation Medicine. St. Louis: CV Mosby; 1988. pp. 298–316. [Google Scholar]
- 78.Mendell JR, et al. Challenges in drug development for muscle disease: a stakeholders’ meeting. Muscle Nerve. 2007;35(1):8–16. doi: 10.1002/mus.20686. [DOI] [PubMed] [Google Scholar]
- 79.FDA; F.a.D.A. Center for Drug Evaluation and Research. Guidance for Industry: Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims. Federal Register; Rockville, MD: 2009. pp. 85132–65133. [Google Scholar]
- 80.Acquadro C, BR, Dubois D, et al. Incorporating the patient’s perspective into drug development and communication: an ad hoc task force report of the Patient-Reported Outcomes (PRO) Harmonization Group meeting at the Food and Drug Administration. Value Health. 2003;6:522–531. doi: 10.1046/j.1524-4733.2003.65309.x. [DOI] [PubMed] [Google Scholar]