

Abstract
A respiratory irritant, acrolein is generated by overheating cooking oils or by domestic cooking using biomass fuels, and is in tobacco smoke, an occupational health hazard in the restaurant workplace. To better understand the metabolic role of the lung and to generate insights into the pathogenesis of acrolein-induced acute lung injury, SM/J (sensitive) and 129×1/SvJ (resistant) inbred mouse strains were exposed and the lung metabolome was integrated with the transcriptome profile. A total of 280 small molecules were identified and mean values (log 2 >0.58 or <−0.58, .p<0.05) were considered different for between-strain comparisons or within-strain responses to acrolein treatment. At baseline, 24 small molecules increased and 33 small molecules decreased in the SM/J mouse lung as compared to 129×1/SvJ mouse lung. Notable among the increased compounds was malonyl carnitine. Following acrolein exposure, several compounds indicative of glycolysis and branched chain amino acid metabolism increased similarly in both strains, whereas SM/J mice were less effective in generating metabolites related to fatty acid β-oxidation. These findings suggest management of energetic stress varies between these strains, and that the ability to evoke auxiliary energy generating pathways rapidly and effectively may be critical in enhancing survival during acute lung injury in mice.
Keywords: ARDS, Smoke Inhalation, Protein Folding, Mitochondrion, Beta-oxidation
Introduction
The array of inbred mouse strains available for biomedical research provides a useful resource to probe the genetics of complex traits. In earlier applications, various mouse strains were found to differ widely in sensitivity to infection with intracellular pathogens such as Mycobacterium and Listeria. Differences between sensitive mouse strains (BALB/cJ, C57BL/6J, NZW/LacJ) vs. resistant strains (129×1/SvJ, CBA/J, A/J) mapped to chromosome 1 and labeled the Bcg/Ict/Lsh locus [1, 2]. Subsequent studies soon identified a null mutation in natural resistance-associated macrophage protein 1 (Nramp1) which latter was named solute carrier family 11 (proton-coupled divalent metal ion transporters), member 1 (Slc11a1) because the protein product transports iron and other divalent metals within the lysosome/phagosome compartment to facilitate bacterial killing [3]. In another example, varying resistance of C3H/HeJ mice to the septic effects of bacterial lipopolysaccharide (endotoxin) can be traced to a functional mutation in the innate immunity sensor, toll-like receptor 4 (Tlr4) [4].
Increased availability of multiple mouse strains with high density single nucleotide polymorphism (SNP) maps has made mouse haplotype mapping analysis a powerful tool to interrogate the genetics of physiology and pathology. Haplotype mapping analysis can be especially useful in studies of conditions with considerable gene-environment interactions. For example, Liu et al. identified multiple candidate tumor suppressor genes using dense single nucleotide polymorphism mapping across mouse strains differing in their sensitivity to chemically-induced lung cancer [5]. Recently, we utilized haplotype mapping analysis to explore the genetics of acrolein-induced acute lung injury [6]. In that study, 40 strains were tested and an approximate 2.5-fold difference was noted in mean survival time. The polar extremes included SM/J as a sensitive strain and 129×1/SvJ as a resistant strain. Several genes, including activin A receptor type 1, were identified as contributing significantly to the phenotype.
The advent of high-content “metabolomics” platforms provides another useful method to probe disease characterization and toxicologic/pathologic responses [7, 8]. The unbiased systematic study of small molecule metabolites in a high-content manner allows a biochemical snapshot of cellular metabolism in its entirety [9]. These small molecules inform on an array of substrates, intermediates, and products involved in a host of diverse anabolic and catabolic metabolic pathways impacted by energetic stress, including nutrient processing and amino acid metabolism.
While the entire human metabolome, containing >7000 compounds, is incompletely characterized, platforms are emerging that allow simultaneous measurement of compounds numbering in the hundreds [10]. Obtaining a metabolomic profile under basal unperturbed conditions depicts the steady-state physiologic condition of the cell/tissue and can be used to screen phenotypic variability between two or more inbred mouse strains. Furthermore, assessing metabolomic responses after challenge with environmental toxins allows evaluation of functional changes in biochemical pathways that present in host response to stress. Again, comparison of these changes between genetically distinct mouse strains with differing susceptibility could provide further insight into possible biochemical mechanisms that alternately exacerbate or protect against injury. Recently, Lundy et al. described 4 patients with inborn metabolic errors of mitochondrial β-oxidation of long-chain fatty acids who presented with acute respiratory distress syndrome [11], although the study of intermediary metabolism has been largely ignored in experimental mechanistic studies into the pathogenesis of acute lung injury.
In this study, we obtained a 280 compound metabolomic profile in normal lung tissue from both the SM/J (acrolein-sensitive) and 129×1/SvJ (acrolein-resistant) mice. Metabolites that quantitatively differed between the strains under basal unperturbed conditions were identified. Next we assessed metabolite changes in lungs of both strains during acrolein exposure sufficient to induce acute lung injury differentially between these strains. Acrolein-induced time-dependent changes in multiple metabolites were observed and several of these alterations were discordant between the sensitive and resistant strains. This information was integrated with microarray transcriptome profiles to better understand the metabolic pathways that may be critical in lung injury
Methods
Experimental Design
This study was performed in accordance with the Institutional Animal Care and Use Committee of the University of Pittsburgh (Pittsburgh, PA) and mice were housed under specific pathogen free conditions. Two inbred mouse strains, SM/J (sensitive) and 129×1/SvJ (resistant) (6-8 wk female, n = 30/strain; The Jackson Laboratory, Bar Harbor, ME), were compared. These strains represented the opposite ends of the phenotypic spectrum of 40 analyzed strains [6]. Mice were exposed to filtered air (0 h, control) or acrolein (10 ppm for 6 or 12 h) as generated and monitored as described previously [12]. To reduce diurnal effects on metabolism, exposure starts were staggered to enable tissue collection at the same time range (2:00 -3:00 PM). Metabolome profiling was performed at Metabolon's laboratory (Durham, NC) as described previously [13, 14]. Briefly, sample tissues were homogenized in a minimal volume of deionized water containing a mixture of recovery standards and metabolites extracted with methanol (80% v/v in deionized water) containing a mixture of internal standards whose purpose is to allow chromatogram alignment (described in [13]). Extracts were separated into 3 aliquots, lyophilized, and prepared for a specific analytical workflow. Samples destined for gas chromatography-mass spectrometry (GC-MS) (Thermo-Finnegan Mat-95 XP, Thermo Fisher, Pittsburgh, PA) were handled as described [14], and samples destined for negative ion mode ultrahigh performance liquid chromatography mass spectrometry/mass spectrometry (uHPLC-MS/MS) or positive ion mode uHPLC-MS/MS platforms (LC: Surveyor; MS/MS: LTQ-FT, Thermo Fisher Corp, Pittsburgh, PA) were handled as described [13]. In contrast to the accurate mass and elution time tags used in shotgun proteomics [15], the metabolomics platform used combines accurate retention times and tandem mass spectrometry to identify metabolites [16]. This platform provided robust performance with exquisite control over both the chromatographic retention times and mass spectrometric fragmentation patterns of over 2400 biochemicals. The high reproducibility of these analytical parameters facilitates a library-based approach to the unambiguous identification of metabolites. To evaluate enzymes related to metabolite formation, re-analysis was performed using previously acquired microarray data with SM/J and 129×1/SvJ mice [6] following the completion of metabolome profiling. Additional details are provided in Supplemental Materials and Methods.
Results
Baseline differences in lung metabolome profile in sensitive (SM/J) mice compared to resistant (129×1/SvJ) mice
A total of 280 small molecules were identified in the lungs of SM/J and 129×1/SvJ mice. Values with log 2 means >0.58 or <−0.58 (i.e. 1.5-fold) (p<0.05) were considered significantly different between strains. Compared to 129×1/SvJ mouse lung, 24 small molecules were enriched and 33 small molecules were reduced in the SM/J mouse lung (Figure 1, Supplemental Table S1). The increased molecules were enriched in the carbohydrates pathway and the lysine metabolism subpathway (Supplemental Table S2). Increased molecules included (a) several sugars (fucose, fructose, ribose, ribulose, xylulose) (b) 1,3-dihydroxyacetone, (c) fructose-1-phosphate (d) N-acetylneuraminate (sialic acid), and (e) 2-aminoadipate. Decreased molecules were enriched in the long chain fatty acid sub-pathway (Supplemental Table S2). Amongst the decreased molecules were (a) several long chain fatty acids (linolenate, palmitate, myristolate, 10-nonadecanoate, among others), (b) the fructose/glucose-1,6-diphosphate isobar and (c) adenosine 3′-monophosphate. The levels of several carnitine derivatives were also discordant between the two strains. Malonyl carnitine and glutarylcarnitine were enriched in the lungs of SM/J mice while isovaleryl-, 2-methylbutyl-, butyryl-, and hexanoylcarnitine were present in much less amounts compared to 129×1/SvJ mice (Figures 1 and 2).
Figure 1. Small molecule metabolites in differential abundance between lungs of SM/J and 129×1/SvJ control untreated mice.
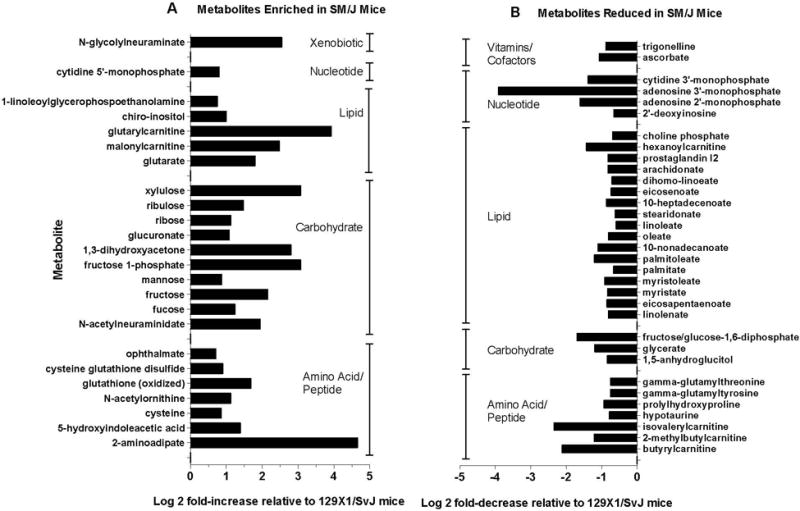
Of the 280 metabolites measured in lung, included compounds are those that differed by more than a factor of 1.5 (log 2 = +/− 0.58) between strains under basal conditions and achieved statistical significance (p < 0.05) by analysis of variance (ANOVA) with an all pairwise comparison procedure (Holm-Sidak method). Panel A: Compounds enriched in SM/J mouse lung compared to 129×1/SvJ mouse lung. Panel B: Compounds reduced in SM/J mouse lung compared to 129×1/SvJ mouse lung. Compound categorization within metabolic pathway was as defined using the Human Metabolome Database (HMDB).
Figure 2. Increased malonylcarnitine levels in lung of sensitive (SM/J) as compared to resistant (129×1/SvJ) mice at baseline and following acrolein exposure.
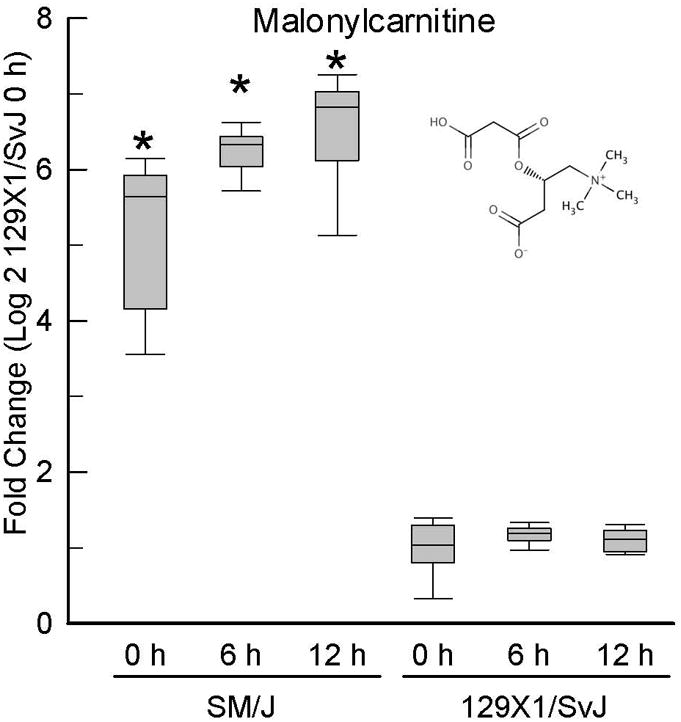
Mice were exposed to filtered air (0 h control) or acrolein (10 ppm) and metabolome profiling performed with lung tissue. Values are normalized to time-matched control 129×1/SvJ (filtered air 0 h) levels and plots indicate the median (line in box) with 25 and 75% confidence intervals (borders of the box) and 95% confidence intervals (error bars). *Significantly different (p <0.00001) from 129×1/SvJ control as determined by analysis of variance (ANOVA) with an all pairwise multiple comparison procedure (Holm-Sidak method).
Lung metabolome profile shared by sensitive (SM/J) mice and resistant (129×1/SvJ) mouse strains with acrolein exposure
The sensitive and resistant mouse strains had several similar changes in small molecule levels following acrolein exposure at 6 or 12 h. Compared to strain-matched control, 15 small molecules increased and 7 small molecules decreased nearly equivalently in the SM/J and 129×1/SvJ mouse lung following 6 h acrolein exposure (Supplemental Figures S1 – S4, Supplemental Table S1). The shared 6 h increased molecules were enriched in energy pathway, and the Krebs cycle sub-pathway (Supplemental Table S2). Individual increased molecules included: (a) citrate, (b) cis-aconitate, (c) putrescine, and (d) glucose. No pathway or sub-pathway was significantly enriched in decreased small molecules. The shared 6 h decreased molecules included: (a) prolylhydroxy-proline, (b) trans-4-hydroxyproline and (c) hypotaurine, among others.
At 12 h acrolein exposure, 15 small molecules increased and 13 small molecules decreased nearly equivalently in the SM/J and 129×1/SvJ mouse lung compared to strain-matched control (Supplemental Figures S1 – S4, Supplemental Table S1). The 12 h shared increased molecules also were enriched in energy pathway, and the Krebs cycle sub-pathway (Supplemental Table S2). Shared increased molecules in addition to citrate, cis-aconitite, and putrescine included (a) N-acetylphenylalanine, and (b) N-acetylleucine. No pathway or sub-pathway was significantly enriched in decreased small molecules. Notable decreased small molecules in addition to trans-4-hydroxy- and prolylhydroxy-proline (which were noted at 6 h) included: (a) glycerophosphorylcholine (GPC), (b) dehydroascorbate, (c) lactate, (d) 1-palmitoylglycerophosphoinositol, and (e) 1,3-dipalmitoylglycerol.
Small molecule metabolites that increased or decreased more in the sensitive (SM/J) mice compared to resistant (129×1/SvJ) mice following acrolein exposure
The sensitive and resistant mouse strains had more differing than similar changes in small molecule levels following 6 or 12 h acrolein exposure. At 6 h, 19 small molecules increased and 5 small molecules decreased more in the SM/J than in the 129×1/SvJ mouse lung compared to strain-matched control (Supplemental Figures S1 – S4, Supplemental Table S1). The increased molecules were enriched in amino acid pathway, and phenylalanine and tyrosine metabolism, valine, leucine and isoleucine metabolism, and bile acid metabolism sub-pathways (Supplemental Table S2). Individual increased molecules in SM/J lung included: (a) isoleucine, (b) phenylacetylglycine, (c) isovalerylcarnitine, and (d) taurocholate. No pathway or sub-pathway was significantly enriched in decreased small molecules. Decreased small molecules in SM/J lung at 6 h included: (a) dehydroascorbate, and (b) 7-alpha-hydroxycholesterol.
At 12 h, 28 small molecules increased and 6 small molecules decreased more in the SM/J and 129×1/SvJ mouse lung compared to strain-matched control (Supplemental Figures S1 – S4, Supplemental Table S1). The increased molecules were enriched in amino acid pathway and phenylalanine and tyrosine metabolism, valine, leucine and isoleucine metabolism, and tryptophan metabolism sub-pathway (Supplemental Table S2). Eleven small molecules that were increased more in SM/J than in 129×1/SvJ at 6 h were also increased more in SM/J than in 129×1/SvJ at 12 h and included: (a) isovalerylglycine, (b) butyrylcarnitine, (c) allantoin, and (d) taurocholate. Additional small molecules increased more in SM/J lung at 12 h included: (a) 3-indoxyl sulfate, and (b) gulono-1,4-lactone. No pathway or sub-pathway was significantly enriched in decreased small molecules. Decreased small molecules in SM/J lung at 12 h included: (a) chiro-inositol, (b) 7-ketocholesterol, and (c) glutarate (pentanedioate). Succinylcarnitine progressively increased in both strains following acrolein, although the response appeared more rapid and of greater magnitude in the SM/J strain (Figure 3).
Figure 3. Increased succinylcarnitine levels develop earlier in lung of sensitive (SM/J) as compared to resistant (129×1/SvJ) mice following acrolein exposure.
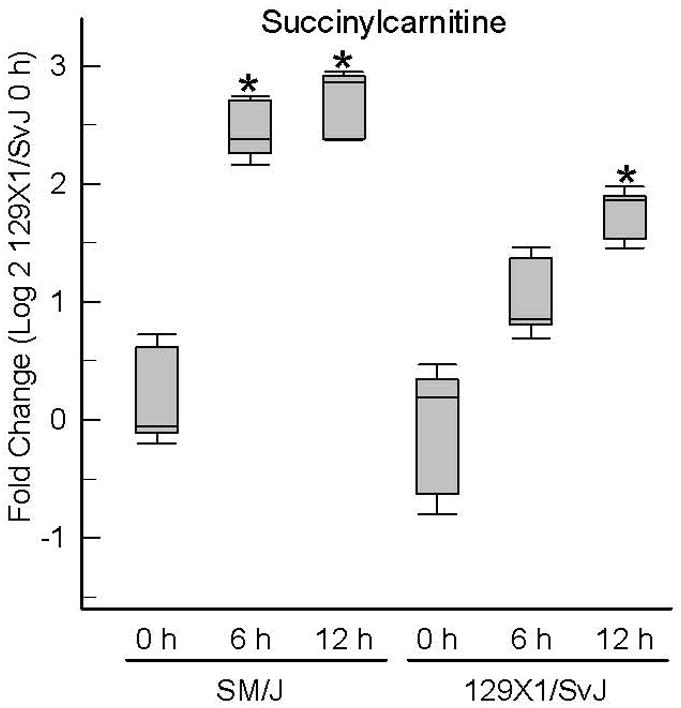
Mice were exposed to filtered air (0 h control) or acrolein (10 ppm) and tissue obtained and metabolome profiling performed. Values are normalized to time-matched control 129×1/SvJ (filtered air 0 h) levels and plots indicate the median (line in box) with 25 and 75% confidence intervals (borders of the box) and 95% confidence intervals (error bars). *Significantly different (p <0.001) from 129×1/SvJ control as determined by analysis of variance (ANOVA) with an all pairwise multiple comparison procedure (Holm-Sidak method).
Small molecule metabolites that increased or decreased more in the resistant (129×1/SvJ) mice compared to sensitive (SM/J) mice following acrolein exposure
The resistant 129×1/SvJ mouse also had metabolomic changes that were greater or less than changes in sensitive SM/J mice following 6 or 12 h acrolein exposure. At 6 h, 5 small molecules increased and 14 small molecules decreased more in the 129×1/SvJ than in the SM/J mouse lung compared to strain-matched control (Supplemental Figures S1 – S4, Supplemental Table S1). The increased molecules were enriched in amino acid and peptide pathway and medium chain fatty acid sub-pathway (Supplemental Table S2). Molecules increased more in the 129×1/SvJ lung included: (a) 3-hydroxybutyrate (BHBA) (Figure 4), (b) fructose, (c) 5-hydroxyindoleacetate and (d) N-acetylmannosamine. The decreased molecules were enriched in glutathione metabolism sub-pathway. At 6h, amongst the decreased small molecules in 129×1/SvJ lung included: (a) glutathione, oxidized (GSSG), (b) cysteine, and (c) gamma-glutamyl glutamate.
Figure 4. Increased 3-hydroxybutyrate (BHBA) levels develop earlier in lung of resistant (129×1/SvJ) as compared to sensitive (SM/J) mice following acrolein exposure.
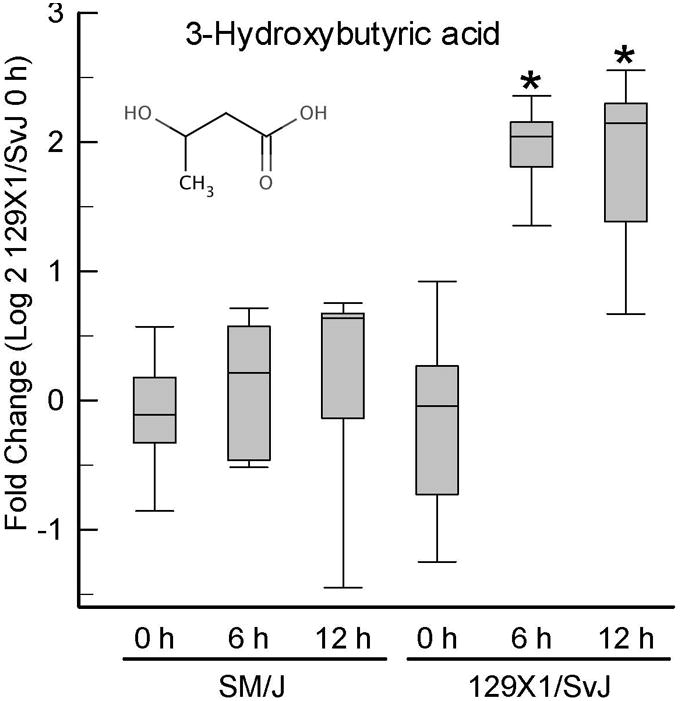
Mice were exposed to filtered air (0 h control) or acrolein (10 ppm) and tissue obtained and metabolome profiling performed. Values are normalized to time-matched control 129×1/SvJ (filtered air 0 h) levels and plots indicate the median (line in box) with 25 and 75% confidence intervals (borders of the box) and 95% confidence intervals (error bars). *Significantly different (p <0.001) from 129×1/SvJ control as determined by analysis of variance (ANOVA) with an all pairwise multiple comparison procedure (Holm-Sidak method).
At 12 h, 9 small molecules increased and 18 small molecules decreased more in the 129×1/SvJ than SM/J mouse lung compared to strain-matched control (Supplemental Figures S1 – S4, Supplemental Table S1). The increased molecules were enriched in fructose, mannose, galactose, starch, and sucrose metabolism sub-pathway (Supplemental Table S2). Two small molecules that were increased more in 129×1/SvJ than SM/J at 6 h were also increased more in 129×1/SvJ than in the SM/J at 12 h and included: (a) 3-hydroxybutyrate (BHBA) (Figure 4), and (b) fructose. Additional small molecules increased more in 129×1/SvJ lung at 12 h included: (a) 2-hydroxybutyrate (AHB), and (b) 2-hydroxy-3-methylbutyric acid (alpha-hydroxyisovalerate). Similar to 6 h, decreased small molecules in 129×1/SvJ lung at 12 h included: (a) glutathione, oxidized (GSSG), (c) cysteine, and (c) gamma-glutamylglutamate. Other decreased small molecules in 129×1/SvJ lung at 12 h were: (a) glutamate, (b) glycine, and (c) ribose.
Baseline differences in lung transcriptome profile in sensitive (SM/J) mice compared to resistant (129×1/SvJ) mice
Values with log 2 means >0.58 or <−0.58 (i.e. 1.5-fold) (p<0.05) were considered significantly different between strains. Before exposure, 720 (Supplemental Table S3) and 712 (Supplemental Table S4) transcripts were increased and decreased, respectively, in SM/J mouse lung as compared to 129×1/SvJ mouse lung. Using pathway analysis, the gene ontogeny categories enriched in increased transcripts included protein folding, carbohydrate binding, and response to wounding (Figure 5). No KEGG pathway was enriched in increased transcripts. The gene ontogeny categories enriched in decreased transcripts included extracellular matrix, immune response, and GTPase regulator activity (Figure 5). Similarly, the KEGG pathways enriched in decreased transcripts included ECM-receptor pathway (Supplemental Figure S5) and B Cell receptor signaling pathway (Supplemental Figure S6).
Figure 5. Metabolome pathways enriched in transcripts in sensitive (SM/J) as compared to resistant (129×1/SvJ) mouse lung.
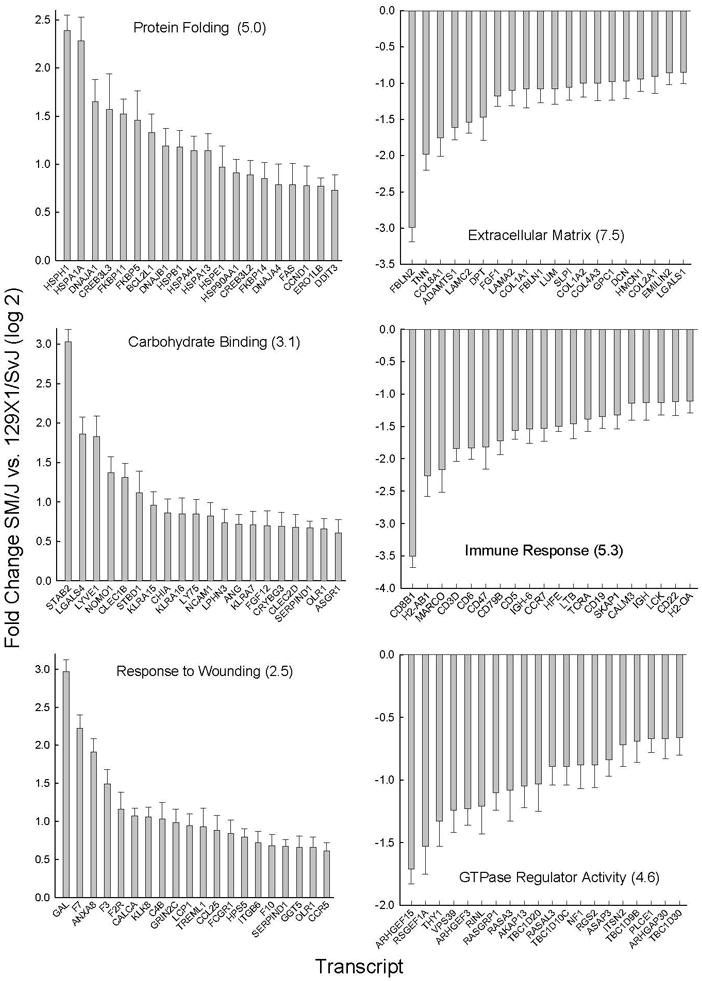
Pathway enrichment was assessed by Database for Annotation, Visualization, and Integrated Discovery (DAVID) using significant (p <0.05 by ANOVA) increased (log 2 mean >0.58) or decreased (log 2 mean <−0.58) microarray transcript levels. The statistically enriched pathways gene ontogeny (GO) categories (−log P value in parenthesis) with (Left) increased transcripts included: (Top) protein folding, (Middle) carbohydrate binding, and (Bottom) response to wounding. Increased transcripts were increased more in SM/J as compared to 129×1/SvJ mouse strains. The statistically enriched pathways gene ontogeny (GO) categories (−log P value in parenthesis) with (Right) decreased transcripts included: (Top) extracellular matrix part, (Middle) immune response, and (Bottom) GTPase regulator activity. Bars: Transcripts with greatest difference between strain (log 2 mean ± SE, n=5 mice/strain, p<0.05). Abbreviations: Current Entrez gene symbols.
Relationship between baseline metabolome and enzyme transcriptome
The increased metabolome molecules enriched in the carbohydrates pathway were accompanied with decreases in related degradation enzymes. For example, fructose is >4-fold higher and the metabolizing enzymes fructose bisphosphatase 1 and 2 transcripts are 2 - 3-fold lower in SM/J mice as compared to the 129×1/SvJ mice. Similarly, glucuronate is 2-fold higher and the metabolizing enzyme glucuronidase, beta transcript is >3-fold lower in SM/J mice as compared to the 129×1/SvJ mice. Amongst the metabolome molecules enriched in SM/J mice as compared to the 129×1/SvJ mice was the lysine metabolism degradation product, 2-aminoadipate (25-fold increased). This was accompanied by an increase in the formation enzyme aminoadipate-semialdehyde synthetase transcript (2.2-fold increased).
Additional differences in the baseline transcriptome included lipid biosynthesis and mitochondrion gene ontogeny categories, which were enriched in decreased transcripts (Figure 6). The decreased transcripts noted in lipid biosynthesis enzyme included stearoyl-Coenzyme A desaturase 1 (SCD1). Interestingly, SCD1 acts on cytoplasmic CoAs and several SCD1-dependent mononunsaturated fatty acid products including 10-nonadecenoate (19:1n9), myristoleate (14:1n5), palmitoleate (16:1n7), and oleate (18:1n9) were amongst the depleted molecules. Other decreased lipid biosynthesis transcripts included carnitine palmitoyltransferase 1c (CPT1C), lysophosphatidylcholine acyltransferase 1 (LPGAT1), acyl-CoA oxidase-like (ACOXL), solute carrier family 27 (fatty acid transporter), member 1 (SLC27A1), and ATP citrate lyase (ACLY).
Figure 6. Decreased transcripts in lipid biosynthesis and mitochondrion gene ontogeny (GO) categories present in sensitive (SM/J) as compared to resistant (129×1/SvJ) mouse lung.
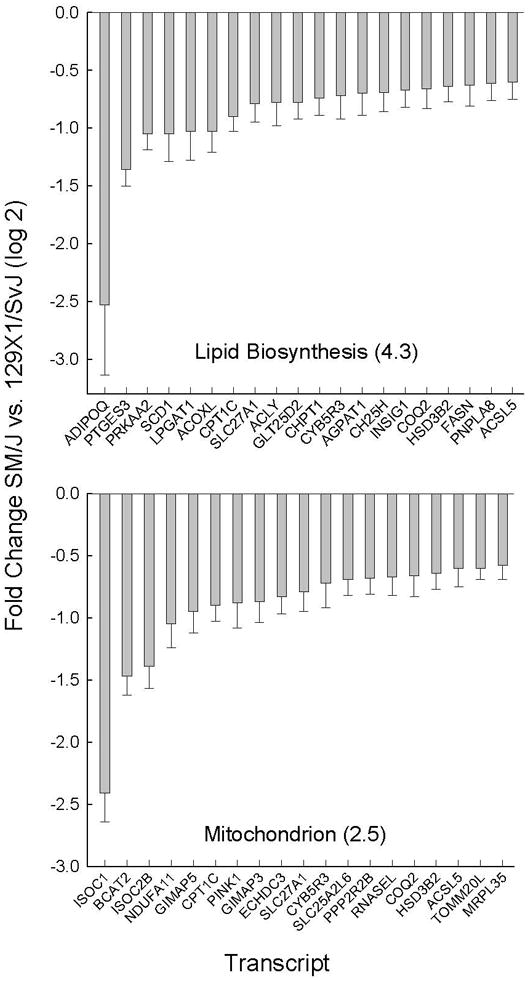
Pathway enrichment was assessed by Database for Annotation, Visualization, and Integrated Discovery (DAVID) using significant (p <0.05 by ANOVA) increased (log 2 mean >0.58) or decreased (log 2 mean <−0.58) microarray transcript levels. GO categories enrichment (−log P value in parenthesis). Bars are transcript levels with the greatest fold difference within each pathway are displayed (log 2 mean ± SE, n=5 mice/strain) that decreased in SM/J as compared to 129×1/SvJ mouse lung. Abbreviations: Current Entrez gene symbols.
Discussion
To our knowledge this is the first experimental study of the lung metabolome in mice. Using 1H-NMR Stringer et al. studied the metabolic consequences of sepsis-induced lung injury in humans [17] and Tang et al. [18] used similar techniques in rats exposed to intratracheal TiO2 nanoparticles. These studies, however, sampled only blood and urine, respectively, and were limited in the number of compounds measured. We were able to quantify 280 small molecules including metabolites associated with such diverse systems as amino acid, carbohydrate, lipid, and nucleotide synthesis/degradation, as well as a variety of vitamins and co-factors. We also compared these pulmonary metabolomic signatures in two genetically distinct inbred strains of mice (SM/J and 129×1/SvJ).
Before we further discuss our findings it is important first to present limitations of this study. First, our approach is essentially descriptive in experimental design and does not, by itself, provide information on a mechanistic basis for acute lung injury. Rather, we sought to obtain high-content information that screened potential metabolic pathways that account for strain-specific differences in response to injury that could then be exploited in mechanistic studies. In addition, the metabolomic profile is limited only to compounds detected in lung tissue, and a better understanding of complex processes such as energy utilization and nutritional homeostasis will require consideration in other systemic compartments (e.g., liver, or adipose tissue). The relationship of the steady-state metabolite level to activation or inhibition of a metabolic pathway is limited, inasmuch as a metabolite accumulation could reflect either activation of an upstream or inhibition of a downstream enzymatic step. Furthermore, steady-state levels determined at any single time could remain unchanged despite significant changes in flux through that pathway. A rigorous interpretation of these results will require ongoing refinement as one acquires new knowledge of precursor/product relationships, biochemical sites of regulation, and rate-limiting steps in multi-enzymatic pathways. In spite of these limitations we feel this approach has yielded productive leads to pursue in understanding of the role of the lung as a metabolic organ and new insights into the pathogenesis and genetic basis of acute lung injury.
Basal differences between SM/J and 129×/1SvJ mice
The inbred mouse strains used in this study display considerable phenotypic variability concerning a variety of traits. The SM/J mouse, for example, is exceptional in reduced post-weanling growth rate [19]. In addition, the SM/J mouse is deficient in the enzyme neuramindase 1 [20] and exhibits altered T Cell and B-cell responsiveness [21]. The latter may reflect the altered transcripts identified in the B Cell receptor signaling pathway (Supplemental Figure S6). The 129×1/SvJ mouse is sensitive to noise-induced hearing loss [22], while resistant to other respiratory insults including hyperoxia [23] and ozone [24]. Comparing the metabolomic profiles in unchallenged mice revealed marked differences in basal pulmonary metabolic signatures. A total of 24 compounds were elevated (≥1.5-fold) and 33 compounds were reduced in SM/J lung as compared to 129×1/SvJ lung, which is about 20% of the 280 compounds identified. Candidate compounds from both the enriched and reduced pools included multiple chemical classes (i.e. amino acids, carbohydrates, lipids, and nucleotides) suggesting that the biochemical differences between these strains are diverse and complex.
Several observations suggest alterations in energetics between these two strains. One striking difference was the level of several carnitine derivatives. Carnitine exists as either free carnitine or as a variety of acyl carnitines that arise via transferase reactions with a corresponding acyl Coenzyme A (acyl CoA) [25, 26]. The acyl groups are organic acid products of varying chain length derived from fatty acids, amino acids, and carbohydrates whose further participation in biochemical reactions require activation by conjugation to CoA as thioesters.
The best understood role of carnitine is facilitation of fatty acid transport into the mitochondria for energy production as substrates for fatty acid β-oxidation [27] (Figure 7). Long chain fatty acyl-CoA in the cytosol cannot permeate the mitochondrial membrane and carnitine acyltransferases (CPT1s) reversibly transfers the acyl group from acyl-CoA to carnitine. The resultant acyl-carnitine is translocated into the mitochondrial matrix where a second carnitine acyl transferase (CPT2) conjugates the fatty acid back to CoA and provides the input for fatty acid β-oxidation. In addition, acetyl-CoA or other acyl-CoA derivatives can accumulate to high levels within the mitochondria and CPT2 may facilitate the formation of acetyl or acyl-carnitine with translocation out of the mitochondria and restoration of the acyl-CoA:free CoA ratio [28].
Figure 7. Malonyl CoA inhibits mitochondrial acyl-CoA transport.
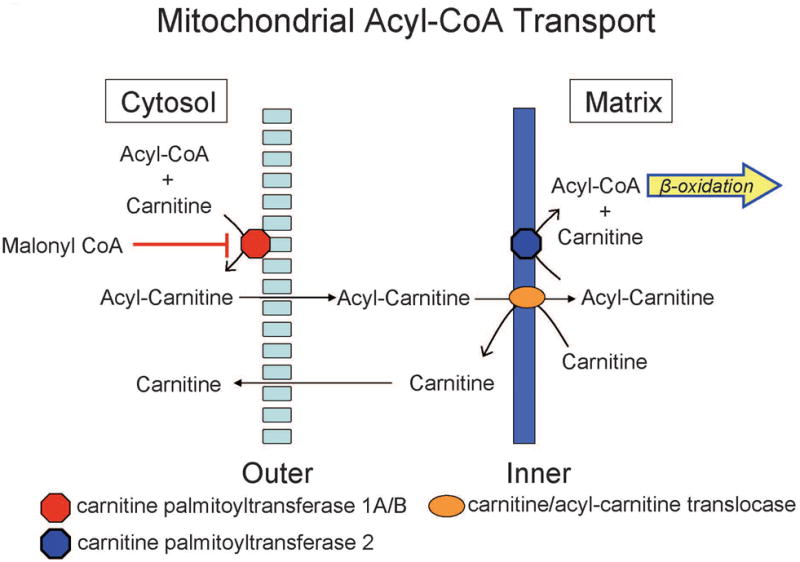
Fatty acids entry into mitochondria can be inhibited by malonyl CoA and diminishes capacity to employ fatty acid beta-oxidation as an energy source during stress induced by acrolein.
Acyl carnitines are biomarkers for specific enzyme deficiencies in certain inborn errors of metabolism presumably by tracking the resulting accumulation of the specific corresponding acyl-CoA metabolite. For example, dehydrogenase deficiencies specific for very long chain acyl CoA, medium-chain acyl CoA, or short-chain acyl CoA can be detected by accumulation of tetradecenoyl carnitine, octanoyl carnitine, and butyryl carnitine, respectively [29].
One intriguing feature is the relatively high lung malonyl carnitine in SM/J mice. Based on the above discussion, we propose this reflects high malonyl CoA levels. Malonyl CoA is formed by the carboxylation of cytoplasmic acetyl-CoA and is recognized as a key regulator of lipid and energy metabolism [30]. Malonyl CoA can impair entry of long-chain acylcarnitine esters into the mitochondria (Figure 7) via inhibition of CPT1 with attendant inhibition of fatty acid β-oxidation and compromise of the mitochondrial respiratory chain [31-33]. Moreover, malonyl CoA serves as the initial carbon source for fatty acid synthesis by fatty acid synthase [34]. The malonyl CoA/acetyl CoA balance is dynamically regulated by the opposing actions of acetyl-CoA carboxylase (ACC) and malonyl-CoA decarboxylase (MCD). Thus, SM/J mice appear ill-prepared to utilize fat as an energy substrate while exhibiting a propensity toward fat synthesis and storage. Despite their small size arising from growth retardation in the post-natal period, adult SM/J mice fed a high-fat diet tend to gain weight (specifically in the form of fat) more rapidly than other strains and thus have been proposed as a model for the study of obesity and metabolic syndrome [35].
Other differences observed include lower levels of isovaleryl carnitine, 2-methylbutyl carnitine, and butyryl carnitine in SM/J compared to 129×1/SvJ. These carnitine intermediates could reflect analogous CoA conjugates obtained during degradation of branched-chain amino acids leucine, isoleucine, and valine, respectively [26, 29, 36, 37]. Reduction of these moieties in SM/J mice suggests an enhanced flux of amino acid-derived substrates through oxidative decarboxylation reactions with subsequent generation of acetyl CoA and succinyl CoA to support the citric acid cycle in compensation for compromised energetics described above. Alternatively, the decrease could reflect decreased branched chain amino transferase 2 (BCAT2) transcripts in lungs of SM/J mice compared to 129×1/SvJ observed in the microarray data. Branched chain amino acids can serve as an energy source in peripheral tissues during stress [38, 39].
These strains also differed in compounds relevant to carbohydrate metabolism. For example, lung fructose-1,6-diphosphate was reduced in SM/J as compared to 129×1/SvJ mouse strain. The enzyme phosphofructokinase, muscle (PFKM: aka 6-phosphofructo-1-kinase), which converts fructose-6-phosphate to fructose-1,6-diphosphate, represents the rate-limiting step in glycolysis [40]. This highly-regulated enzyme is allosterically inhibited by ATP and cytoplasmic citrate while AMP and ADP enhance enzymatic activity [41]. A complex role for fructose-1,6-diphosphate in acute lung injury has emerged inasmuch as fructose-1,6-diphosphate attenuated endotoxin-induced edema in mouse lung [42] but augmented paraquat-induced edema in isolated dog lung [43], [44]. SM/J mice also seem to have impaired glucose tolerance [45] and phosphofructokinase C (Pfkp) has been identified as a positional candidate gene accounting for gene-diet-interactions identified in independent QTL analyses of obesity-like traits [46].
Several steps discussed above are specific targets for AMP kinase (AMPK), which appears to have a master role in regulation of cellular energetics [47]. AMPK senses specific energy requirements and regulates the balance between anabolic and catabolic processes. For example, malonyl CoA (and hence regulation of lipolysis/lipogenesis) is a central target for AMPK which phosphorylates both ACC and MCD resulting in inhibition and activation, respectively. This relationship would decrease malonyl CoA (and presumably malonyl carnitine) [48-50] and increase energy derivation via fatty acid β-oxidation. Analogous to its effects on the malonyl CoA/acetyl CoA balance, the PFKM step in glycolysis is another control point by AMP kinase, albeit indirectly via 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 1 (PFKFB1) modulation, which produces fructose-2,6-diphosphate, a powerful positive allosteric regulator of PFKM [51]. This explains, in part, an AMPK-dependent stimulation of glycolysis [52]. A possible contribution of AMPK to strain-dependent differences was also supported by the decreased PRKAA2 transcript that encodes alpha 2 catalytic subunit of AMPK in SM/J compared to 129×1/SvJ mouse lung (Figure 6).
Other differences in specific metabolites may be associated with differences of relevant transcripts revealed by microarray analysis. For example, the robust accumulation of 2-aminoadipate in lungs of SM/J mice could be a consequence of decreased transcripts for aminoadipate-semialdehyde synthease, which utilizes 2-aminoadipate as a metabolite in the lysine pathway. SM/J mice display significant reductions in both isoforms of fructose bisphosphatase (FBP1, FBP2) (Supplementary data Table S4). While this might be expected to change fructose-1,6-diphosphate in the opposite direction from the observed increase in SM/J, it may, instead, represent a compensatory alteration geared to promote glycolytic flux in a compromised pathway.
One common component that serves to connect the multiple strain-dependent differences observed in energetics described above is Coenzyme A. CoA is a co-factor necessary for activation of organic acids derived from fatty acids, glycolysis, and amino acids to facilitate their oxidation and subsequently deliver energy-yielding electrons to the respiratory chain. Transcripts that encode enzymes involved in CoA utilization increased in SM/J compared to 129×1/SvJ included: acyl-CoA dehydrogenase family member 9 (ACAD9), acyl CoA thioesterase (ACOT1), malonyl CoA decarboxylase (MLYCD), methylmalonyl CoA mutase (MUT) (Supplemental Table S3). Transcripts that encode enzymes involved in CoA utilization decreased in SM/J compared to 129×1/SvJ included: ATP citrate lyase (ACLY), acyl CoA synthetase long-chain family member 5 (ACSL5), acyl CoA oxidase-like (ACOCL), 2-4-dienoyl-CoA reductase 2 (DECR2), enoyl CoA hydrolase domain-containing 3 (ECHDC3), carnitine palmitoyltransferase 1C (CPT1C), fatty acid synthase (FASN) (Supplemental Table S4). In addition, transmembrane transport of energy substrates might differ between strains because of differential expression of the transcripts encoding members of solute carrier protein family. SM/J mice show decreased SLC16A13 (monocarboxylic acids), SLC27A1 (fatty acids), SLC38A1 (neutral amino acids) and increased SLC25A29 (mitochondrial, palmitoylcarnitine), SLC5A12 (Na+/glucose co-transporter), SLC7A8 (cationic amino acids) (Supplemental Tables S3, S4). Together, these findings point to significant differences between the SM/J and 129×1/SvJ strains of mice in a number of basic pathways pertinent to energetics.
Metabolomic response to acrolein-induced lung injury
The tissue response to injury is undeniably complex and simultaneously reflects alterations that arise from multiple processes including damage to cellular constituents, inflammatory responses, tissue repair, and potential reprogramming of energy metabolism. Therefore, it is not unexpected that many metabolites were altered after acrolein exposure. In addition, the temporal course of changes varied considerably with many metabolites. Some response patterns like the changes in metabolites involved in histidine metabolism and Krebs cycle appeared relatively conserved between strains. Krebs cycle intermediates, citrate and cis-aconitate, were rapidly and robustly increased following acrolein exposure. In general, increased carbohydrate substrates included glucose, fructose, and maltose, among others, that may serve as energy sources through glycolysis. Other compounds, (e.g., phenylalanine/tyrosine metabolites) changed in similar directions but differed in magnitude and/or speed of response and may reflect the differential sensitivity to succumbing to acute lung injury.
Increased branched chain amino acids and corresponding keto acids in both strains suggest that the lung might be able to utilize these energy sources during injury. However, only the SM/J mice demonstrate persistent elevation of several of the carnitine derivatives during lung injury. Beyond its simple role as an intermediate metabolite, isovalerylcarnitine can induce calpain proteases [53] and modulate immune function [54]. Acrolein-induced elevation in glycine conjugates, isovaleryl glycine and phenylacetylglycine (PAG), in the SM/J mice are also presumably derived from high levels of the respective CoA derivatives. This again points to dysfunctional CoA conjugation required for activation of the organic acids facilitating their further oxidation and energy-yielding potential. Elevated urinary and tissue PAG is related to phospholipid accumulation during experimental phospholipidosis [55].
The sensitive SM/J mice had a surprising accumulation of bile acid metabolites (e.g., taurocholate, taurodeoxycholate, or tauro-beta-muricholate) as early as 6 h, little change smaller increase taurocholate at 12 h or even decreases were observed in 129×1/SvJ. The origin of these moieties, normally associated with liver, remains obscure. Nonetheless, it is tempting to speculate that these cytotoxic compounds could further amplify lung damage from acrolein. Bile acids within the lung are thought to play a role in acute lung injury observed in aspiration pneumonia [56] or in infants born to mothers with intrahepatic cholestasis [57]. In brown adipose tissue and muscle, bile acids are sensed by G protein-coupled bile acid receptor 1 which triggers an increase in energy expenditure and attenuates diet-induced obesity [58].
Lipid metabolism during injury was clearly different between strains. For example, lung 3-hydroxybutyrate increased robustly during injury in resistant 129×1/SvJ mice but was unchange in sensitive SM/J mice (Figure 4). A terminal product of the fatty acid β-oxidation, increased 3-hydroxybutyrate typically reflects fat utilization as an energy source. This response is likely to compensate for the increased energetic demands following tissue damage that require auxiliary pathways for ATP-derived energy. The sensitive SM/J mice lack this response and, thus, would be compromised in meeting an enhanced energy demand. The high basal level of the negative-regulator of fatty acid transport/oxidation, malonyl CoA, in SM/J mice was maintained throughout acrolein exposure and provides a potential mechanism for β-oxidation inhibition. The increase in several medium chain, long chain, and essential fatty acids early in SM/J lung compared to 129×1/SvJ lung also suggests that the flux of fatty acid-derived substrates into the β-oxidation path might be impaired in the sensitive strain. A schematic model outlining these effects is provided in Figure 7. Previously, we identified lipid metabolism as a pathway enriched in transcripts that were modulated differentially following acrolein exposure [6].
In summary, the acrolein-induced acute lung injury is accompanied by a metabolome pattern of energetic stress. A mouse strain sensitive to acute lung injury exhibited diminished ability to generate metabolites of fatty acid β-oxidation before and during acrolein exposure. Together our findings suggest that the ability to evoke auxiliary energy generating pathways rapidly and effectively may be critical in enhancing survival.
Supplementary Material
Acknowledgments
This study was supported by the NIH: (G.D.L): ES015675, HL077763, HL085655, (M.M.): HG003749, LM009662, (N.K.): HL084932, HL095397
Footnotes
Conception and design: KB, MM, NK, GDL
Analysis and interpretation: JPF, MM, DCA, JM, VJC, KB, ASJ, LJV, KAB, HPV, RAD, KG, SU, QL, ZH, NK, GDL
Drafting the manuscript for important intellectual content: JPF, KB, GDL
References
- 1.Malo D, Vogan K, Vidal S, Hu J, et al. Haplotype Mapping and sequence analysis of the mouse Nramp gene predict suseceptibility to infection with intracellular parasites. Genomics. 1994;23:51–61. doi: 10.1006/geno.1994.1458. [DOI] [PubMed] [Google Scholar]
- 2.Skamene E, Schurr E, Gros P. Infection genomics: Nramp1 as a major determinant of natural resistance to intracellular infections. Ann Rev Med. 1998;49:275–287. doi: 10.1146/annurev.med.49.1.275. [DOI] [PubMed] [Google Scholar]
- 3.Jaboda N, Jankowski A, Dougaparsad S, Picard V, et al. Natural resistance to intracellular infections: natural resistance-associated protein 1 (NRAMP1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J Exp Med. 2000;192:1237–1247. doi: 10.1084/jem.192.9.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poltorak A, He X, Smirnova I, Liu MY, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScSr mice: Mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 5.Liu P, EWang Y, Vikis H, Maciag A, et al. Candidate lung tumor susceptibility genes identified through whole-genome association analyses in inbred mice. Nat Genet. 2006;38:888–895. doi: 10.1038/ng1849. [DOI] [PubMed] [Google Scholar]
- 6.Leikauf GD, Concel VJ, Liu P, Bein K, et al. Haplotype association mapping of acute lung injury in mice implicates activin A receptor, type 1. Am J Respir Crit Care Med. 2011 doi: 10.1164/rccm.201006-0912OC. [Epub ahead of print) PMID: 21297076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.German JB, Hammock BD, Watkins SM. Metabolomics: building on a century of biochemistry to guide human health. Metabolomics. 2005;1:3–9. doi: 10.1007/s11306-005-1102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson JK, Connelly J, Lindon JC, Holmes E. Metabolomics: a platform for studying drg toxicity and gene function. Nat Rev Drug Discov. 2002;1:153–161. doi: 10.1038/nrd728. [DOI] [PubMed] [Google Scholar]
- 9.Roessner U, Browne J. What is metabolomics all about? Biotechniques. 2009;46:363–365. doi: 10.2144/000113133. [DOI] [PubMed] [Google Scholar]
- 10.Wishart DS, Knox C, Guo AC, Eisner R, et al. HMDB: a knowledgebase of the human metabolome. Nucleic Acids Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundy CT, Shield JPH, Kvittingen EA, Vinorum OJ, et al. Acute respiratory distress syndrome in long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial trifunctional protein deficiencies. J Inherit Metab Dis. 2003;26:537–541. doi: 10.1023/a:1025995813914. [DOI] [PubMed] [Google Scholar]
- 12.Leikauf GD, Leming LM, O'Donell JR, Doupnik CA. Bronchial responsiveness and inflammation in guinea pigs exposed to acrolein. J Appl Physiol. 1989;66:171–178. doi: 10.1152/jappl.1989.66.1.171. [DOI] [PubMed] [Google Scholar]
- 13.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6657. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 14.Lawton KA, Berger A, Mitchell M, Milgram KE, et al. Analysis of the adult plasma metabolome. Pharmacogenonmics. 2008;9:383–397. doi: 10.2217/14622416.9.4.383. [DOI] [PubMed] [Google Scholar]
- 15.Strittmatter EF, Ferguson PL, Tang K, Smith RD. Proteome analyses using accurate mass and elution time peptide tags with capillary LC time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 2003;14:980–991. doi: 10.1016/S1044-0305(03)00146-6. [DOI] [PubMed] [Google Scholar]
- 16.Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform. 2010;2:9. doi: 10.1186/1758-2946-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stringer KA, Serkova NJ, Karnovsky A, Guire K, et al. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma 1H-nuclear magnetic resonance quantitative metabolomics and computional analysis. Am J Physiol Lung Cell Molec Physiol. 2011;300:L4–L11. doi: 10.1152/ajplung.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang M, Zhang T, Xue Y, Wang S, et al. Dose-dependent in vivo metabolic characteristics of titanium dioxide nanoparticles. J Nanosci Nanotechnol. 2010;10:8575–8583. doi: 10.1166/jnn.2010.2482. [DOI] [PubMed] [Google Scholar]
- 19.Kramer MG, Vaughn TT, Pletscher LS, King-Ellison E, et al. Genetic variation in body weight growth and composition in the intercross Large (LG/J and Small (SM/J) inbred strains of mice. Genet Mol Biol. 1998;21:211–218. [Google Scholar]
- 20.Womack JE, Yan DL, Potier M. Gene for neuraminidase activity on mouse chromosome 17 near h-2: pleiotroic effects on multiple hydrolases. Science. 1981;212:63–65. doi: 10.1126/science.7209520. [DOI] [PubMed] [Google Scholar]
- 21.Engel D, Clark EA, Held L, Kimball H, Clagett J. Immune responsiveness of SM/J mice. Cellular characteristics and genetic analysis of hyperresponsiveness to B cell mitogens. J Exp Med. 1981;154:726–736. doi: 10.1084/jem.154.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gratton MA, Eleftheriadou A, Garcia J, Verduzco E, et al. Noise-induced changes in gene expression in the cochlaeae of mice differing in their susceptibility to noise damage. Hear Res. 2010 doi: 10.1016/j.heares.2010.12.014. In Press, Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prows DR, Hafertepen AP, Gibbons WJ, Jr, Winterberg AV, Nick TG. A genetic mouse model to investigate hyperoxic acute lung injury survival. Physiol Genomics. 2007;30:262–270. doi: 10.1152/physiolgenomics.00232.2006. [DOI] [PubMed] [Google Scholar]
- 24.Vancza EM, Galdanes K, Gunnison A, Hatch G, Gordon T. Age, strain, and gender as factors for increased sensitivity of the mouse lung to inhaled ozone. Toxicol Sci. 2009;107:535–543. doi: 10.1093/toxsci/kfn253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma S, Black SM. Carnitine homeostasis, mitochondrial function, and cardiovascular disease. Drug Discov Today Dis Mech. 2009;6:e-31–e-39. doi: 10.1016/j.ddmec.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bremer J. Carnitine - metabolism and function. Physiol Rev. 1983;63:1420–1480. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- 27.Kerner J, Hoppel C. Fatty acid import into mitochondria. Biochim Biophys Acta. 2000;1486:1–17. doi: 10.1016/s1388-1981(00)00044-5. [DOI] [PubMed] [Google Scholar]
- 28.Lysiak W, Lilly K, diLisa F, Toth PP, Bieber LL. Quantification of the effect of L-carnitine on the levels of acid soluble short-chain acyl CoA and CoA in rat heart and liver mitochondria. J Biol Chem. 1988;269 [PubMed] [Google Scholar]
- 29.Rinaldo P, Cowan TM, Matern D. Acylcarnitine profile analysis. Genet Med. 2008;10:151. doi: 10.1097/GIM.0b013e3181614289. [DOI] [PubMed] [Google Scholar]
- 30.Saggerson D. Malonyl-CoA, a Key signaling molecule in mammalian cells. Ann Rev Nutr. 2008;28:253–272. doi: 10.1146/annurev.nutr.28.061807.155434. [DOI] [PubMed] [Google Scholar]
- 31.Paulson DJ, Ward KM, Shug AL. Malonyl CoA inhibition of carnitine palmitoyltrasferase in rat heart mitochondria. FEBS Lett. 1984;176:381–384. doi: 10.1016/0014-5793(84)81201-6. [DOI] [PubMed] [Google Scholar]
- 32.Rasmussen BB, Holmback UC, Volpi E, Morio-Liondore B, et al. Malonyl coenzyme A and regulation of functional carnitine palmitoyltransferase-1 activity and fat oxidation in human skeletal muscle. J Clin Invest. 2002;110:1687–1693. doi: 10.1172/JCI15715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saggerson D. Carnitine acyltransferase activities in rat liver and heart measured with palmitoyl-CoA and octanoyl-CoA. Latency, effects of K+, bivalent metal ions, and malonyl CoA. Biochem J. 1982;202:397–405. doi: 10.1042/bj2020397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cinti DL, Cook L, Nagi MN, Suneja SK. The fatty acid chain elongation system in mammalian endoplasmic reticulum. Prog Lipid Res. 1992;31:1–51. doi: 10.1016/0163-7827(92)90014-a. [DOI] [PubMed] [Google Scholar]
- 35.Cheverud J, Pletscher LS, Vaughn TT, Marshall B. Differential response to dietary fat in large (LG/J) and small (SM/J) inbred mouse strains. Physiol Genomics. 1999;1:33–39. doi: 10.1152/physiolgenomics.1999.1.1.33. [DOI] [PubMed] [Google Scholar]
- 36.Paul HS, Adipi SA. Effect of carnitine on branched chain amino acid oxidation. J Clin Chem Biochem. 1978;234:E494–E499. doi: 10.1152/ajpendo.1978.234.5.E494. [DOI] [PubMed] [Google Scholar]
- 37.Platell C, Kong SE, McCauley R, Hall JC. Branched-chain amino acids. J Gastroenterol Hepatol. 2000;15:706–717. doi: 10.1046/j.1440-1746.2000.02205.x. [DOI] [PubMed] [Google Scholar]
- 38.Ferrando AA, Williams BD, Stuart CA, Lane HW, Wolfe RR. Oral branched-chain amino acids decrease whole-body proteolysis. J Parenteral Enteral Nutr. 1995;19:47–54. doi: 10.1177/014860719501900147. [DOI] [PubMed] [Google Scholar]
- 39.Shinnick FL, Harper AE. Branched-chain amino acid oxidation in isolated rat tissue preparations. Biochim Biophys Acta. 1976;437:477–486. doi: 10.1016/0304-4165(76)90016-7. [DOI] [PubMed] [Google Scholar]
- 40.Lehninger AL. Biochemistry: The Molecular Basis of Cell Structure and Function. Worth Publishers, Inc.; New York: 1975. [Google Scholar]
- 41.Kemp RG, Foe LG. Allosteric regulatory properties of muscle phosphofructokinase. Mol Cell Biochem. 1983;57:147–154. doi: 10.1007/BF00849191. [DOI] [PubMed] [Google Scholar]
- 42.Yin H, Jin XB, Gong Q, Yang H, et al. Fructose-1,6-diphosphate attenuates acute lung injury induced by lipopolysaccharide in mice. Int Immunolpharmacol. 2008;8:1842–1847. doi: 10.1016/j.intimp.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Shibamoto T, Parker JC. Fructose-1,6-diphosphate augments paraquat injury in isolated dog lungs. J Appl Physiol. 1991;71:1830–1835. doi: 10.1152/jappl.1991.71.5.1830. [DOI] [PubMed] [Google Scholar]
- 44.Sillero MAG, Sillero A, Sols A. Enzymes involved in fructose metabolism in liver and the glyceraldehyde metabolic crossroads. Eur J Biochem. 1969;10:345–350. doi: 10.1111/j.1432-1033.1969.tb00696.x. [DOI] [PubMed] [Google Scholar]
- 45.Ehrich TH, Kenney-Hunt JP, Pletscher LS, Cheverud JM. Genetic variation and correlation of dietary response in an advanced intercross mouse line produced from divergent growth lines. Genet Res Camb. 2005;85:211–222. doi: 10.1017/S0016672305007603. [DOI] [PubMed] [Google Scholar]
- 46.Ehrich TH, Hrbek T, Kenney-Hunt JP, Pletscher LS, et al. Fine-mapping gene-by-diet interactions on chromosome 13 in a LG/J × SM/J murine model of obesity. Diabetes. 2005;54:1863. doi: 10.2337/diabetes.54.6.1863. [DOI] [PubMed] [Google Scholar]
- 47.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 48.Park H, Kaushik V, Constant S, Prentki M, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to excercise. J Biol Chem. 2002;277:32571–32577. doi: 10.1074/jbc.M201692200. [DOI] [PubMed] [Google Scholar]
- 49.Ruderman N, Park H, Kaushik V. AMPK as a metabolic switch in rat muscle, liver, and adipose tissue after excercise. Acta Physiol Scand. 2003;178:435–442. doi: 10.1046/j.1365-201X.2003.01164.x. [DOI] [PubMed] [Google Scholar]
- 50.Saha AK, Schwarsin AJ, Roduit R, Masse F, et al. Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and AMP-activated protein kinase activator 5-aminoimidazole-4-carboxaminde-a-beta-D-ribofuranoside. J Biol Chem. 2000;275:24279–24283. doi: 10.1074/jbc.C000291200. [DOI] [PubMed] [Google Scholar]
- 51.Chesney J. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphotase and tumor cell glycolysis. Curr Opin Clin Nutr Metab Care. 2006;9:535–539. doi: 10.1097/01.mco.0000241661.15514.fb. [DOI] [PubMed] [Google Scholar]
- 52.Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycoloysis through AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- 53.Pontremoli S, Melloni E, Viotti PL, Michetti M, et al. Isovalerylcarnitine is a specific activator of high calcium requireing calpain forms. Biochem Biophys Res Commun. 1990;167:373–380. doi: 10.1016/0006-291x(90)91775-n. [DOI] [PubMed] [Google Scholar]
- 54.Ferrara F, Bertelli A, Falchi M. Evaluation of carnitine, acetylcarnitine, and isovalerylcarnitine on immune function and apoptosis. Drugs Exp Clin Res. 2005;31:109–114. [PubMed] [Google Scholar]
- 55.Delaney J, Neville WA, Swain A, Miles A, et al. Phenylacetylglycine, a putative biomarker of phospholipidosis: its origins and relevance to phospholipid accumulation using amiodarone treated rats as a model. Biomarkers. 2004;9:271–290. doi: 10.1080/13547500400018570. [DOI] [PubMed] [Google Scholar]
- 56.Wu YC, Hsu PK, Su KC, Liu LY, et al. Bile acid aspiration in suspected ventilator-associated pneumonia. Chest. 2009;136:118–124. doi: 10.1378/chest.08-2668. [DOI] [PubMed] [Google Scholar]
- 57.Zecca E, De Luca D, Baroni S, Vento G, et al. Bile acid-induced lung injury in newborn infants: a bronchoalveolar lavage fluid study. Pediatrics. 2008;121:e146–149. doi: 10.1542/peds.2007-1220. [DOI] [PubMed] [Google Scholar]
- 58.Thomas C, Gioiello A, Noriega L, Strehle A, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.