

Abstract
The recently discovered enzyme lysine-specific demethylase 1 (LSD1) plays an important role in the epigenetic control of gene expression, and aberrant gene silencing secondary to LSD1 dysregulation is thought to contribute to the development of cancer. We reported that (bis)guanidines, (bis)biguanides and their urea- and thiourea isosteres are potent inhibitors of LSD1, and induce the re-expression of aberrantly silenced tumor suppressor genes in tumor cells in vitro. We now report a series of small molecule amidoximes that are moderate inhibitors of recombinant LSD1, but that produce dramatic changes in methylation at the histone 3 lysine 4 (H3K4) chromatin mark, a specific target of LSD1, in Calu-6 lung carcinoma cells. In addition, these analogues increase cellular levels of secreted frizzle-related protein (SFRP) 2, H-cadherin (HCAD) and transcription factor GATA4. These compounds represent leads for an important new series of drug-like epigenetic modulators with the potential for use as antitumor agents.
Keywords: Epigenetics, lysine-specific demethylase 1, histones, histone demethylase, amidoxime, gene expression, enzyme inhibitor, secreted frizzle-related protein, H-cadherin, GATA4, Calu-6 human anaplastic non-small cell lung carcinoma
Introduction
Chromatin architecture is a key determinant in the regulation of gene expression, and is strongly influenced by post–translational modification of histones.1, 2 Histones, which are the chief protein component of chromatin, are complex macromolecules comprised of core protein subunits termed H2a, H2b, H3, and H4. Histone tails, consisting of up to 40 amino acid residues, protrude through the DNA strand, and lysines, arginines, serines, threonines and tyrosines on this tail act as sites for post-translational modification. These modifications, including methylation, ubiquitination, sumoylation, ADP-ribosylation and acetylation of histone lysine residues, and phosphorylation of histone serine, threonine and tyrosine,3 allow alteration of higher order nucleosome structure, and thus contribute to structural changes in chromatin.4, 5 In cancer, DNA promoter CpG island hypermethylation in combination with other chromatin modifications, including decreased activating marks and increased repressive marks on histone proteins 3 and 4, have been associated with the silencing of tumor suppressor genes.6, 7
Histone lysine methylation/demethylation represents a critical epigenetic modification cycle,5, 8, 9 and dysregulation of this cycle can lead to aberrant silencing of tumor suppressor genes. Histone lysine methylation has been shown to be a dynamic process regulated by the addition of methyl groups by a number of specific histone methyltransferases, and removal of methyl groups from mono-, di, and trimethyllysines by lysine specific demethylase 1 (LSD1)8 and by specific Jumonji C (JmjC) domain-containing demethylases.8–12 To date, 17 lysine residues and 7 arginine residues on histone proteins have been shown to undergo methylation,12–16 and lysine methylation on histones can signal transcriptional activation or repression, depending on the specific lysine residue involved.17–19
A key transcription-activating chromatin mark found associated with promoters of active genes is histone 3 dimethyllysine 4 (H3K4).20, 21 The flavin adenine dinucleotide (FAD)-dependent amine oxidase LSD1, also known as BHC110 and KDM1,8, 22 catalyzes the oxidative demethylation of histone 3 methyllysine 4 (H3K4me1) and histone 3 dimethyllysine 4 (H3K4me2), leading to transcriptional repression.8 Aberrant demethylation of H3K4me1 and H3K4me2 is known to prevent expression of tumor suppressor genes important in human cancer.23 Thus, LSD1 and other histone demethylases have emerged as important new targets for the development of specific inhibitors as a new class of antitumor drugs.24
To date, only a few small molecule inhibitors of LSD1 have been identified. The classical monoamine oxidase (MAO) inhibitors phenelzine and tranylcypromine inhibit the recombinant LSD1/CoRest complex, and increase global levels of H3K4me2.22, 25 Other recently described synthetic demethylase inhibitors include peptidic substrate mimics featuring modified lysine residues26 and trans-2-arylcyclopropylamine analogues.27–29 All of the above are based either on the peptide substrate, making them unlikely candidates for drug development, or on a tranylcypromine scaffold. Among the most effective LSD1 inhibitors is the tranylcypromine-based inhibitor (1S, 2R)-NCL-1 (IC50 1.6 μM),30, 31 and the (bis)-3,3-(diphenyl)propylbiguanide known as 2d.32, 33 A comprehensive review of inhibitors of lysine demethylases has recently been published.34
We previously reported the synthesis of a novel series of (bis)guanidines and (bis)biguanides35 that act as potent inhibitors of LSD1.23 The two most potent LSD1 inhibitors, 1c and 2d (Figure 1) produced significant global increases in both H3K4me1 and H3K4me2 in HCT116 colon tumor cells in vitro, and promoted the re-expression of multiple, aberrantly silenced genes important in the development of colon cancer, including members of the secreted frizzle-related proteins (SFRPs) and the GATA family of transcription factors. Compound 2d has also been shown to produce a synergistic effect on tumor growth when combined with 5-azacytidine in nude mice bearing HCT116 human colon carcinoma xenografts.33 These studies confirm that the use of histone demethylase inhibitors in combination with a DNA methyltransferase inhibitor represents a highly promising and novel approach to cancer chemotherapy.
Figure 1.

Structures of previously discovered LSD1 inhibitors 1c and 2d, the monoamine oxidase inhibitors tranylcypromine 3 and pargyline 4, and virtual screening hits 5–7.
Because of the promising antitumor effects of 1c and 2d, we sought to find additional lead compounds for use as epigenetic modulators. Other groups have already pursued novel analogues related to the known monoamine oxidase inhibitors tranylcypromine 3 and pargyline 4 (Figure 1), however, we were interested in finding previously unknown scaffolds for use in LDS1 inhibitor design. Thus a virtual screen strategy was undertaken to examine the Maybridge Hitfinder 5 compound library, with the goal of identifying potential lead compounds. The crystal structure of LSD1 (Protein Database #2V1D) was prepared for the virtual screen using PrepWizard, and SiteMap was then used to assess the “druggability” of the LSD1 histone-binding pocket, and to map the active site. The active site features a hydrophilic pocket near the FAD cofactor, which appears to be the binding area for H3K4me2. Notably, the active site also contains a large hydrophobic pocket that could serve as a target for the structure-based design of inhibitor molecules. For the virtual screen, compounds in the Maybridge database were converted from 2D to 3D, and the lowest energy conformers were determined. These conformers were then docked in the LSD1 active site using Glide. A total of 10 hits were identified, each of which had a GlideScore lower than −7.5 kcal/mol. Each of the identified molecules contained a hydrophobic moiety that appeared to be situated near the FAD cofactor, and a hydrophobic substituent that was bound in the hydrophobic pocket (data not shown). We examined these hits, and selected three that seemed amenable to structural optimization, compounds 5, and 6 and 7, for our initial studies (Figure 1).
Chemistry
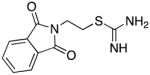
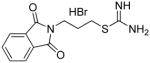
Compounds 5–7 and analogues 8–33 could be readily synthesized using the routes described in Schemes 1–3. As was mentioned above, compounds 5–7 were among the virtual hits identified from the Maybridge Hitfinder 5 database, as was compound 12, while the remaining analogues were largely unreported. The synthesis of carbamimidothioates 5, 8 and 9 could be completed in a single step, as shown in Scheme 1. The appropriate bromoalkylphthalimide 34a, b or c was treated with thiourea 3536 to afford the desired products 5, 8 and 9, respectively, in 62–79% yield. In each case, the product precipitated from the reaction mixture as a white amorphous solid.
Scheme 1.

Scheme 3.

Compounds 6, 10 and 11 were also accessible in a single step using an adaptation of a previously published procedure,37 as shown in Scheme 2. Treatment of 1,3-dinitro-2-chloro-5-trifluoromethylbenzene 35 with the appropriate diamine 36a–c in the presence of triethylamine at 10°C afforded the desired target molecules 6, 10 and 11 in 68 to 87% yield.
Scheme 2.

The remaining target molecules 7 and 12–33 were synthesized as outlined in Scheme 3. Thus substituted anilines 37–59 (see Table 1 for identities of R1, R2 and R3) were treated with cyanoacetic acid in the presence of phosphorus pentachloride in dichloromethane,38 resulting in the formation of the corresponding amides 60–82. Yields for this transformation were largely between 85–95%, although in a few cases they were as low as 52%. These amides in turn were reacted with hydroxylamine and sodium bicarbonate in dichloromethane39 to yield the desired amidoximes 7 and 12–33. In this step, yields varied widely between 23 and 78%.
Table 1.
Inhibition of purified recombinant LSD1 following treatment with 10 μM 3–33, and fold increase in global H3K4me2 levels in Calu-6 human lung adenocarcinoma cells following treatment with carbamimidothioates 5, 8 and 9, aromatic amines 6, 10 and 11 and amidoximes 7 and 12–33 at 10 μM. Each data point is the average of three determinations that in each case differed by less than 5%. ND = not determined.
| Structure | Compound | % inhibition of LSD1 at 10 μM | H3K4me2 Fold Increase (48 h) |
|---|---|---|---|
| 2d | 2d | 82.9 | 5.1 |
 tranylcypromine |
3 | 18.4 | ND |
 pargyline |
4 | 68.8 | 1.29 |
|
5 | 24.0 | 1.94 |
|
8 | 31.8 | 1.16 |
|
9 | 17.3 | 1.16 |
|
6 | 0.1 | 1.1 |
|
10 | 5.5 | 1.1 |
|
11 | 13.4 | 0.8 |
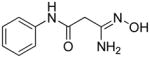
|
7 | 31.7 | 0.62 |
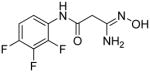
|
12 | 10.4 | 1.6 |
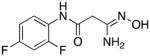
|
13 | 0 | 7.1 |

|
14 | 0 | 0.2 |
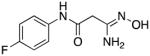
|
15 | 25.3 | 1.2 |

|
16 | 0 | 21.4 |
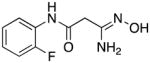
|
17 | 5.6 | 12.44 |
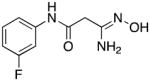
|
18 | 0 | 0.6 |
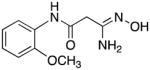
|
19 | 15.4 | 1.4 |
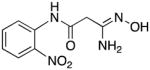
|
20 | 2.0 | 0.4 |
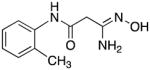
|
21 | 0 | 6.2 |
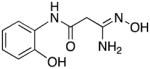
|
22 | 30.1 | 3.7 |
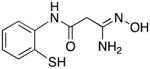
|
23 | 8.6 | 2.1 |
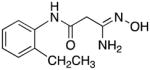
|
24 | 0 | 5.4 |
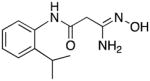
|
25 | 0 | 0.8 |
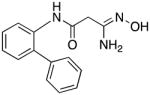
|
26 | 0 | 7.7 |
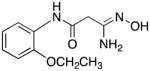
|
27 | 10.4 | 13.4 |
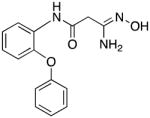
|
28 | 4.8 | 3.1 |
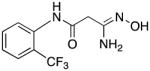
|
29 | 0 | 13.1 |
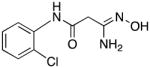
|
30 | 0 | 49.0 |
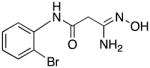
|
31 | 0.2 | 6.7 |
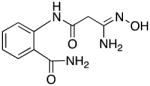
|
32 | 2.1 | 11.5 |
|
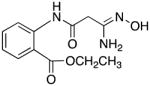
33 | 16.6 | 3.1 |
Biological Evaluation
In order for synthetic histone demethylase inhibitors to be effective at the cellular level, any observed decreases in cellular LSD1 activity should be accompanied by an increase in global H3K4me1 and H3K4me2 content. The fortuitous decision to measure methylation changes prior to evaluating the analogues as LSD1 inhibitors was made based on our preliminary findings that suggested that fluorinated amidoximes such as 14 and 15 produced significant changes in H3K4me2 levels in the Calu-6 human lung adenocarcinoma cell line. Thus, the ability of compounds 5–33 to produce increases in global H3K4me2 levels at both 24 and 48 hours was measured as previously described.8 Although indicative of changes in gene transcription, increases in H3K4 methylation levels measured at 24 hours are determined when cells are in log phase growth. Increases in H3K4 methylation at 48 hours give a more realistic picture of changes in gene transcription, since they are measured when cells are nearing confluence. Thus, only the results of these studies at 48 hours are shown in Table 1, while H3K4me2 levels at 24 hours appear in the supplemental material (Table S1). Increases in H3K4me1 and H3K4me2 levels promoted by carbamimidothioates 5, 8 and 9 and the 2,5-dinitro-4-trifluoromethylaniline derivatives 6, 10 and 11 were unremarkable at both 24 and 48 hours. Among the fluorinated amidoximes 12–18, compounds 16 and 17 promoted 21.4- and 12.4-fold increases at 48 hours, respectively. Compounds 14 and 15 produced a 7.1- and 7.0-fold increase in H3K4me2 levels at 24 hours, respectively, but this effect was not observed at 48 hours. Compounds 12, 13 and 18 had no effect on H3K4me2 at this time point. Compound 17, which featured a 2-fluoro substituent, produced a significant increase in H3K4me2 levels (12.4-fold) and was easier to access synthetically, and as such a series of 2-substituted aromatic amidoximes 19–33 was synthesized. Among these analogues, changes in the levels of H3K4me2 at 24 hours ranged between 2.1-fold and 7658.7-fold, with the exception of compounds 19–21, which had no significant effect at 24 hours. At the 48-hour time point, significant increases in H3K4me2 levels between 2.1- and 49-fold were observed, with the exception of compounds 19, 20 and 25, which did not promote increases at the 48-hour time point.
The ability of the target amidoximes 5–33 to inhibit LSD1 was determined in an assay procedure utilizing the recombinant human enzyme. Expression and purification of LSD1 were conducted as previously reported.8, 23 Enzymatic activity of LSD1 in the presence of target compounds was determined using luminol-dependent chemiluminescence to measure the production of H2O2, as previously described.23, 40 The results of these experiments are summarized in Table 1. Compound 2d inhibited purified recombinant LSD1 by 82.9% at 10 μM, and initiated a 5-fold increase in cellular H3K4me2 levels in the Calu-6 cell model system, as previously reported.32, 41 By contrast, the monoamine oxidase inhibitors tranylcypromine 3 and pargyline 4 inhibited the enzyme by 18.4 and 68.8% at 10 μM, respectively. Carbamimidothioates 5, 8 and 9 were modest inhibitors of LSD1, producing 24.0, 31.8 and 17.3% inhibition at 10 μM, but this inhibition did not correlate with an increase in H3K4me2 levels, as described above. The aminoalkylaniline derivatives 6, 10 and 11 were essentially inactive as inhibitors of recombinant LSD1, and also had no effect on lysine methylation in the Calu-6 cell line. Surprisingly, despite promoting dramatic increases in cellular H3K4me2 levels at either 24 or 48 hours, compounds 14–17, 23–27, 29, 31 and 33 were poor to modest inhibitors of LSD1. The most effective amidoxime inhibitor, 22, exhibited a 30.1% inhibition of the enzyme at 10 μM and an IC50 value of 16.8 μM, which correlated with an 837.5-fold increase in H3K4me2 levels at 24 hours. The parent compound 7 was a slightly better inhibitor (IC50 15.6 μM), but remarkably, it did not produce any significant changes in H3K4 methylation. It is well established that the specificity of LSD1 for H3K4 demethylation is observed when the enzyme is bound to the CoREST.42, 43 Thus one possible interpretation of the lack of correlation between LSD1 inhibition and increase in methylation is that the recombinant LSD1 used in the inhibition assay is not bound to the CoREST, a functional corepressor required for gene expression.44 It is possible that amidoximes related to 22 are poor inhibitors of uncomplexed recombinant LSD1 in vitro, but are more effective inhibitors of the LSD1/CoREST complex in cultured Calu-6 cells, a hypothesis that is supported by the molecular modeling studies described below.
The compounds that produced the most dramatic increases in global H3K4me2 levels, 22–27, 29 and 31, were next examined for their ability to promote the re-expression of aberrantly silenced tumor suppressor genes in the Calu-6 cell line. The soluble modulator of Wnt signaling secreted frizzle-related protein (SFRP) 2,45 the membrane-bound mediator of calcium-dependent cell-cell adhesion H-cadherin (HCAD, CDH13),46 and the zinc-finger transcription factor GATA447 were chosen because they are known to be under expressed in human lung cancer, and because their silencing is thought to play a role in tumorigenesis.41 Cells were treated for 24 or 48 hours with a 5 μM concentration of 2d or the polyamine analogue PN11144,48 or a 10 μM concentration of each amidoxime analogue, after which the levels of SFRP2, HCAD and GATA4 were determined by quantitative PCR (qPCR).32, 41 In the case of each re-expressed protein, a fold increase of ≥1.5 was considered significant. Four of the amidoxime analogues evaluated, 25, 27, 29 and 31 produced increases in SFRP2 levels (25: 2.29-fold at 24 hours, 1.45-fold at 48 hours; 27: 1.69-fold at 24 hours; 29: 1.55-fold at 48 hours; 31: 1.89-fold at both 24- and 48 hours, Figure 2). However, none of these increases were of the same magnitude as the increases promoted by the positive control 2d. Aminoguanidine at 1 mM (AG) and the polyamine analogue PG1114448 had no effect on SFRP2 levels. In the case of HCAD (Figure 3), significant increases were observed with 10 μM 23 (1.6-fold at 48 hours), 26 (1.6-fold at 48 hours), 27 (1.58 fold at 48 hours), 29 (1.8-fold at 48 hours) and 31 (2.15-fold at 48 hours). By contrast, 2d increased HCAD expression by 1.62-fold at 24 hours and 3.83-fold at 48 hours. PG11144 also had an effect at 10 μM (1.47-fold) at 24 hours. The most dramatic effects were observed in the case of the transcription factor GATA4 (Figure 4), where increased expression was observed following treatment with 23 (1.69-fold at 48 hours), 26 (1.57-fold at 24 hours, 1.77-fold at 48 hours), 27 (2.06-fold at 24 hours), 29 (1.66-fold at 24 hours) and 31 (4.22-fold at 48 hours). These effects were comparable to 2d (2.07-fold at 24 hours, 1.94-fold at 48 hours) for 27 (24 hours) and 31, which had twice the effect of 2d at 48 hours.
Figure 2.

Fold increase in the expression of SFRP2 in Calu-6 anaplastic epithelial lung carcinoma cells. Cells were treated with either 1 mM aminoguanidine (AG), 5 μM PN11144, 5 μM 2d or 10 μM of the indicated test compound for 24 or 48 hours. Each data point is the average of three determinations that in no case differed by more than 5%.
Figure 3.

Fold increase in the expression of HCAD Calu-6 anaplastic epithelial lung carcinoma cells. Cells were treated with either 1 mM aminoguanidine (AG), 5 μM PN11144, 5 μM 2d or 10 μM of the indicated test compound for 24 or 48 hours. Each data point is the average of three determinations that in no case differed by more than 5%.
Figure 4.

Fold increase in the expression of GATA4 in Calu-6 anaplastic epithelial lung carcinoma cells. Cells were treated with either 1 mM aminoguanidine (AG), 5 μM PN11144, 5 μM 2d or 10 μM of the indicated test compound for 24 or 48 hours. Each data point is the average of three determinations that in no case differed by more than 5%.
Importantly, the magnitudes of the observed increases in H3K4 methylation induced by amidoximes 22–27, 29 and 31 do not correlate well with the re-expression of the aberrantly silenced gene products chosen for this study (SFRP2, HCAD and GATA4). However, the dramatic nature of the observed increases in H3K4me2 methylation suggest that significant changes in cellular gene expression must be taking place. As such, we are currently undertaking a DNA microarray strategy to determine which genes are affected in tumor cells following treatment with amidoximes 22–27, 29 and 31.
In Silico Analysis
In order to provide a rationale for the observed biological activity of the amidoxime inhibitors, we performed an in silico docking experiment using the GOLD software package.49 At present, there are 27 published X-ray structures of LSD1, including those crystallized with and without the CoREST complex, and with and without a bound ligand. For our purposes, we selected PDB structure 2V1D, because it featured the necessary components (the LSD1 binding pocket, a co-crystallized inhibitor as ligand, free FAD and the associated CoREST complex),50 and compound 22, which was the best LSD1 inhibitor in vitro (Table 1). The inhibitor in the 2V1D complex is a 21-mer pLys4Met H3 peptide in which a methionine takes the place of the lysine corresponding to H3K4.51 The pLys4Met H3 peptide exhibited a 30-fold greater binding affinity (Ki = 0.05 μM) as compared to the wild-type peptide (Ki = 1.8 μM). Since the LSD1 binding pocket and the 21-mer co-crystal peptide ligand are relatively large, we included only the residues that lie within 6 Å around the ligand in our analysis. The pLys4Met H3 ligand was removed, and the energy-minimized amidoxime 22 was substituted and docked in the binding site of LSD1/CoREST (see the Experimental section). The resulting poses were scored using ChemScore.49
Visualization of the lowest energy binding mode of 22 in the LSD1/CoREST binding site revealed that 22 binds to the same region as that occupied by the pThr3 and pMet4 of the pLys4Met H3 peptide, as shown in Figure 5. This result supports the hypothesis that 22 and related amidoximes occupy the active site of LSD1. The distance between the enolic hydroxyl group of 22 and N5 of the FAD is about 6.3 Å, whereas the distance between the sulfur of pMet4 and N5 of the FAD is about 4.9 Å, demonstrating that the amidoxime moiety of 22 lies in proximity to the FAD binding-domain. The phenyl group in 22 is situated in a hydrophobic cavity surrounded by residues Phe 560, Tyr 807 and His 812. Interestingly, this is the region that is occupied by the side-chain of the p3 Thr of pLys4Met H3 peptide. As shown in Figure 5, the phenolic hydroxyl group of the amidoxime forms a hydrogen bond with the side-chain hydroxyl group of Thr 335, while the primary amine group of the amidoxime moiety forms hydrogen bond with the backbone carbonyl of Ala 539. This interaction is analogous to the hydrogen bond formed between the amide nitrogen of pAla1 in the pLys4Met H3 ligand and the backbone carbonyl of Ala339, underscoring the importance of this interaction. Finally, the enolic hydroxyl of the amidoxime forms a hydrogen bond with both the backbone carbonyl of Ala 539 and the 4-hydroxyl group of Tyr 761. These docking results suggest that 22 binds to the LSD1/CoREST active site in exactly the same region as both H3K4 of the histone tail and pMet4 of the pLys4Met H3 peptide. A combination of both hydrogen bonding and hydrophobic interactions stabilize the LSD1/CoREST complex with 22 (Figure 6) positioning it directly in front of the FAD cofactor and blocking the access of the substrate to the FAD. A similar docking experiment involving 22 and native LSD1 lacking the CoREST cofactor (PDB index 2H94) revealed that these specific interactions between 22 and the amino acids Thr 335, Ala 539 and Tyr 761 are not present (Supplementary materials, Figure S1). In this model, the amidoxime moiety of 22 forms a hydrogen bond with N5 of the flavin ring, but no other electrostatic associations could be detected. Although the models shown in Figures 6 and S1 are preliminary, they support the contention that 22 acts as an inhibitor of LSD1 only when the CoREST co-repressor is present. These data could also explain the observation that large increases in H3K4me2 levels following treatment with 22 and its homologues do not correlate with inhibition of LSD1 in the assay, which does not contain CoREST. Importantly, the in silico model shown in Figure 6 suggest modifications to 22 which could lead to more potent analogues.
Figure 5.

Graphic representation of binding mode of amidoxime 22 in the LSD1-CoREST complex active site. Panel A: Distance between the amidoxime functionality of 22 and N-5 of the flavin ring of FAD. Panel 3: Distance between the MET residue of the Forneris inhibitor and N-5 of the flavin ring of FAD. Panel B: Hydrogen bonding distances between 22 and amino acid residues THR 335, ALA 539 and TYR 761.
Figure 6.
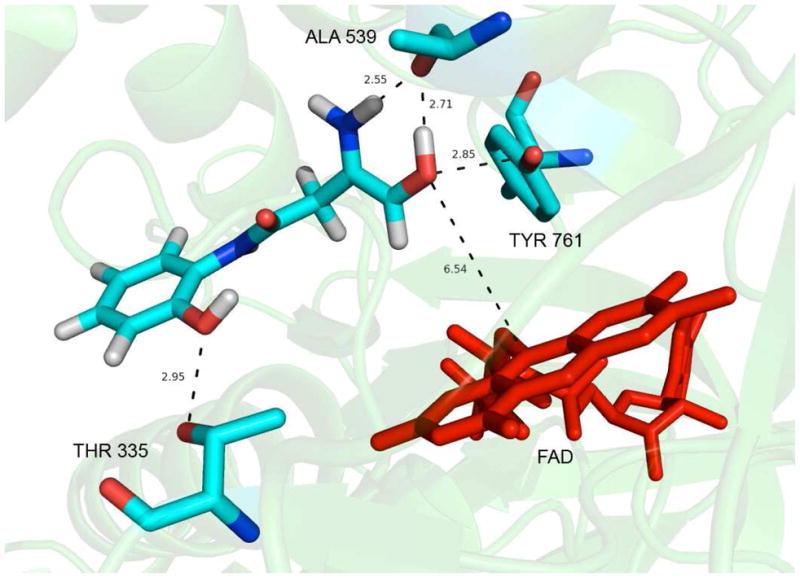
Results of in silico docking of amidoxime 22 in the active site of the LSD1-CoREST complex. The FAD cofactor is shown in red, and distances are in angstroms.
Discussion
Our virtual screening strategy resulted in the identification of 10 potential LSD1 inhibitors that have desirable drug-like properties and can be readily synthesized. Many of the inhibitors of LSD1 described in the recent literature are based either on the tranylcypromine scaffold, or are oligopeptides that are unlikely to be used as therapeutic agents. More recently, a series of small molecules was described that inhibit LSD1, enhance H3K4 methylation and promote the re-expression of aberrantly silenced tumor suppressor genes. These analogues were also shown to selectively inhibit the growth of tumor cells with pluripotent stem cell properties.52 However, these analogues are structurally related to the (bis)guanidine, (bis)biguanide, (bis)urea and (bis)thiourea analogues previously described in our laboratories,32, 41, 53 which produce similar cellular effects in non-pluripotent tumor cell lines and exhibit in vivo activity.33 Compared to the known LSD1 inhibitors described above, compound 22 and its homologues possess more favorable drug-like properties54 (CLogP 0.37, MW 208.21, 5 H-bond acceptors, 4 H-bond donors), and can be readily synthesized in two steps. Amidoximes 22–27, 29 and 31 produce dramatic increases in global H3K4me2 levels, and also promote the re-expression of aberrantly silenced tumor suppressor genes in the Calu-6 cell line. Thus, these compounds, and in particular 22, can be considered lead compounds within a new structural class of histone demethylase inhibitors. We have initiated lead optimization studies, and will report the data from these studies in a future manuscript. Additional studies are being performed to determine which aberrantly silenced genes are reactivated secondary to the dramatic increases in H3K4 methylation observed for amidoximes 22–27, 29 and 31. In addition, amidoximes 22–27, 29 and 31 are being evaluated in other tumor cell lines, alone and in combination with the DNA methyltransferase inhibitor 5-azacytidine, as well as in a murine xenograft model.
Experimental Section
All reagents and dry solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI), Sigma Chemical Co. (St. Louis, MO) or Acros Chemical (Chicago, IL) and were used without further purification except as noted below. Pyridine was dried by passing it through an aluminum oxide column and then stored over KOH. Triethylamine was distilled from potassium hydroxide and stored in a nitrogen atmosphere. Methanol was distilled from magnesium and iodine under a nitrogen atmosphere and stored over molecular sieves. Methylene chloride was distilled from phosphorus pentoxide and chloroform was distilled from calcium sulfate. Tetrahydrofuran was purified by distillation from sodium and benzophenone. Dimethyl formamide was dried by distillation from anhydrous calcium sulfate and was stored under nitrogen. Preparative scale chromatographic procedures were carried out using E. Merck silica gel 60, 230–440 mesh. Thin layer chromatography was conducted on Merck precoated silica gel 60 F-254. Ion exchange chromatography was conducted on Dowex 1X8-200 anion exchange resin. Compounds 41a–c 35, 55 were synthesized as previously described.
All 1H- and 13C-NMR spectra were recorded on a Varian Mercury 400 mHz spectrometer, and all chemical shifts are reported as δ values referenced to TMS or DSS. In all cases, 1H-NMR, 13C-NMR and MS spectra were consistent with assigned structures. Mass spectra were recorded by LC/MS on a Waters autopurification liquid chromatograph with a 3100 mass spectrometer detector. Prior to biological testing procedures, target molecules 5–33 were determined to be >95% pure by UPLC chromatography (95% H2O/5% acetonitrile to 20% H2O/80% acetonitrile over 10 minutes) using a Waters Acquity H-series ultrahigh-performance liquid chromatograph fitted with a C18 reversed-phase column (Acquity UPLC BEH C18 1.7 μM, 2.1 × 50 mm).
Synthetic H3K4me2 peptides were purchased from Millipore (Billerica, MA). Calu-6 cells were maintained in RPMI medium, both supplemented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA) and grown at 37°C in 5% CO2 atmosphere.
General procedure for preparation carbamimidothioates 5, 8 and 9.36
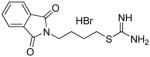
2-[N-(phthalimido)]ethyl carbamimidothioate hydrochloride 5
A 3.0 g (11.6 mmol) portion of N-(2-bromoethyl)phthalimide 34a and 1.77 g (12.1 mmol, 2.0 equiv.) of thiourea was dissolved in 10 mL of anhydrous ethanol, and the mixture was allowed to reflux overnight. The reaction mixture was allowed to cool until a white crystalline precipitate formed, and the solid was collected by filtration, washed with acetone and ether and dried. Recrystallization from 1.0 M HCl/ethanol then afforded 2.05 g (62%) of pure 5 as a white crystalline solid. Melting point 380–381°C; UPLC retention time 3.47 min; 1H NMR (CD3OD): δ 3.54 (t, J= 6.4 Hz, 2H), 4.05 (t, J = 6.4 Hz, 2H), 7.90 (m, 4H).
2-[N-(phthalimido)]propyl]carbamimidothioate hydrochloride 8
Compound 8 was prepared from 3.0 g (11.2 mmol) of N-(3-bromopropyl)phthalimide 34b and 1.70 g (22.4 mmol, 2.0 equiv.) of thiourea in 68% yield exactly as described for the preparation of compound 5. Melting point 362–363°C; UPLC retention time 4.45 min; 1H NMR (CD3OD): δ 2.11 (quint, J = 6.8 Hz, 2H), 3.22 (t, 3.22, J = 6.8 Hz, 2H), 3.85 (t, J=6.8 Hz, 2H), 7.87 (m, 4H).
2-[N-(phthalimido)]butyl]carbamimidothioate hydrochloride 9
Compound 9 was prepared from 1.5 g (5.2 mmol) of N-(4-bromobutyl)phthalimide 34c and 0.79 g (10.4 mmol, 2.0 equiv.) of thiourea in 79% yield exactly as described for the preparation of compound 5. Melting point 358–359°C; UPLC retention time 5.32 min; 1H NMR (CD3OD): δ 1.82 (m, 4H), 3.22 (t, J = 7.3 Hz, 2H), 3.75 (t, J = 6.7 Hz, 2H), 7.86 (m, 4H).
General procedure for the preparation of compounds 6, 10 and 11.37
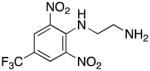
N1-(2,6-dinitro-4-[(trifluoromethyl)phenyl]ethane-1,2-diamine hydrochloride 6
A solution of ethylene diamine 36a (6.0 g, 100 mmol, 6.7 mL, 20 equiv.) and triethylamine (0.51 g, 0.70 mL, 5.0 mmol) in 40 mL of benzene was cooled to 10°C, and a solution of 4-chloro-3,5-dinitrobenzotrifluoride 35 (1.36 g, 5.00 mmol) in 40 mL of benzene was added dropwise over 2 hours. The reaction was stirred overnight and concentrated in vacuo to yield an orange liquid. This liquid was dissolved in 200 mL of ethyl acetate, and the organic layer was washed with two 25 mL portions of water and then 25 mL of saturated aqueous NaCl. The ethyl acetate solution was dried over anhydrous MgSO4, filtered and concentrated, and the crude mixture was purified by column chromatography on silica gel (with a gradient of 1:1 hexanes:AcOEt to AcOEt to 3:1 AcOEt:MeOH). Fractions containing the desired product (Rf = 0.39) were pooled and concentrated to give an orange solid, which was dissolved in ethyl acetate. A 1.0 M solution of HCl in ethyl acetate was added until no more product dropped out of solution. The product was filtered and washed with ethyl acetate to afford pure 6 as the yellow solid dihydrochloride salt (1.55 g, 84.7% yield). Melting point 324–326°C (dec.); UPLC retention time 6.22 min; 1H NMR (400MHz, CDCl3) δ 9.45 (bs, 1H), 8.41 (s, 2H), 3.10–2.95 (m, 4H), 1.27 (bs, 2H). 19F NMR (376MHz, CDCl3) δ −62.36 (s, 3F).
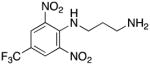
N1-(2,6-dinitro-4-[(trifluoromethyl)phenyl]propane-1,2-diamine hydrochloride 10
Compound 10 was prepared from 7.41 g (100.0 mmol) of 1,3-propanediamine 36b and 0.79 g of 4-chloro-3,5-dinitrobenzotrifluoride 35 (5.00 mmol) in 87% yield exactly as described for the preparation of compound 6. Melting point 380–381°C (dec.); UPLC retention time 6.69 min; 1H NMR (400MHz, CDCl3) δ 9.52 (bs, 1H), 8.39 (s, 2H), 3.14 (s, 2H), 2.94 (t, J = 6.0 Hz, 2H), 1.78 (quint, J = 6.0 Hz, 2H), 1.33 (bs, 2H). 19F NMR (376MHz, CDCl3) δ −62.36 (s, 3F).
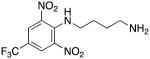
N1-(2,6-dinitro-4-[(trifluoromethyl)phenyl]butane-1,2-diamine hydrochloride 11
Compound 11 was prepared from 8.81 g (100.0 mmol) of 1,4-butanediamine 36c and 0.79 g of 4-chloro-3,5-dinitrobenzotrifluoride 35 (5.00 mmol) in 42% yield exactly as described for the preparation of compound 6. Melting point 374–376°C (dec.); UPLC retention time 7.05 min; 1H NMR (400MHz, D2O) δ 8.48 (s, 2H), 2.94 (t, J = 6.4 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H), 1.70–1.50 (m, 4H). 19F NMR (376MHz, D2O) δ −62.51 (s, 3F).
General procedure for the preparation of cyano-N-phenylacetamides 60 – 82.38
2-Cyano-N-phenylacetamide 60
A 0.96 g portion (11.1 mmol) of cyanoacetic acid was added to a mixture of PCl5 (2.35 g, 11.1 mmol) and 200 mL of dichloromethane, and the mixture refluxed for 30 minutes. After cooling, 1.03 g of aniline (11.1 mmol) was added and the solution was refluxed for 2hrs. The solution was then concentrated, H2O was added and the solid was collected and washed with NaHCO3 solution, H2O and dried. The intermediate 60 was isolated in 92% yield, and was of sufficient purity to use in the subsequent reaction without further purification. 1H NMR (400 MHz, Acetone-d6) δ 9.58 (s, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.33 (t, J = 8.0 Hz, 2H), 7.11 (t, J = 7.2 Hz, 1H), 3.82 (s, 2H).
2-Cyano-N-[(2,3,4-trifluoro)phenyl]acetamide 61
Compound 61 was synthesized in 90% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, Acetone-d6) δ 9.60 (s, 1H), 7.89–7.83 (m, 1H), 7.29–7.14 (m, 1H), 3.97 (s, 2H). 19F NMR (376 MHz, Acetone-d6) δ −141.75 (m, 1F), −147.85 (m, 1F), −162.75 (m, 1F).
2-Cyano-N-[(2,4-(difluoro)phenyl]acetamide 62
Compound 62 was synthesized in 76% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, DMSO-d6) δ 10.14 (s, 1H), 7.84–7.77 (m, 1H), 7.37–7.32 (m, 1H), 7.12–7.05 (m, 1H), 3.96 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −114.33 (m, 1F), −119.95 (s, 1F).
2-Cyano-N-[2,3-(difluoro)phenyl]acetamide 63
Compound 63 was synthesized in 83% yield exactly as described for the preparation of compound 60. Yellow solid: 1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 7.66 (s, 1H), 7.24–7.14 (m, 2H), 3.99 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −138.69 (m, 1F), −149.64 (m, 1F).
2-Cyano-N-[4-(fluoro)phenyl]acetamide 64
Compound 64 was synthesized in 83% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 7.55–7.53 (m, 2H), 7.20–7.13 (m, 2H), 3.88 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −118.87 (s, 1F).
2-Cyano-N-[3,4-(difluoro)phenyl]acetamide 65
Compound 65 was synthesized in 94% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 7.76–7.64 (m, 1H), 7.45–7.30 (m, 1H), 7.25–7.20 (m, 1H), 3.89 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −137.20 (m, 1F), −144.36 (m, 1F).
2-Cyano-N-[2-(fluoro)phenyl]acetamide 66
Compound 66 was synthesized in 85% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 7.87 (t, J = 8.8 Hz, 1H), 7.35–7.13 (m, 3H), 3.99 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −126.08 (m, 1F).
2-Cyano-N-[3-(fluoro)phenyl]acetamide 67
Compound 67 was synthesized in 68% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 7.52 (dt, J = 11.6 Hz, 2.0 Hz, 1H), 7.41–7.34 (m, 1H), 7.28–7.23 (m, 1H), 6.93 (td, J = 6.0 Hz, 2.4 Hz, 1H), 3.93 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −112.15 (m, 1F).
2-Cyano-N-[2-(methoxy)phenyl]acetamide 68
Compound 68 was synthesized in 94% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 8.34 (bs, 1H), 8.25 (dd, J = 8.0, 2.0 Hz, 1H), 7.12 (td, J = 8.0, 1.6 Hz, 1H), 6.97 (dt, J = 8.0, 1.2 Hz, 1H), 6.91 (dd, J = 8.0, 1.2 Hz, 1H), 3.91 (s, 3H), 3.56 (s, 2H).
2-Cyano-N-[2-(nitro)phenyl]acetamide 69
Compound 69 was synthesized in quantitative yield exactly as described for the preparation of compound 60. Tan solid: 1H NMR (400 MHz, CDCl3) δ 10.92 (bs, 1H), 8.68 (dd, J = 8.4, 1.2 Hz, 1H), 8.27 (dd, J = 8.4, 1.6 Hz, 1H), 7.71 (dt, J = 8.4, 1.6 Hz, 1H), 7.30 (dt, J = 8.0, 1.2 Hz, 1H), 3.67 (s, 2H).
2-Cyano-N-[2-(methyl)phenyl]acetamide 70
Compound 70 was synthesized in 94% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 7.73 (bs, 1H), 7.66 (d, J = 7.6 Hz, 1H), 7.26–7.11 (m, 3H), 3.56 (s, 2H), 2.28 (s, 3H).
2-Cyano-N-[2-(hydroxyl)phenyl]acetamide 71
Compound 71 was synthesized in 52% yield exactly as described for the preparation of compound 60. Pale purple-white solid: 1H NMR (400 MHz, Acetoned6) δ 9.09 (bs, 1H), 7.94 (dd, J = 8.0, 1.2 Hz, 1H), 6.99 (dt, J = 8.4, 1.2 Hz, 1H), 6.92 (dd, J = 8.0, 1.2 Hz, 1H), 6.84 (dt, J = 8.0, 1.2 Hz, 1H), 3.98 (s, 2H). 13C NMR (100 MHz, Acetone-d6) δ 162.1, 148.1, 126.9, 126.1, 122.4, 120.6, 116.6, 115.8, 27.1.
2-Cyano-N-[2-(mercapto)phenyl]acetamide 72
Compound 72 was synthesized in 66% yield exactly as described for the preparation of compound 60, except that the material was further purified by dissolving the residue in dichloromethane and filtering through silica gel plug. Light yellow solid: 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.45 (t, J = 8.0 Hz, 1H), 4.24 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 158.4, 152.8, 135.5, 126.7, 126.0, 123.3, 121.8, 115.1, 23.2.
2-Cyano-N-[2-(ethyl)phenyl]acetamide 73
Compound 73 was synthesized in 81% yield exactly as described for the preparation of compound 60. Off-white solid: 1H NMR (400 MHz, CDCl3) δ 7.74 (bs, 1H), 7.68 (dd, J = 7.6, 1.6 Hz, 1H), 7.26–7.17 (m, 3H), 3.57 (s, 2H), 2.63 (q, J = 7.6 Hz, 2H), 1.26 (t, J = 7.6 Hz, 3H).
2-Cyano-N-[2-(isopropyl)phenyl]acetamide 74
Compound 74 was synthesized in quantitative yield exactly as described for the preparation of compound 60, except that the oil was dissolved in 100 mL of ethyl acetate, and the organic layer was washed with 1.0 N sodium bicarbonate and saturated aqueous NaCl. Filtration and evaporation of the solvent then afforded pure 74 as an orange oil that solidified at RT: 1H NMR (400 MHz, CDCl3) δ 7.85 (bs, 1H), 7.52 (dd, J = 7.6, 1.2 Hz, 1H), 7.32 (dd, J = 7.6, 1.2 Hz, 1H), 7.29–7.17 (m, 2H), 3.54 (s, 2H), 3.02 (septet, J = 6.8 Hz, 1H), 1.24 (d, J = 6.8 Hz, 6H).
2-Cyano-N-[2-(phenyl)phenyl]acetamide 75
Compound 75 was synthesized in 97% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 8.4 Hz, 1H), 7.79 (bs, 1H), 7.55–7.23 (m, 9H), 3.39 (s, 2H).
2-Cyano-N-[2-(ethoxy)phenyl]acetamide 76
Compound 76 was synthesized in 81% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 8.52 (bs, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 6.97 (t, J = 7.6 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 4.13 (q, J = 6.8 Hz, 2H), 3.57 (s, 2H), 1.49 (t, J = 6.8 Hz, 3H).
2-Cyano-N-[2-(phenoxy)phenyl]acetamide 77
Compound 77 was synthesized in quantitative yield exactly as described for the preparation of compound 60. Light yellow solid: 1H NMR (400 MHz, CDCl3) δ 8.32 (dd, J = 8.0, 1.6 Hz, 1H), 8.29 (bs, 1H), 7.41–7.34 (m, 2H), 7.20–7.01 (m, 5H), 6.87 (dd, J = 8.0, 1.2 Hz, 1H), 3.53 (s, 2H).
2-Cyano-N-[2-(trifluoromethyl)phenyl]acetamide 78
Compound 78 was synthesized in 89% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.4 Hz, 1H), 8.00 (bs, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 3.61 (s, 2H). 19F NMR (376 MHz, CDCl3) δ −60.98 (s).
2-Cyano-N-[2-(chloro)phenyl]acetamide 79
Compound 79 was synthesized in 90% yield exactly as described for the preparation of compound 60. Off-white solid: 1H NMR (400 MHz, CDCl3) δ 8.25 (dd, J = 8.0, 1.2 Hz, 1H), 8.24 (bs, 1H), 7.41 (dd, J = 8.0, 0.8 Hz, 1H), 7.31 (td, J = 8.0, 1.2 Hz, 1H), 7.13 (td, J = 7.6, 1.2 Hz, 1H), 3.62 (s, 2H).
2-Cyano-N-[2-(bromo)phenyl]acetamide 80
Compound 80 was synthesized in 95% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 8.24 (dd, J = 8.0, 1.2 Hz, 1H), 8.24 (bs, 1H), 7.58 (dd, J = 8.0, 1.2 Hz, 1H), 7.35 (td, J = 8.0, 1.2 Hz, 1H), 7.07 (td, J = 7.6, 1.2 Hz, 1H), 3.62 (s, 2H).
2-(2-Cyanoacetamido)benzamide 81
Compound 81 was synthesized in 97% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, DMSO-d6) δ 11.83 (bs, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.31 (s, 1H), 7.80 (dd, J = 8.0, 1.2 Hz, 1H), 7.78 (s, 1H), 7.52 (td, J = 8.0, 1.2 Hz, 1H), 7.18 (td, J = 7.6, 1.2 Hz, 1H), 4.07 (s, 2H).
Ethyl 2-(2-cyanoacetamido)benzoate 82
Compound 82 was synthesized in 96% yield exactly as described for the preparation of compound 60. White solid: 1H NMR (400 MHz, CDCl3) δ 11.75 (bs, 1H), 8.61 (d, J = 8.0 Hz, 1H), 8.09 (dd, J = 8.0, 1.6 Hz, 1H), 7.58 (td, J = 8.0, 1.6 Hz, 1H), 7.17 (td, J = 7.6, 1.2 Hz, 1H), 4.42 (q, J = 7.2 Hz, 2H), 3.61 (s, 2H), 1.42 (t, J = 7.2 Hz, 3H).
General procedure for the preparation of amidoximes 7 and 12 – 33.39
3-Amino-3-(hydroxyimino)-N-phenylpropanamide 7
A 0.9 g portion of NH2OH·HCl (12.8 mmol) was added to a mixture of sodium carbonate (1.36 g, 12.8 mmol) in 5 mL of water, and the solution was diluted with 50 mL of MeOH. A 1.64 g portion (10.2 mmol) of compound 60 was then added, and the mixture refluxed for 2 hours. The mixture was cooled, the solvent was removed in vacuo, and the resulting solid was mixed with hot 3:1 AcOEt:MeOH. The insoluble material was then removed by filtration of the hot mixture, and the filtrate was concentrated and purified by chromatography on silica gel (gradient of AcOEt → 20:1 AcOEt:MeOH). Fractions containing the product were pooled and the solvent was removed, and the resulting solid was recrystallized from MeOH/EtOAc to afford 0.67 g (34%) of pure 7 as a white amorphous solid. An analytical sample was prepared by recrystallization from 1.0 M HCl/ethanol. Melting point 312–313°C (dec.), UPLC retention time 2.29 min; 1H NMR (400 MHz, Acetone-d6) δ 9.53 (s, 1H), 8.70 (s, 1H), 7.65–7.63 (m, 2H), 7.31–7.27 (m, 2H), 7.07–7.04 (m, 1H), 5.39 (s, 2H), 3.17 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2,3,4-(trifluoro)phenyl]propanamide 12
Compound 12 was synthesized from 61 in 26% yield exactly as described for the preparation of compound 7. White crystals. Melting point 310–312°C (dec.), UPLC retention time 6.07 min; 1H NMR (400 MHz, Acetone-d6) δ 9.72 (s, 1H), 8.79 (s, 1H), 7.99–7.92 (m, 1H), 7.19–7.12 (m, 1H), 5.45 (s, 2H), 3.28 (s, 2H). 19F NMR (376 MHz, Acetone-d6) δ −143.11 (m, 1F), −148.97 (m, 1F), −163.25 (m, 1F).
3-Amino-3-(hydroxyimino)-N-[2,4-(difluoro)phenyl]propanamide 13
Compound 13 was synthesized from 62 in 59% yield exactly as described for the preparation of compound 7. White solid. Melting point 314–315°C (dec.), UPLC retention time 2.36 min; 1H NMR (400 MHz, DMSO-d6) δ 9.83 (s, 1H), 9.06 (d, J = 6.8 Hz, 1H), 7.93–7.85 (m, 1H), 7.33–7.27 (m, 1H), 7.06–7.03 (m, 1H), 5.48 (s, 2H), 3.12 (d, J = 0.64 Hz, 2H). 19F NMR (376 MHz, DMSO-d6) δ −115.49 (d, 1F), −120.73 (bs, 1F).
3-Amino-3-(hydroxyimino)-N-[2,3-(difluoro)phenyl]propanamide 14
Compound 13 was synthesized from 63 in 43% yield exactly as described for the preparation of compound 7. White solid. Melting point 290–292°C (dec.), UPLC retention time 2.67 min; 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 9.05 (s, 1H), 7.75–7.72 (m, 1H), 7.19–7.09 (m, 2H), 5.48 (s, 2H), 3.14 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −139.10 (m, 1F), −150.53 (m, 1F).
3-Amino-3-(hydroxyimino)-N-[4-(fluoro)phenyl]propanamide 15
Compound 15 was synthesized from 64 in 53% yield exactly as described for the preparation of compound 7. White solid. Melting point 299–300°C (dec.), UPLC retention time 2.76 min; 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 9.02 (s, 1H), 7.61–7.57 (m, 2H), 7.15–7.10 (m, 2H), 5.44 (bs, 2H), 3.03 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ - 119.87 (m, 1F).
3-Amino-3-(hydroxyimino)-N-[3,4-(difluoro)phenyl]propanamide 16
Compound 16 was synthesized from 65 in 68% yield exactly as described for the preparation of compound 7. White solid. Melting point 320–322°C (dec.), UPLC retention time 3.87 min; 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 9.12–9.04 (m, 1H), 7.81–7.76 (m, 1H), 7.40–7.34 (m, 1H), 7.33–7.28 (m, 1H), 5.47 (s, 2H), 3.06–3.01 (m, 2H). 19F NMR (376 MHz, DMSO-d6) δ −137.60 (m, 1F), −145.33 (s, 1F).
3-Amino-3-(hydroxyimino)-N-[2-(fluoro)phenyl]propanamide 17
Compound 17 was synthesized from 66 in 51% yield exactly as described for the preparation of compound 7. White solid. Melting point 294–297°C (dec.), UPLC retention time 1.95 min; 1H NMR (400 MHz, DMSO-d6) δ9.79 (s, 1H), 9.05 (s, 1H), 7.97–7.88 (m, 1H), 7.25–7.05 (m, 3H), 5.48 (s, 1H), 3.13 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −126.09 (m, 1F).
3-Amino-3-(hydroxyimino)-N-[3-(fluoro)phenyl]propanamide 18
Compound 18 was synthesized from 67 in 53% yield exactly as described for the preparation of compound 7. White solid. Melting point 338–340°C (dec.), UPLC retention time 3.32 min; 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 9.04 (s, 1H), 7.58 (d, J = 13.2 Hz, 1H), 7.33–7.66 (m, 2H), 6.88–6.84 (m, 1H), 5.46 (s, 1H), 3.06 (s, 1H). 19F NMR (376 MHz, DMSO-d6) δ - 112.45 (m, 1F).
3-Amino-3-(hydroxyimino)-N-[2-(methoxy)phenyl]propanamide 19
Compound 19 was synthesized from 68 in 67% yield exactly as described for the preparation of compound 7. White crystals. Melting point 277–278°C (dec.), UPLC retention time 2.94 min; 1H NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 9.13 (s, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.08–7.00 (m, 2H), 6.90 (dt, J = 8.0, 1.6 Hz, 1H), 5.60 (s, 2H), 3.83 (s, 3H), 3.14 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(nitro)phenyl]propanamide 20
Compound 20 was synthesized from 69 in 72% yield exactly as described for the preparation of compound 7. Orange-yellow crystals. Melting point 294–295°C (dec.), UPLC retention time 2.09 min; 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 9.14 (s, 1H), 8.08 (dd, J = 8.0, 1.2 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.72 (dt, J = 8.0, 0.8 Hz, 1H), 7.36 (dt, J = 8.0, 0.8 Hz, 1H), 5.55 (s, 2H), 3.13 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(methyl)phenyl]propanamide 21
Compound 21 was synthesized from 70 in 78% yield exactly as described for the preparation of compound 7. White crystals. Melting point 290–291°C (dec.), UPLC retention time 2.55 min; 1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 9.09 (s, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 7.2 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1H), 5.53 (s, 2H), 3.11 (s, 2H), 2.21 (s, 3H).
3-Amino-3-(hydroxyimino)-N-[2-(hydroxyl)phenyl]propanamide 22
Compound 22 was synthesized from 71 in 53% yield exactly as described for the preparation of compound 7. Off-white crystals. Melting point 308–310°C (dec.), UPLC retention time 1.17 min; 1H NMR (400 MHz, DMSO-d6) δ 9.85 (s, 1H), 9.29 (s, 1H), 9.12 (s, 1H), 7.88 (dd, J = 8.0, 1.6 Hz, 1H), 6.92 (dt, J = 7.6, 1.6 Hz, 1H), 6.85 (dd, J = 8.0, 1.2 Hz, 1H), 6.75 (dt, J = 8.0, 1.6 Hz, 1H), 5.60 (s, 2H), 3.12 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(mercapto)phenyl]propanamide 23
Compound 23 was synthesized from 72 in 65% yield exactly as described for the preparation of compound 7. Light grey crystals. Melting point 324–325°C (dec.), UPLC retention time 3.32 min; 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.05 (dd, J = 8.0, 0.8 Hz, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.49 (dt, J = 7.6, 1.2 Hz, 1H), 7.41 (dt, J = 7.6, 1.2 Hz, 1H), 5.70 (s, 2H), 3.83 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(ethyl)phenyl]propanamide 24
Compound 24 was synthesized from 73 in 74% yield exactly as described for the preparation of compound 7. Off-white crystals. Melting point 266–268°C (dec.), UPLC retention time 3.89 min; 1H NMR (400 MHz, DMSO-d6) δ 9.43 (s, 1H), 9.10 (s, 1H), 7.50 (d, J = 7.6 Hz, 1H), 7.21 (dd, J = 7.2, 1.2 Hz, 1H), 7.19–7.08 (m, 2H), 5.55 (s, 2H), 3.11 (s, 2H), 2.58 (q, J = 7.6 Hz, 2H), 1.12 (t, J = 7.6 Hz, 3H).
3-Amino-3-(hydroxyimino)-N-[2-(isopropyl)phenyl]propanamide 25
Compound 25 was synthesized from 74 in 64% yield exactly as described for the preparation of compound 7. Tan crystals. Melting point 228–230°C (dec.), UPLC retention time 4.87 min; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.08 (s, 1H), 7.38 (dd, J = 8.0, 2.4 Hz, 1H), 7.30 (dd, J = 8.0, 2.8 Hz, 1H), 7.20–7.12 (m, 2H), 5.53 (s, 2H), 3.14 (septet, J = 6.8 Hz, 1H), 3.10 (s, 2H), 1.14 (d, J = 6.8 Hz, 6H).
3-Amino-3-(hydroxyimino)-N-[2-(phenyl)phenyl]propanamide 26
Compound 26 was synthesized from 75 in 77% yield exactly as described for the preparation of compound 7, except that the compound was isolated as the hydrochloride salt by dissolving it in ethyl acetate and adding HCl in ether. Off-white solid. Melting point 284–285°C (dec.), UPLC retention time 6.01 min; 1H NMR (400 MHz, DMSO-d6) δ 12.71 (bs, 1H), 11.09 (s, 1H), 9.85 (s, 1H), 9.02 (bs, 1H), 8.73 (bs, 1H), 7.53–7.30 (m, 9H), 3.48 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(ethoxy)phenyl]propanamide 27
Compound 27 was synthesized from 76 in 59% yield exactly as described for the preparation of compound 7. Off-white crystals. Melting point 372–374°C (dec.), UPLC retention time 3.85 min; 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 9.14 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.04–7.00 (m, 2H), 6.92–6.86 (m, 1H), 5.67 (s, 2H), 4.07 (q, J = 6.8 Hz, 2H), 3.14 (s, 2H), 1.14 (t, J = 6.8 Hz, 3H).
3-Amino-3-(hydroxyimino)-N-[2-(phenoxy)phenyl]propanamide 28
Compound 28 was synthesized from 77 in 79% yield exactly as described for the preparation of compound 7. White crystals. Melting point 275–277°C (dec.), UPLC retention time 6.40 min; 1H NMR (400 MHz, DMSO-d6) δ9.63 (s, 1H), 9.05 (s, 1H), 8.13 (dd, J = 8.0, 1.2 Hz, 1H), 7.39 (t, J = 8.0 Hz, 2H), 7.17–7.00 (m, 5H), 6.87 (dd, J = 8.0, 1.2 Hz, 1H), 5.51 (s, 2H), 3.11 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(trifluoromethyl)phenyl]propanamide 29
Compound 29 was synthesized from 78 in 67% yield exactly as described for the preparation of compound 7. Pale green crystals. Melting point 280–281°C (dec.), UPLC retention time 3.31 min; 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.12 (s, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.70–7.65 (m, 2H), 7.45–7.39 (m, 1H), 5.56 (s, 2H), 3.13 (s, 2H). 19F NMR (376 MHz, DMSO-d6) δ −59.83 (s, 3F).
3-Amino-3-(hydroxyimino)-N-[2-(chloro)phenyl]propanamide 30
Compound 30 was synthesized from 79 in 69% yield exactly as described for the preparation of compound 7. Off-white crystals. Melting point 291–293°C (dec.), UPLC retention time 2.66 min; 1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, 1H), 9.16 (s, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 1H), 7.15 (t, J = 7.6 Hz, 1H), 5.63 (s, 2H), 3.18 (s, 2H).
3-Amino-3-(hydroxyimino)-N-[2-(bromo)phenyl]propanamide 31
Compound 31 was synthesized from 80 in 77% yield exactly as described for the preparation of compound 7. White crystals. Melting point 278–280°C (dec.), UPLC retention time 2.87 min; 1H NMR (400 MHz, DMSO-d6) δ 9.74 (s, 1H), 9.15 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 5.63 (s, 2H), 3.16 (s, 2H).
2-(3-amino-3-(hydroxyimino)propanamido)benzamide 32
Compound 32 was synthesized from 81 in 50% yield exactly as described for the preparation of compound 7. Tan crystals. Melting point 320–321°C (dec.), UPLC retention time 0.94 min; 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 9.09 (s, 1H), 8.44 (d, J = 8.4 Hz, 1H), 8.23 (s, 1H), 7.77 (dd, J = 8.0, 1.2 Hz, 1H), 7.67 (s, 1H), 7.48 (dt, J = 8.0, 1.2 Hz, 1H), 7.12 (dt, J = 7.6, 0.8 Hz, 1H), 5.52 (s, 2H), 3.06 (s, 2H).
2-(3-amino-3-(hydroxyimino)propanamido)benzoate 33
Compound 33 was synthesized from 82 in 69% yield exactly as described for the preparation of compound 7. Tan crystals. Melting point 282–283°C (dec.), UPLC retention time 5.45 min; 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 9.16 (s, 1H), 8.36 (d, J = 8.4 Hz, 1H), 7.94 (dd, J = 8.0, 1.6 Hz, 1H), 7.61 (dt, J = 8.0, 1.6 Hz, 1H), 7.19 (dt, J = 7.6, 1.2 Hz, 1H), 5.59 (s, 2H), 4.33 (q, J = 7.2 Hz, 2H), 3.12 (s, 2H), 1.33 (t, J = 7.2 Hz, 3H).
Expression and Purification of Recombinant Proteins and the Histone Demethylase Assay
Full-length human LSD1 cDNA was subcloned into the pET15b bacterial expression vector (Novagen, Madison, WI) in frame with an N-terminal 6× HIS-tag and transformed into the BL21(DE3) strain of Escherichia coli. Following selection, expression and purification of recombinant LSD1 protein were performed as previously described.8 Briefly, expression of LSD1-HIS protein was induced by 1 mM isopropyl β-D-1-thiogalactopyranoside for 6 h at 25 °C. The HIS-tagged protein was purified using Ni-NTA affinity purification resin and column as recommended by the manufacturer (Qiagen, Valencia, California). Bound protein was eluted by imidazole and the eluate was dialyzed in PBS at 4°C. Enzymatic activity of LSD1 was examined using luminol-dependent chemiluminescence to measure the production of H2O2, as previously described.40 In brief, LSD1 activity was assayed in 50 mM Tris, pH 8.5, 50 mM KCl, 5 mM MgCl, 5 nmol luminol, and 20 μg/ml horseradish peroxidase with the indicated concentrations of H3K4me2 (1–21 aa) peptide as substrate. The integral values were calibrated against standards containing known concentrations of H2O2, and the activities expressed as pmols H2O2/mg protein/min. Reaction mixtures were incubated with or without 5 μg purified LSD1 in 50 mM Tris, pH 8.5, 50 mM KCl, 5 mM MgCl, 0.5% BSA, and 5% glycerol for 3 hr at 37°C. This reaction mixture was analyzed by Western blotting using antibodies (Millipore) that specifically recognize the dimethyl group of H3K4.
Western Blotting
Cytoplasmic and nuclear fractions were prepared for Western blot analysis using the NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Pierce, Rockford, IL). Primary antibodies against H3K4me2 were from Millipore. The pCNA monoclonal antibody was purchased from Oncogene Research Products (Cambridge, MA). Dye-conjugated secondary antibodies were used for quantification of Western blot results using the Odyssey Infrared Detection system and software (LI-COR Biosciences, Lincoln, NE). Determination of H3K4me2, SFRP2, HCAD and GATA4 levels in the Calu-6 cell line were determined as previously reported.32, 41
RNA isolation and qPCR
RNA was extracted using TRIzol reagents (Invitrogen, Carlsbad, CA). First-strand cDNA was synthesized using SuperScript III reverse transcriptase with an oligo(dT) primer (Invitrogen). qPCR was performed using the following primers: SFRP2 sense, 5′AAG CCT GCA AAA ATA AAA ATG ATG; SFRP2 antisense, 5′TGT AAA TGG TCT TGC TCT TGG TCT (annealing at 57.4°C); GATA4 sense, 5′GGC CGC CCG ACA CCC CAA TCT; GATA4 antisense, 5′ ATA GTG ACC CGT CCC ATC TCG (annealing at 64°C). qPCR was performed in a MyiQ single color real-time PCR machine (Bio-Rad, Hercules, CA) with GAPDH as an internal control.
Determination of cell viability
Calu-6 human anaplastic non-small cell lung carcinoma cells were maintained in culture using RPMI medium plus 10% fetal bovine serum. For the (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) reduction assay, 4000 cells/well were seeded in 100 μl medium in a 96-well plate and the cells were allowed to attach at 37°C in 5% CO2 for one day. The medium was aspirated and cells were treated with 100 μl of fresh medium containing appropriate concentrations of each test compound. The cells were incubated for 4 days at 37°C in 5% CO2. After 4 days 20 μL of the MTS reagent solution (Promega CellTiter 96 Aqueous One Solution Cell Proliferation Assay) was added to the medium. The cells were incubated for another 2 hours at 37°C under 5% CO2 environment. Absorbance was measured at 490 nm on a microplate reader equipped with SOFTmax PRO 4.0 software to determine the cell viability.
Molecular modeling studies
All molecular modeling studies were performed using the GOLD software package, version 5.1 (Cambridge Crytallographic Data Centre, Cambridge, UK). The X-ray coordinates of LSD1 (PDB code 2V1D) were downloaded from the Protein Databank,50 and the active site was defined as a sphere enclosing residues within 6 Å around the substrate-like peptide inhibitor co-crystal. The 3D structure of 22 was built using Chem 3D Pro software (version 12.0) and was energy minimized using MM2 force field for 1000 iterations and a convergence value of 0.001 kcal/mol/Å as the termination criterion. The energy minimized amidoxime 22 was docked in the binding site of LSD1 and scored using ChemScore.49 All poses generated by the program were visualized; however the pose with the highest fitness score was used for elucidating the binding characteristics of 22 in the LSD1 active site. The numbering sequence of amino acid residues in 2V1D is preserved throughout this paper.
Supplementary Material
Acknowledgments
The research described in this manuscript was supported by NIH Grant 1R01CA149095 (PMW) 1RO1CA98454 (RAC) and 1RO1CA51085 (RAC) and by grants from the Samuel Waxman Cancer Research Foundation (RAC, PMW) and the Susan B. Komen for the Cure Foundation (RAC). In addition, a portion of this work was supported by a grant from Progen Pharmaceuticals, Ltd., Brisbane, Queensland, Australia (PMW, RAC). RAC also serves as a consultant to Progen Pharmaceuticals.
ABBREVIATIONS
- LSD1
lysine-specific demethylase 1
- H3K4me1
histone 3 methyllysine 4
- H3K4me2
histone 3 dimethyllysine 4
- JmjC
Jumonji C domain-containing demethylase
- FAD
flavin adenine dinucleotide
- MAO
monoamine oxidase
- CoREST
RE1 silencing factor/neural restrictive silencing factor co-repressor protein
- SFRP
secreted frizzle-related protein
- HCAD
h-cadherin
- qPCR
quantitative polymerase chain reaction
- PCNA
proliferating cell nuclear antigen
References
- 1.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13(6):477–483. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Eugeni EE, Parthun MR, Freitas MA. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma. 2003;112(2):77–86. doi: 10.1007/s00412-003-0244-6. [DOI] [PubMed] [Google Scholar]
- 4.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 5.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1(4):287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 6.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 7.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125(3):467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 10.Tsukada Y, Zhang Y. Purification of histone demethylases from HeLa cells. Methods. 2006;40(4):318–326. doi: 10.1016/j.ymeth.2006.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huarte M, Lan F, Kim T, Vaughn MW, Zaratiegui M, Martienssen RA, Buratowski S, Shi Y. The fission yeast JMJ2 reverses histone H3 lysine 4 tri-methylation. J Biol Chem. 2007 doi: 10.1074/jbc.M703897200. [DOI] [PubMed] [Google Scholar]
- 12.Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochimica et biophysica acta. 2011;1815(1):75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Bannister AJ, Kouzarides T. Reversing histone methylation. Nature. 2005;436(7054):1103–1106. doi: 10.1038/nature04048. [DOI] [PubMed] [Google Scholar]
- 14.Jenuwein T. The epigenetic magic of histone lysine methylation. Febs J. 2006;273(14):3121–3135. doi: 10.1111/j.1742-4658.2006.05343.x. [DOI] [PubMed] [Google Scholar]
- 15.Schneider R, Bannister AJ, Kouzarides T. Unsafe SETs: histone lysine methyltransferases and cancer. Trends Biochem Sci. 2002;27(8):396–402. doi: 10.1016/s0968-0004(02)02141-2. [DOI] [PubMed] [Google Scholar]
- 16.Wilson JR, Jing C, Walker PA, Martin SR, Howell SA, Blackburn GM, Gamblin SJ, Xiao B. Crystal structure and functional analysis of the histone methyltransferase SET7/9. Cell. 2002;111(1):105–115. doi: 10.1016/s0092-8674(02)00964-9. [DOI] [PubMed] [Google Scholar]
- 17.Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12(2):198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 18.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15(18):2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 20.Liang G, Lin JC, Wei V, Yoo C, Cheng JC, Nguyen CT, Weisenberger DJ, Egger G, Takai D, Gonzales FA, Jones PA. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101(19):7357–7362. doi: 10.1073/pnas.0401866101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6(1):73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 22.Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13(6):563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A. 2007;104(19):8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stavropoulos P, Hoelz A. Lysine-specific demethylase 1 as a potential therapeutic target. Expert Opin Ther Targets. 2007;11(6):809–820. doi: 10.1517/14728222.11.6.809. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46(14):4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 26.Culhane JC, Szewczuk LM, Liu X, Da G, Marmorstein R, Cole PA. A mechanism-based inactivator for histone demethylase LSD1. J Am Chem Soc. 2006;128(14):4536–4537. doi: 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- 27.Shao GB, Ding HM, Gong AH. Role of histone methylation in zygotic genome activation in the preimplantation mouse embryo. In Vitro Cell Dev Biol Anim. 2008;44(3–4):115–120. doi: 10.1007/s11626-008-9082-4. [DOI] [PubMed] [Google Scholar]
- 28.Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J Am Chem Soc. 2009;131(48):17536–17537. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- 29.Binda C, Valente S, Romanenghi M, Pilotto S, Cirilli R, Karytinos A, Ciossani G, Botrugno OA, Forneris F, Tardugno M, Edmondson DE, Minucci S, Mattevi A, Mai A. Biochemical, Structural, and Biological Evaluation of Tranylcypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD2. J Am Chem Soc. 2010;132 doi: 10.1021/ja101557k. ePub. [DOI] [PubMed] [Google Scholar]
- 30.Ogasawara D, Suzuki T, Mino K, Ueda R, Khan MN, Matsubara T, Koseki K, Hasegawa M, Sasaki R, Nakagawa H, Mizukami T, Miyata N. Synthesis and biological activity of optically active NCL-1, a lysine-specific demethylase 1 selective inhibitor. Bioorganic & Medicinal Chemistry. 2011;19(12):3702–3708. doi: 10.1016/j.bmc.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 31.Ueda R, Suzuki T, Mino K, Tsumoto H, Nakagawa H, Hasegawa M, Sasaki R, Mizukami T, Miyata N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. Journal of the American Chemical Society. 2009;131(48):17536–17537. doi: 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Greene E, Stewart TM, Goodwin AC, Baylin SB, Woster PM, Casero RA. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. PNAS. 2007;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y, Stewart TM, Wu Y, Baylin SB, Marton LJ, Perkins B, Jones RJ, Woster PM, Casero RA., Jr Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin Cancer Res. 2009;15(23):7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki T, Miyata N. Lysine demethylases inhibitors. Journal of medicinal chemistry. 2011;54(24):8236–8250. doi: 10.1021/jm201048w. [DOI] [PubMed] [Google Scholar]
- 35.Bi X, Lopez C, Bacchi CJ, Rattendi D, Woster PM. Novel alkylpolyaminoguanidines and alkylpolyaminobiguanides with potent antitrypanosomal activity. Bioorg Med Chem Lett. 2006;16(12):3229–3232. doi: 10.1016/j.bmcl.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 36.Karginov VA, Nestorovich EM, Yohannes A, Robinson TM, Fahmi NE, Schmidtmann F, Hecht SM, Bezrukov SM. Search for cyclodextrin-based inhibitors of anthrax toxins: synthesis, structural features, and relative activities. Antimicrobial Agents and Chemotherapy. 2006;50(11):3740–3753. doi: 10.1128/AAC.00693-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji JS, Chen MJ, Wang QW, Sheng W. 1,3-Dinitro-2-chloro-5-trifluorotoluene and its bridged derivatives I. An investigation on the displacement reactions of bidentate nucleophiles towards aromatic rings. Chinese Journal of Chemistry. 1991;9:343–350. [Google Scholar]
- 38.Fecher A, Fretz H, Hilpert K, Riederer M. PCT Int’l Application WO 2005094816. Preparation of indolyl acetic acid derivatives as CRTH2 receptor inhibitors. 2005
- 39.Hyatt JA. Neber rearrangement of amidoximesulfonates. Synthesis of 2-amino-1-azirines. J Org Chem. 1981;46:3953–3955. [Google Scholar]
- 40.Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster PM, Casero RA., Jr Properties of purified recombinant human polyamine oxidase, PAOh1/SMO. Biochem Biophys Res Commun. 2003;304(4):605–611. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 41.Sharma S, Wu Y, Steinbergs N, Crowley M, Hanson A, Casero RAJ, Woster P. (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J Med Chem. 2010;53(14):5197–5212. doi: 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc Natl Acad Sci U S A. 2006;103(38):13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437(7057):432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- 44.Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(17):9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang GX, Liu W, Cui YF, Zhong XY, Tai S, Wang ZD, Shi YG, Li CL, Zhao SY. Reconstitution of secreted frizzled-related protein 1 suppresses tumor growth and lung metastasis in an orthotopic model of hepatocellular carcinoma. Digestive diseases and sciences. 2010;55(10):2838–2843. doi: 10.1007/s10620-009-1099-3. [DOI] [PubMed] [Google Scholar]
- 46.Toyooka S, Toyooka KO, Harada K, Miyajima K, Makarla P, Sathyanarayana UG, Yin J, Sato F, Shivapurkar N, Meltzer SJ, Gazdar AF. Aberrant methylation of the CDH13 (H-cadherin) promoter region in colorectal cancers and adenomas. Cancer research. 2002;62(12):3382–3386. [PubMed] [Google Scholar]
- 47.Wu LP, Wang X, Li L, Zhao Y, Lu S, Yu Y, Zhou W, Liu X, Yang J, Zheng Z, Zhang H, Feng J, Yang Y, Wang H, Zhu WG. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Molecular and cellular biology. 2008;28(10):3219–3235. doi: 10.1128/MCB.01516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Y, Murray-Stewart T, Wu Y, Baylin SB, Marton LJ, Woster PM, Casero JRA. Novel oligoamine analogues inhibit lysine-specific demethylase 1 (LSD1) induce re-expression of epigenetically silenced genes. Cancer Chemo Pharmacol. 2009;15:7217–7228. doi: 10.1158/1078-0432.CCR-09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones G, Willett P, Glen RC. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J Mol Biol. 1995;245(1):43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 50.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007;282(28):20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 51.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007 doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, Zhang H. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer research. 2011;71(23):7238–7249. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma SK, Hazeldine S, Crowley ML, Hanson A, Beattie R, Varghese S, Sennanayake TMD, Hirata A, Hirata F, Huang Y, Wu Y, Steinbergs N, Murray-Stewart T, Bytheway I, Casero J, Woster PMRA. Polyamine-based small molecule epigenetic modulators. MedChemComm. 2011 doi: 10.1039/c1031md00220a. ePub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 55.Bellevue FH, III, Boahbedason M, Wu R, Woster PM, Casero JRA, Rattendi D, Lane S, Bacchi CJ. Structural comparison of alkylpolyamine analogues with potent in vitro antitumor or antiparasitic activity. Bioorganic & Medicinal Chemistry Letters. 1996;6(22):2765–2770. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.