

Abstract
Burkholderia cenocepacia is an opportunistic pathogen that causes chronic infection and induces progressive respiratory inflammation in cystic fibrosis patients. Recognition of bacteria by mononuclear cells generally results in the activation of caspase-1 and processing of IL-1β, a major proinflammatory cytokine. In this study, we report that human pyrin is required to detect intracellular B. cenocepacia leading to IL-1β processing and release. This inflammatory response involves the host adapter molecule ASC and the bacterial type VI secretion system (T6SS). Human monocytes and THP-1 cells stably expressing either small interfering RNA against pyrin or YFP–pyrin and ASC (YFP–ASC) were infected with B. cenocepacia and analyzed for inflammasome activation. B. cenocepacia efficiently activates the inflammasome and IL-1β release in monocytes and THP-1. Suppression of pyrin levels in monocytes and THP-1 cells reduced caspase-1 activation and IL-1β release in response to B. cenocepacia challenge. In contrast, overexpression of pyrin or ASC induced a robust IL-1β response to B. cenocepacia, which correlated with enhanced host cell death. Inflammasome activation was significantly reduced in cells infected with T6SS-defective mutants of B. cenocepacia, suggesting that the inflammatory reaction is likely induced by an as yet uncharacterized effector(s) of the T6SS. Together, we show for the first time, to our knowledge, that in human mononuclear cells infected with B. cenocepacia, pyrin associates with caspase-1 and ASC forming an inflammasome that upregulates mononuclear cell IL-1β processing and release.
Chronic bacterial colonization and sustained inflammation of the airways by opportunistic bacteria is the primary cause of lung tissue damage and mortality in cystic fibrosis (CF) patients. CF is the most frequently inherited, lethal disease of Caucasian individuals and is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on human chromosome 7 (1). These mutations result in malfunction of the CFTR protein leading to multiorgan dysfunction (2). However, nearly 85% of deaths in CF patients are associated with progressive respiratory inflammation (3). In response to the airway colonization by opportunistic bacteria, activated macrophages induce a proinflammatory cytokine storm that contributes to inflammation and lung tissue destruction (4-7). Among the bacteria infecting CF patients, members of the multidrug-resistant Burkholderia cepacia complex and in particular Burkholderia cenocepacia are important risk factors associated with poor prognosis. These bacteria are extraordinarily resistant to almost all clinically useful antibiotics, and some can cause a rapid and severe deterioration of the lung function known as the cepacia syndrome (8). Therefore, identifying the host factors involved in bacterial recognition and elicitation of profuse inflammatory cytokine responses is an urgent need to devise new ways to treat infected CF patients.
B. cenocepacia survives and proliferates within eukaryotic cells such as amoebae, epithelial cells, and human macrophages (9-11). Bacterial intracellular survival occurs in a membrane-bound compartment that shows a maturation delay resulting in a slow fusion with lysosomes and delayed assembly of the NADPH oxidase complex (12-15). Macrophages possess various Nod-like receptors (NLRs) in the cytoplasm that interact with molecules from the pathogens. Upon microbial recognition, NLRs initiate assembly of a multiprotein complex called inflammasome (16). Protein partners in the inflammasome possess either a caspase recruitment domain (CARD) or a pyrin domain (PYD) and assemble via CARD–CARD and PYD–PYD interactions (17, 18). The prototypical inflammasome consists of a CARD containing caspase-1, a CARD and PYD-containing adapter molecule, ASC, and an NLR sensor of pathogen- or danger-associated molecular patterns (PAMPs or DAMPs). Depending on the presence of CARDs or PYDs, NLR sensors are subdivided as NLRC or NLRP family members, respectively (19). Because caspase-1 is the central protein of the inflammasome and ASC is present in the majority of them, inflammasomes are named based on the participating pattern recognition receptor (NLR or other CARD or PYD containing protein). To date, several NLR-based inflammasome structures have been described: NLRP1 (16), NLRP3 (20), NLRC4 (IPAF) (21), and the NLRC5 (22). However, non-NLR proteins with a PYD may also initiate the assembly of inflammasomes like RIG-I (23), AIM2 (24-27), and pyrin (28, 29). Specific inflammasomes assembled in response to the detection of intracellular Salmonella, Legionella, Listeria, Francisella, Shigella, and Pseudomonas have been described (22, 29-39), but the inflammasome platform activated upon Burkholderia infection is unknown.
Inflammasome assembly results in caspase-1 activation, which subsequently cleaves biologically inactive pro–IL-1β to its active 17-kDa form (40, 41). IL-1β is the major proinflammatory cytokine associated with the initiation of inflammatory reaction, tissue destruction, and pyroptosis (42). To produce IL-1β, macrophages need two signals: one through TLR ligands that induce gene transcription and another through NLR agonists that activate caspase-1 via the inflammasome complex (43). We have previously reported that B. cenocepacia induces pro–IL-1β synthesis via LPS detection by TLR4 in murine macrophages (44). The subsequent conversion of pro–IL-1β to the mature form depends on caspase-1 activation, as caspase-1 knockout mice did not release IL-1β in response to B. cenocepacia (44). In this study, we show that human pyrin is responsible for intracellular detection of B. cenocepacia to induce IL-1β processing and release. This inflammatory response requires the host adapter molecule ASC and the bacterial type VI secretion system (T6SS).
Materials and Methods
Bacterial strains
B. cenocepacia strains (K56-2 clinical isolate and mutants) were cultured as previously described (44). The bacterial type III secretion system (T3SS) mutant JRL2 has an insertion inactivating bcsV, a critical gene of the type III secretion apparatus. The T6SS mutant DFA2 carries a deletion of the icmF gene (45). OD at 600 nm was used to calculate the multiplicity of infection, which was further confirmed by plating serial dilutions of cultures on Luria–Bertani agar and counting CFUs (46). Salmonella enterica serovar Typhimurium (S. typhimurium), used as a control, was cultured as described previously (47). For some experiments, bacteria were killed by heat at 95°C for 10 min.
Mononuclear phagocytes
THP-1 cells were purchased from American Type Culture Collection (lot 385653) and used to generate derivatives stably overexpressing or down-regulating desired inflammasome proteins. Human monocytes were isolated from fresh blood or buffy coat (local Red Cross) of healthy volunteers by Histopaque-1077 (Sigma-Aldrich) followed by CD14-positive selection (Miltenyi Biotec). This procedure consistently resulted in a ≥98% pure population of CD14+ cells, as confirmed by flow cytometry. Purified monocytes were used immediately after isolation. THP-1 cells and monocytes were cultured in RPMI 1640 (MediaTech) supplemented with 10% heat-inactivated FBS (Atlanta Biologicals).
In knockdown experiments, small interfering RNA (siRNA; Dharmacon) was delivered in monocytes by nucleofection (Lonza). To knock down pyrin, we used an ON-TARGETplus set of four siRNAs, each individually verified to suppress MEFV expression: 5′-GCAGGCCCUUCGAAGUGUA-3′, 5′-GCCCGCAAAUCCAGAAAUU-3′, 5′-GCAUAUGACACCCGCGUAU-3′, 5′-GCUACUGGGUGGUGAUAAU-3′. In control nucleofections, we used siGenome control siRNA. For nucleofection, 107 monocytes were resuspended in 100 μl of nucleofection solution containing 150 pmol siRNA. Nucleofection was performed with the Y-01 program. Immediately after nucleofection, monocytes were resuspended in 5 ml Lonza medium supplemented with 10% FBS and left to recover overnight in polypropylene blue snap cap culture test tubes (Fisher Health Care) to avoid adherence. The next morning, monocytes were counted with trypan blue (Sigma) showing that 90% of cells were viable. Monocytes were transferred to polypropylene tubes and infected with B. cenocepacia at a multiplicity of infection (MOI) of 10 for 6 and 24 h and with S. typhimurium at 2.5 MOI for 4 h. In selected experiments, monocytes were pretreated with 5 μg/ml actin polymerization inhibitor cytochalasin D (Sigma-Aldrich) for 30 min to block phagocytosis of bacteria. Released cytokines were detected in cell culture medium while cells were lysed and analyzed for protein and RNA.
Ethics statement
The procedure to isolate monocytes from blood samples of healthy donors and the respective consent forms were approved by the Institutional Review Board for human subject research at The Ohio State University (IRB protocol number 2011H0059). All healthy donors provided written consent for the collection of samples and subsequent analysis.
Construction of lentiviruses and generation of stable cell lines
To overexpress desired proteins, human ASC and pyrin were cloned into the pLenti6/V5 TOPO vector (Invitrogen Life Technologies). For visualization of the expressed proteins and sorting expressing cells, ASC and pyrin were fused with C terminus of yellow fluorescent protein (YFP), as it was described earlier (29). To express siRNA, we used the same pLenti6/V5 TOPO vector containing either green or red fluorescent proteins, where the H1 promoter was inserted in front of the CMV promoter. Pyrin siRNA (siPyrin) (5′-CAGGGCAGCCATTCAGGAATA-3′ and 5′-GACCACTCCTCAAGAGATAAA-3′) or control siRNA (siControl) (5′-AAGCTGACCCTGAAGTTCA-3′) oligonucleotides were ligated into the siRNA cassette [5′-CTAGCCC(sense)TTCAAGAGA(antisense)TTTTTGGAATT-3′ and 5′-GGG(antisense) TCTCTTGAAT(sense) AAAAACCTTTAAGC-3′]. Plasmids were verified by DNA sequencing. Each of these siPyrin constructs suppressed pyrin levels by 70%. Packaging cell line HEK293-FT (Invitrogen Life Technologies) was transfected with pLenti and helper plasmids pCMVΔR8.2 and pMD.G to produce lentivirus. Cell culture medium containing virus was harvested at 48 and 72 h posttransfection and concentrated at 3200 × g for 30 min (Centricon C-20 columns, 100 000 MWCO; Amicon), resulting in a titer 1 × 107 to 2 × 107 TU/ml. Cells were transduced with virus at 2–5 MOI in the presence of 6 μg/ml polybrene. After lentiviral transduction, 10–15% THP-1 cells expressed fluorescent protein. Stably transduced cells were selected with blasticidin (Invitrogen Life Technologies) for 10 d followed by two rounds of flow sorting using FACSAria (Becton Dickinson), resulting in nearly homogeneous yield of stably transduced THP-1 cells. All cells were regularly checked for the absence of Mycoplasma contamination (48).
Preparation of cell lysate, immunoblots, and ELISA
Cells were lysed in TN1 buffer [50 mM Tris-HCl (pH 8), 125 mM NaCl, 10 mM EDTA, and 1% Triton X-100] supplemented with complete protease inhibitor mixture (Sigma), 1 mM PMSF, and 100 μM N-(methoxysuccinyl)-Ala-Ala-Pro-Val chloromethyl ketone. The protein concentrations were determined using Bio-Rad Dc protein Lowry assay (Bio-Rad). After SDS-PAGE gel separation, samples were transferred to a nitrocellulose membrane, probed with the Ab of interest, and developed by ECL (Amersham Biosciences). Rabbit polyclonal Abs against IL-1β, pyrin, ASC, and caspase-1 were developed in our laboratory as described (49, 50). Other Abs were monoclonal anti-actin (clone C4; MP Biomedicals), monoclonal anti-enhanced GFP (EGFP) (clone JL-8; Clontech), and monoclonal anti-ASC (MBL).
For immunoprecipitation, we used two approaches: standard pull-down with Abs and with μMACS anti-EGFP magnetic beads (Miltenyi Biotec). In pull-down experiments, ASC was immunoprecipitated with the rabbit polyclonal anti-ASC Ab and then blotted with monoclonal anti-ASC, antipyrin, and monoclonal anti-EGFP (if we used YFP-fused protein). For the other approach, we used cell lines stably expressing either pyrin or ASC fused with enhanced YFP. Cells were lysed, and magnetic beads conjugated with anti-EGFP Abs were added. Magnetically labeled proteins were retained on a column, washed, and eluted. Eluate was subjected to immunoblot to detect proteins bound to the protein of interest.
Released IL-1β and caspase-1 were detected by immunoblot of cell culture medium with our anti–IL-1β and caspase-1 Ab. In addition, IL-1β and IL-8 in the cell culture medium were quantified using ELISA from R&D Systems according to the manufacturer’s protocol.
Cell death detection by quantification of lactate dehydrogenase release in cell culture media
Lactate dehydrogenase (LDH) release into cell culture medium was used as an indicator of cell death using NAD+ reduction assay (Roche Applied Science). Cells were plated in a 12-well plate at the density 1 × 106, and 10 MOI of B. cenocepacia or 2.5 MOI of S. typhimurium was added. Cell culture medium was collected 4, 6, and 24 h postinfection, clarified from floating bacteria by centrifugation, and used for LDH assay. To determine spontaneous cell death (negative control), we collected medium from cells incubated at the same time without bacteria. Total LDH content in cells (positive control) was measured in cells lysed with Triton X-100 (1% final concentration). Cell culture medium alone was used as a blank, and OD values were subtracted from readings of samples and positive control. LDH concentration in the medium was detected at wavelength 490 nm. Cell death was calculated by the formula [cytotoxicity (%) = (sample/positive control) × 100], as described earlier (51).
Real-time PCR
Cells were lysed in TRIzol (Invitrogen Life Technologies), and total RNA was converted into cDNA by ThermoScript RT system (Invitrogen Life Technologies). Quantitative PCR was done in the StepOne Plus machine using SYBR Green PCR mix (Applied Biosystems). The values of target genes were normalized to the values of two housekeeping genes, GAPDH and CAP-1, and expressed as relative copy number, as described previously (52-54).
Microscopy
For transmission electron microscopy, monocytes were fixed overnight in 2.5% glutaraldehyde in 0.1 M phosphate buffer with 0.1 M sucrose, washed, and resuspended in 2% agarose to produce a solid matrix containing the cell. The cells in agarose were postfixed in 1% osmium tetraoxide in phosphate buffer and then en bloc-stained with 2% uranyl acetate in 10% ethanol, dehydrated in a graded series of ethanol, and embedded in Eponate 12 epoxy resin (Ted Pella). Ultrathin sections were cut on a Leica EM UC6 ultramicrotome (Leica Microsystems, Wetzlar, Germany), collected on copper grids, and then stained with lead citrate and uranyl acetate. Images were acquired with an FEI Technai G2 Spirit transmission electron microscope (FEI) and a Macrofire (Optronics) digital camera and AMT image capture software. For live time-lapse confocal microscopy, THP-1 cells were stimulated with PMA at concentration 200 nM for 3 h. Then, cells were washed three times and plated in 35-mm glass-bottom culture dishes (MatTek) for 48 h, then infected with the K56-2 clinical isolate of B. cenocepacia that expresses mRFP. Plates were transferred to a special incubation chamber that provides 37°C and 5% carbon dioxide and were kept for 30 min for accommodation to the chamber atmosphere before the image recording started. Images were taken using an Olympus Fluoview FV10i microscope with ×60 magnification. Pictures were taken at 1-min intervals for 2–6 h.
Statistical analysis
All experiments were performed a minimum of three (or four) independent times and expressed as mean values ± SD. Comparison of groups for statistical difference were done using Student t test. A p value ≤0.05 was considered significant.
Results
Depletion of pyrin in primary human monocytes and in the immortal THP-1 cells results in reduced inflammasome activation upon B. cenocepacia infection
To identify whether pyrin plays a role in the macrophage response to B. cenocepacia, we modulated pyrin expression level in primary human monocytes and evaluated their ability to release mature IL-1β upon B. cenocepacia infection. siRNA against MEFV-coding pyrin and control siRNA were introduced into primary human monocytes by nucleofection. As expected, mRNA and pyrin protein levels were reduced after nucleofection with siPyrin, but not with siControl (Fig. 1A). After overnight recovery, nucleofected monocytes were evaluated for viability, seeded in polypropylene tubes, and infected with B. cenocepacia for 6 and 24 h and S. typhimurium for 4 h. Postinfection, IL1B and IL8 gene expression and corresponding released cytokines were evaluated. Uninfected nucleofected monocytes did not release detectable amounts of IL-1β and showed minimal release of IL-8. Manipulation of pyrin levels did not affect cytokine gene expression (Fig. 1B, 1E). Because gene expression is independent of the inflammasome-induced processing and release of IL-1β, we evaluated inflammasome activity by measuring the release of mature IL-1β and active caspase-1 from control and knockdown cells. In contrast to the equal mRNA induction, the release of mature IL-1β in response to B. cenocepacia was significantly lower in cells with reduced pyrin levels (Fig. 1C). Active caspase-1 self-cleavage also diminished in the cells with decreased pyrin levels (Fig. 1D) whereas the inflammasome-independent IL-8 release was unaffected (Fig. 1F). In contrast, S. typhimurium-induced caspase-1 activation and IL-1β release by human monocytes was pyrin independent (Fig. 1B–F). Thus, pyrin-regulated inflammasome activation was specific to B. cenocepacia.
FIGURE 1.

Human monocytes with knocked-down pyrin release less IL-1β and active caspase-1 in response to B. cenocepacia. (A) Human monocytes, either noninfected or infected with B. cenocepacia, show decreased pyrin expression at mRNA (MEFV relative copy numbers) and protein levels after nucleofection with siPyrin RNA compared with siControl. (B) B. cenocepacia and S. typhimurium induce similar IL-1β mRNA expression in human monocytes nucleofected with siControl and siPyrin RNA. Inflammasome-dependent release of mature IL-1β (C) and active caspase-1 (D) was significantly lower for siPyrin monocytes infected with B. cenocepacia but not with S. typhimurium. Inflammasome-independent IL-8 mRNA synthesis (E) and cytokine release (F) was similar between siControl and siPyrin monocytes infected with B. cenocepacia and S. typhimurium. Data are expressed as mean ± SD; n = 5 independent experiments. B.c., B. cenocepacia; NT, noninfected; siCtr, siControl; S.t., S. typhimurium.
To provide further evidence that pyrin promotes IL-1β processing and release in response to mononuclear cells infected with B. cenocepacia, we used a human THP-1 cell line stably depleted of pyrin (29) (Supplemental Fig. 1). Depletion of pyrin resulted in significant decrease of IL-1β release after B. cenocepacia infection (Fig. 2A). Inflammasome-independent IL-8 release in response to B. cenocepacia was independent of pyrin levels (Fig. 2B). Similarly, THP-1 response to S. typhimurium infection was not affected by pyrin levels as no difference in IL-1β and IL-8 release was observed between siControl and siPyrin THP-1 cells (Fig. 2C, 2D). These results confirm our observation with primary human monocytes that decreased pyrin expression level results in reduced inflammasome-dependent IL-1β release in response to B. cenocepacia infection.
FIGURE 2.

Pyrin downregulation decreases IL-1β release by THP-1 cells infected with B. cenocepacia. THP-1 cells, stably expressing siControl or siPyrin, were infected with B. cenocepacia, and cell culture medium was collected 6 and 24 h later for cytokine detection by ELISA. (A) THP-1 stably suppressing pyrin (siPyrin) showed significantly lower release of inflammasome-dependent IL-1β at 24 h postinfection compared with the THP-1 expressing siControl. (B) Inflammasome-independent IL-8 release was equal between siControl- and siPyrin-expressing THP-1 cells. (C and D) The same cells as in (A) were infected with S. typhimurium for 4 h. There was no difference in IL-1β and IL-8 release between siControl- and siPyrin-expressing THP-1 cells. Data are expressed as mean ± SD; n = 4 independent experiments. B.c., B. cenocepacia; NT, noninfected; siCtr, siControl; S.t., S. typhimurium.
Pyrin overexpression enhances IL-1β secretion by THP-1 in response to B. cenocepacia
To better understand the role of pyrin in the innate immune response to B. cenocepacia infection, we tested THP-1 cells stably overexpressing YFP–pyrin (Supplemental Fig. 1). Mononuclear cells were infected with B. cenocepacia or S. typhimurium, and IL-1β and IL-8 release was measured. YFP–pyrin–expressing THP-1 cells released significantly more IL-1β compared with control THP-1 cells (Fig. 3A). This process is inflammasome-dependent because no difference in inflammasome-independent IL-8 release was observed between these two cell types infected with B. cenocepacia (Fig. 3B). The majority of IL-1β release by THP-1 cells overexpressing YFP–pyrin was observed at early time points (6 h postinfection with B. cenocepacia) compared with 24 h (Fig. 3A). At 6 h, active caspase-1 (p20) accumulated in supernatants concurrently with IL-1β but only in cells overexpressing pyrin, whereas there was no difference at 24 h (Supplemental Fig. 2). Caspase-1 p20 release correlated with mature IL-1β release and was enhanced by pyrin expression levels (Supplemental Fig. 2).
FIGURE 3.
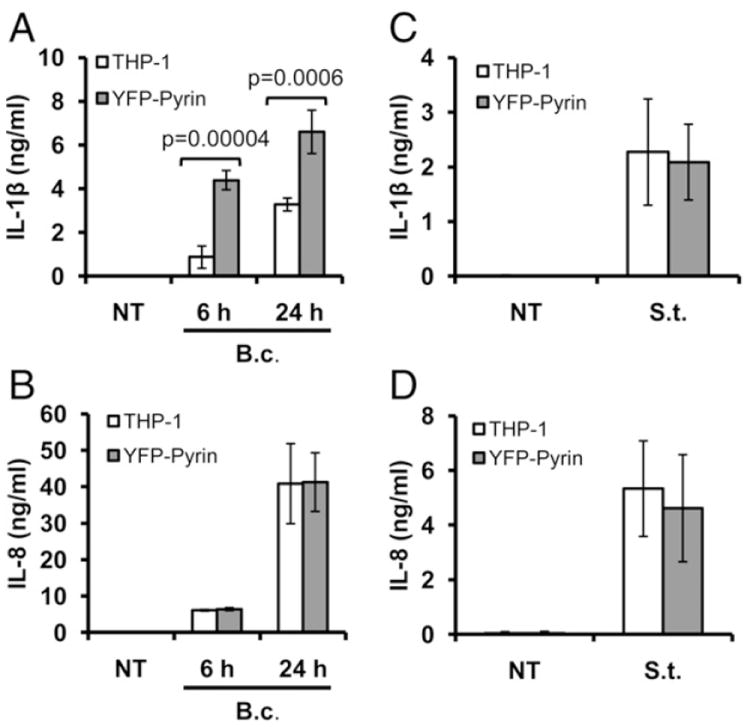
Pyrin overexpression enhances IL-1β release in response to B. cenocepacia. (A) THP-1 cells, stably expressing YFP–pyrin, showed significantly higher inflammasome-dependent IL-1β release in response to B. cenocepacia compared with the control THP-1 cells. (B) Inflammasome-independent IL-8 release was similar between control and YFP–pyrin–overexpressing THP-1 cells infected with B. cenocepacia. (C and D) The same cells were infected with S. typhimurium for 4 h. There was no difference in IL-1β and IL-8 release between THP-1 varying in pyrin levels. Data are expressed as mean ± SD; n = 4 independent experiments. B.c., B. cenocepacia; NT, noninfected; S.t., S. typhimurium.
Because pro–IL-1β and mature IL-1β are indistinguishable by ELISA, we analyzed supernatants by immunoblots (Supplemental Fig. 2). Cells overexpressing YFP–pyrin released more processed IL-1β at 6 h postinfection with B. cenocepacia compared with THP-1 cells. In contrast, YFP–pyrin–expressing cells infected with S. typhimurium did not show augmented inflammasome-dependent IL-1β or inflammasome-independent IL-8 release (Fig. 3C, 3D). These data suggest that pyrin-augmented inflammasome activation is pathogen specific and results from B. cenocepacia but not from S. typhimurium infection.
Host cell death in response to B. cenocepacia correlates with pyrin expression levels
To test whether caspase-1 activation and IL-1β accumulation in the cell culture medium was associated with pyroptosis, we measured LDH release by THP-1 cells expressing different pyrin levels. LDH release by THP-1 cells expressing YFP–pyrin was significantly elevated at 6 and 24 h after infection with B. cenocepacia compared with that of control THP-1 cells (Fig. 4A). However, pyrin knockdown did not reduce the percent of cell death in response to B. cenocepacia in comparison with control THP-1 cells (Fig. 4B). This discrepancy suggests that pyrin levels may have a threshold relationship with pyroptosis but a more uniform linear relationship with IL-1β processing. At the same time, S. typhimurium-induced cell death was massive and did not correlate with the pyrin expression levels (Fig. 4C, 4D). Hence, pyrin overexpression promotes early LDH release in response to B. cenocepacia, which correlates with early inflammasome activation and IL-1β release as discussed earlier.
FIGURE 4.

THP-1 cells overexpressing pyrin show higher cell death in response to B. cenocepacia infection. THP-1 cells with different levels of pyrin expression were left untreated or infected with B. cenocepacia for 6 and 24 h and with S. typhimurium for 4 h. Cell culture medium was used to measure percentage of LDH release relative to the total LDH content in the cell. (A) Pyrin overexpression leads to a significant increase in LDH release 6 and 24 h postinfection with B. cenocepacia compared with the control THP-1 cells. In contrast, pyrin knockdown did not affect LDH release by THP-1 cells infected with B. cenocepacia. (B). LDH release in response to S. typhimurium infection was equally high between control THP-1 cells overexpressing pyrin (C) and knockdown of pyrin (D). Data are expressed as mean ± SD; n = 3 independent experiments. B.c., B. cenocepacia; NT, noninfected; siCtr, siControl; S.t., S. typhimurium.
ASC overexpression is associated with enhanced IL-1β release by THP-1 cells in response to B. cenocepacia
Inflammasome activation usually requires the presence of the universal adapter protein, ASC, which links the PYD of intracellular sensors of pathogens with the CARD of caspase-1 (55). To check whether enhancement of IL-1β release in response to B. cenocepacia infection of monocytes is determined by inflammasome activation and requires ASC, we used two lines of THP-1 cells differing in ASC levels: one expressing endogenous ASC only and the other stably overexpressing YFP–ASC. YFP–ASC–expressing THP-1 cells showed significantly higher IL-1β release in response to B. cenocepacia (Fig. 5A). This ASC effect is independent of NF-κB signaling, as IL-8 release in response to B. cenocepacia was comparable in both cell types (Fig. 5B).
FIGURE 5.

ASC important to B. cenocepacia response. THP-1 cells, plain and overexpressing YFP–ASC, were infected with B. cenocepacia for 6 and 24 h; cell culture medium was collected and analyzed for the IL-1β and IL-8 release by ELISA. Data are expressed as mean ± SD; n = 4 independent experiments. (A) Inflammasome-dependent IL-1β release in response to B. cenocepacia was significantly higher in cells overexpressing ASC. (B) Inflammasome-independent IL-8 release in response to B. cenocepacia infection was similar between cells differing in ASC expression. (C) ASC is colocalized with pyrin after infection with B. cenocepacia. THP-1 cells were immunoprecipitated with anti-ASC Ab and immunoblotted for ASC and pyrin. (D) Pyrin is colocalized with ASC after infection with B. cenocepacia. YFP–pyrin from stably transfected THP-1 cells was captured on magnetic beads conjugated with anti-EGFP Ab, washed, and eluted. Cell extract (CE), flow through (F), and eluate from untreated (−) or B. cenocepacia treated (+) cells were immunoblotted. ASC was bound to YFP–pyrin only after infection with B. cenocepacia. B.c., B. cenocepacia; NT, noninfected.
To determine whether the ASC-dependent inflammasome contains pyrin upon B. cenocepacia infection, we studied ASC–pyrin interactions in B. cenocepacia-infected cells at 6 h. Pyrin coprecipitated with ASC after monocytes were infected with B. cenocepacia, but no coprecipitation was detected in uninfected cells (Fig. 5C, 5D).
YFP–ASC and YFP–pyrin colocalize with B. cenocepacia-containing phagosomes
Electron microscopy documented that B. cenocepacia infects mononuclear phagocytes and resides in a phagosome (Fig. 6A). We used confocal time-lapse microscopy to follow the redistribution of YFP–pyrin or YFP–ASC in THP-1 cells infected with B. cenocepacia expressing RFP (Fig. 6B, 6C; Supplemental Videos 1 and 2). Homogeneous cytoplasmic YFP–pyrin or YFP–ASC rapidly concentrated into a single speck containing intracellular RFP-expressing B. cenocepacia. However, the initial patterns of pyrin oligomerization are different from ASC speck formation. Pyrin first appears to form oligomers concentrated on the cell membrane (Supplemental Video 1). Notably, YFP–ASC–expressing THP-1 cells also released microvesicles that contain ASC (and IL-1β, data not shown). After phagocytosis, both YFP–pyrin and YFP–ASC colocalized with the B. cenocepacia-containing phagosome generating a single ASC speck (Fig. 6B, 6C), which was immediately followed by cell dissolution (presumed cell death) (Supplemental Video 2). Together, biochemical and microscopy experiments indicate that pyrin and ASC are involved in the formation of the speck, which is accompanied by the production of IL-1β and cell pyroptosis in response to B. cenocepacia.
FIGURE 6.

ASC and pyrin are colocalized with intracellular B. cenocepacia. (A) Transmission electron microscope imaging of human monocytes infected with B. cenocepacia (B.c.) shows that this bacteria is taken up intracellularly (black arrow). (B) THP-1 cells stably expressing YFP–pyrin were infected with an RFP-expressing K56.2 clinical isolate of B. cenocepacia. Intracellular RFP-Burkholderia (B.c.) was colocalized with YFP–pyrin (white arrow) just prior to microscopic evidence of cell death. This is a representative image from Supplemental Video 1. (C) THP-1 cells stably expressing YFP–ASC were infected with RFP-B. cenocepacia. Intracellular B. cenocepacia (B.c.) induced rapid pyroptosis after colocalization with YFP–ASC (white arrow). This is a representative image from Supplemental Video 2.
B. cenocepacia type VI (T6SS) but not type III (T3SS) secretion is essential for inflammasome activation and IL-1β and caspase-1 release by mononuclear phagocytes
NLRs sense bacteria or their PAMPs in the cytoplasm of infected cells. If the pathogen resides in a vacuole, the PAMPs could be sensed by NLRs after bacterial molecules reach the cytoplasm, which will require bacterial secretion systems. B. cenocepacia harbors several secretion systems including type III, IV, and VI (11, 56). To determine which secretion system is required to engage the Burkholderia inflammasome leading to IL-1β production, human monocytes were infected with the parental B. cenocepacia strain K56-2 or with mutants in genes encoding structural components of the type III and VI secretion systems. K56-2, T3SS, and T6SS mutants show equal induction of pro–IL-1β synthesis in human monocytes (Fig. 7A) and THP-1 cells (Fig. 7B). B. cenocepacia K56-2 and the T6SS mutant efficiently engulfed bacteria (Supplemental Fig. 3). However, IL-1β and active caspase-1 release were reduced in cells infected with the T6SS mutant (Fig. 7A, 7B). We concluded that detection of B. cenocepacia by pyrin and activation of the inflammasome requires a functional bacterial T6SS.
FIGURE 7.

T6SS is important in inflammasome activation in response to B. cenocepacia. Human monocytes (HM) (A) and THP-1 cells (B) were infected with B. cenocepacia WT and with mutants of type VI and type III secretion systems, respectively (T6SS and T3SS). Cells were lysed (CE) for detection of the levels of pro–IL-1β synthesis upon bacterial infection, and cell culture medium was used to determine whether mature IL-1β and active caspase-1 are released, as a signature of inflammasome activation. All bacteria types equally induce pro–IL-1β synthesis in human monocytes (A) and THP-1 cells (B). However, mutation of T6SS reduces inflammasome activation and caspase-1 and IL-1β release into cell culture medium. NT, noninfected.
To validate further the role of T6SS in inflammasome activation, we infected human monocytes with live and heat-killed wild-type (WT) and T6SS mutant B. cenocepacia. There was no difference in NF-κB signaling between WT and T6SS mutant based on IL1B and IL8 RNA expression and inflammasome-independent IL-8 release (Fig. 8A, 8D, 8E). Abrogation of bacteria internalization with cytochalasin D did not affect NF-κB signaling. However, IL-1β release, which depends on inflammasome activation, was significantly lower for the T6SS mutant, cytochalasin D, and heat-killed bacteria (Fig. 8B) despite adequate presence of precursor IL-1β (Fig. 8C). Finally, LDH release correlated well with mature IL-1β release (Fig. 8F).
FIGURE 8.

Internalization of live B. cenocepacia is important for T6SS-dependent inflammasome activation. Human monocytes were infected with live or heat-killed B. cenocepacia for 6 h. To inhibit bacteria internalization, monocytes were pretreated for 30 min with 5 μg/ml cytochalasin D. NF-κB activation, measured by mRNA expression for IL1B (A), IL8 (D), and IL-8 release (E), shows no difference between WT and T6SS mutants of B. cenocepacia. Pro–IL-1β synthesis was also equal between experimental groups, based on immunoblot of cell lysates (C). Inflammasome-dependent IL-1β release was significantly decreased in monocytes infected with T6SS mutant, and also when bacteria internalization was inhibited by cytochalasin D or when bacteria were killed (B). Cell death, measured by LDH release, correlates well with IL-1β release (F). Data are expressed as mean ± SD; n = 3 independent experiments. *p < 0.05, **p < 0.005. cD, cytochalasin D; K, heat-killed B. cenocepacia; L, live B. cenocepacia; NT, noninfected.
Discussion
B. cenocepacia induces severe inflammation in the lung of CF patients possibly by direct activation of macrophages, which results in strong proinflammatory responses (7, 57). Intense inflammation compromises lung tissue integrity and leads to progressive respiratory failure or cepacia syndrome, resulting in death. High levels of IL-1β, the caspase-1 substrate, are detected in the serum and bronchoalveolar lavages of B. cenocepacia-infected CF patients (6, 58, 59). In support of this model, we have recently shown that CFTR-defective macrophages produce more IL-1β than normal macrophages during B. cenocepacia infection (44).
Macrophage activation is initiated by B. cenocepacia LPS and depends on CD14, TLR4, and MyD88 in human and murine models (44, 60). This results in secretion of many proinflammatory cytokines. However, murine macrophages stimulated with highly purified B. cenocepacia LPS produce pro–IL-1β but do not release mature IL-1β (61). Cleavage and release of the major proinflammatory cytokine IL-1β depends on the activation of caspase-1, which is tightly regulated by the inflammasome (17). To stimulate pro–IL-1β cleavage and release, B. cenocepacia must be detected intracellularly. In this study, we demonstrate for the first time, to our knowledge, that pyrin regulates caspase-1 activation in human monocytes in response to B. cenocepacia infection.
Pyrin is a protein in which several conserved mutations result in familial Mediterranean fever and belongs to the tripartite motif family of proteins, all of which share structural homology (62). Although pyrin has an N-terminal PYD, it was not initially recognized as a member of the NLR family of intracellular sensors of pathogens. However, we have recently shown that modulation of intracellular pyrin levels in human mononuclear cells infected with Francisella novicida also affects caspase-1 activation and IL-1β release (29, 63). Pyrin interacts via its PYD with the adapter protein ASC (64), and the two proteins colocalize in cellular sites rich in polymerizing actin (65). The CARD of the ASC molecule can interact directly with caspase-1 via its N-terminal CARD, and, as a result, these three proteins may assemble into an inflammasome (28). During pyrin activation the PYD is unmasked, allowing activated pyrin to interact with ASC and facilitate ASC oligomerization (66). Then, the ASC oligomer serves as a molecular platform for recruiting and activating caspase-1 (67, 68). Our data suggest that the pyrin–ASC interaction is important in intracellular B. cenocepacia detection and inflammasome or pyroptosome activation. The pyroptosome is a single (one per cell) 1- to 2-μm supramolecular complex that contains ASC and caspase-1. Pyroptosome assembly occurs within minutes of the ASC speck complex formation and is associated with host cell death (68). This type of cell death is uniquely dependent on caspase-1 activation and named pyroptosis because of its ability to induce IL-1β–dependent fever (69). The inflammasome forms a ring-like structure with an outer diameter of ~13 nm and an inner diameter of ~4 nm (70). Using time-lapse microscopy, we observed the formation of pyrin and ASC specks within infected macrophages. We also observed the formation of pyrin and ASC complexes in the vicinity of the B. cenocepacia-containing vacuole. To our knowledge, this is the first documented interaction of pyrin with a bacterium.
The current work adds B. cenocepacia to the list of potential pyrin targets. Because intracellular B. cenocepacia resides in a phagosome, cytoplasmic activation of the inflammasome implies escape or active secretion of bacterial molecules into the cytosol. Recently, it was shown that P. aeruginosa activates the inflammasome by secreting pilin, the major structural protein of type IV pili, via a T3SS (71). In this study, we determined that T6SS of B. cenocepacia is responsible for pyrin-dependent caspase-1 and IL-1β processing and release by human mononuclear phagocytes. The specific bacterial effector molecules initiating activation of the pyrin inflammasome is a topic for future studies.
Supplementary Material
Supplementary Figure 1. Comparison between different stable THP-1 cells, used in the study. THP-YFP-Pyrin show expression of the endogenous pyrin and YFP-pyrin. THP-1 with stable pyrin knock-down (siPyrin) show decrease in the endogenous pyrin expression. All used cells express ASC and show induction of proIL-1β upon stimulation.
Supplementary Figure 2 B. cenocepacia more efficiently activate inflammasome in THP-1 cells expressing YFP-Pyrin. THP-1 and THP-YFP-Pyrin, differing in level of pyrin expression, were infected with B. cenocepacia. Cells over-expressing YFP-Pyrin released more active caspase-1 and processed IL-1β at 6 h post-infection with B. cenocepacia, compared to THP-1 cells.
Supplementary Figure 3. Similar numbers of wild type and T6SS B. cenocepacia are recovered from THP1 cells at early stage of infection. THP1 cells were infected with either WT or T6SS B. cenocepacia at an MOI of 10. Cells were lysed and plated for colony forming units (CFUs) within 1 h of infection.
Concentration of pyrin specks around intracellular B. cenocepacia containing phagosome is prior to the cell pyroptosis. Images have been taken for 3 h at one minute interval using 60X magnification.
ASC speck formation and cell pyroptosis is initiated around intracellular B. cenocepacia containing phagosome. Images have been taken for 2 h at one minute interval using 60X magnification.
Acknowledgments
We thank Austin Duprey and Amy Gross for assistance with preliminary experiments.
This work was supported by National Institutes of Health Grants HL089440, HL76278, HL102724 (to M.D.W.), HL094586, and AI083871, the American Lung Association (to A.O.A.), and Cystic Fibrosis Canada (to M.A.V.). M.A.V. holds a Canada Research Chair in Infectious Diseases and Microbial Pathogenesis and the Zeller’s Senior Scientist Award from Cystic Fibrosis Canada.
Abbreviations used in this article
- CARD
caspase recruitment domain
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- DAMP
danger-associated molecular pattern
- EGFP
enhanced GFP
- LDH
lactate dehydrogenase
- MOI
multiplicity of infection
- NLR
Nod-like receptor
- PAMP
pathogen-associated molecular pattern
- PYD
pyrin domain
- siControl
control siRNA
- siPyrin
pyrin siRNA
- siRNA
small interfering RNA
- T3SS
bacterial type III secretion system
- T6SS
bacterial type VI secretion system
- WT
wild-type
- YFP
yellow fluorescent protein
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
Contributor Information
Mikhail A. Gavrilin, Email: Mikhail.Gavrilin@osumc.edu.
Mark D. Wewers, Email: Mark.Wewers@osumc.edu.
Amal O. Amer, Email: Amal.Amer@osumc.edu.
References
- 1.Magni A, Giordano A, Mancini C, Pecoraro C, Varesi P, Quattrucci S, Trancassini M. Emerging cystic fibrosis pathogens: incidence and antimicrobial resistance. New Microbiol. 2007;30:59–62. [PubMed] [Google Scholar]
- 2.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 3.Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L. Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest. 2009;136:1554–1560. doi: 10.1378/chest.09-0132. [DOI] [PubMed] [Google Scholar]
- 4.Desjardins M, Celis JE, van Meer G, Dieplinger H, Jahraus A, Griffiths G, Huber LA. Molecular characterization of phagosomes. J Biol Chem. 1994;269:32194–32200. [PubMed] [Google Scholar]
- 5.Desjardins M, Nzala NN, Corsini R, Rondeau C. Maturation of phagosomes is accompanied by changes in their fusion properties and size-selective acquisition of solute materials from endosomes. J Cell Sci. 1997;110:2303–2314. doi: 10.1242/jcs.110.18.2303. [DOI] [PubMed] [Google Scholar]
- 6.Bonfield TL, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- 7.Chmiel JF, Berger M, Konstan MW. The role of inflammation in the pathophysiology of CF lung disease. Clin Rev Allergy Immunol. 2002;23:5–27. doi: 10.1385/CRIAI:23:1:005. [DOI] [PubMed] [Google Scholar]
- 8.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 9.Sajjan US, Yang JH, Hershenson MB, LiPuma JJ. Intracellular trafficking and replication of Burkholderia cenocepacia in human cystic fibrosis airway epithelial cells. Cell Microbiol. 2006;8:1456–1466. doi: 10.1111/j.1462-5822.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 10.Lamothe J, Thyssen S, Valvano MA. Burkholderia cepacia complex isolates survive intracellularly without replication within acidic vacuoles of Acanthamoeba polyphaga. Cell Microbiol. 2004;6:1127–1138. doi: 10.1111/j.1462-5822.2004.00424.x. [DOI] [PubMed] [Google Scholar]
- 11.Saldías MS, Valvano MA. Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology. 2009;155:2809–2817. doi: 10.1099/mic.0.031344-0. [DOI] [PubMed] [Google Scholar]
- 12.Lamothe J, Huynh KK, Grinstein S, Valvano MA. Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell Microbiol. 2007;9:40–53. doi: 10.1111/j.1462-5822.2006.00766.x. [DOI] [PubMed] [Google Scholar]
- 13.Lamothe J, Valvano MA. Burkholderia cenocepacia-induced delay of acidification and phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective macrophages. Microbiology. 2008;154:3825–3834. doi: 10.1099/mic.0.2008/023200-0. [DOI] [PubMed] [Google Scholar]
- 14.Keith KE, Hynes DW, Sholdice JE, Valvano MA. Delayed association of the NADPH oxidase complex with macrophage vacuoles containing the opportunistic pathogen Burkholderia cenocepacia. Microbiology. 2009;155:1004–1015. doi: 10.1099/mic.0.026781-0. [DOI] [PubMed] [Google Scholar]
- 15.Huynh KK, Plumb JD, Downey GP, Valvano MA, Grinstein S. Inactivation of macrophage Rab7 by Burkholderia cenocepacia. J Innate Immun. 2010;2:522–533. doi: 10.1159/000319864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 17.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 18.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 19.Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 21.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 22.Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol. 2011;186:1333–1337. doi: 10.4049/jimmunol.1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschläger N, Schlee M, Rothenfusser S, Barchet W, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 24.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 25.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 28.Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, Sagara J, Fernandes-Alnemri T, Alnemri ES. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13:236–249. doi: 10.1038/sj.cdd.4401734. [DOI] [PubMed] [Google Scholar]
- 29.Gavrilin MA, Mitra S, Seshadri S, Nateri J, Berhe F, Hall MW, Wewers MD. Pyrin critical to macrophage IL-1beta response to Francisella challenge. J Immunol. 2009;182:7982–7989. doi: 10.4049/jimmunol.0803073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arlehamn CS, Pétrilli V, Gross O, Tschopp J, Evans TJ. The role of potassium in inflammasome activation by bacteria. J Biol Chem. 2010;285:10508–10518. doi: 10.1074/jbc.M109.067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 32.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Núñez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26:433–443. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA. 2010;107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdelaziz DH, Amr K, Amer AO. Nlrc4/Ipaf/CLAN/CARD12: more than a flagellin sensor. Int J Biochem Cell Biol. 2010;42:789–791. doi: 10.1016/j.biocel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdelaziz DH, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, Abdulrahman BA, Grandhi J, Hassan ZA, Marsh C, et al. Apoptosis-associated speck-like protein (ASC) controls Legionella pneumophila infection in human monocytes. J Biol Chem. 2011;286:3203–3208. doi: 10.1074/jbc.M110.197681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Fernandes-Alnemri T, Alnemri ES. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J Clin Immunol. 2010;30:693–702. doi: 10.1007/s10875-010-9425-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Núñez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. 2007;37:3030–3039. doi: 10.1002/eji.200737532. [DOI] [PubMed] [Google Scholar]
- 40.Yamin TT, Ayala JM, Miller DK. Activation of the native 45-kDa precursor form of interleukin-1-converting enzyme. J Biol Chem. 1996;271:13273–13282. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 41.Dinarello CA. Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann N Y Acad Sci. 1998;856:1–11. doi: 10.1111/j.1749-6632.1998.tb08307.x. [DOI] [PubMed] [Google Scholar]
- 42.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Netea MG, van de Veerdonk FL, Kullberg BJ, Van der Meer JW, Joosten LA. The role of NLRs and TLRs in the activation of the inflammasome. Expert Opin Biol Ther. 2008;8:1867–1872. doi: 10.1517/14712590802494212. [DOI] [PubMed] [Google Scholar]
- 44.Kotrange S, Kopp B, Akhter A, Abdelaziz D, Abu Khweek A, Caution K, Abdulrahman B, Wewers MD, McCoy K, Marsh C, et al. Burkholderia cenocepacia O polysaccharide chain contributes to caspase-1-dependent IL-1beta production in macrophages. J Leukoc Biol. 2011;89:481–488. doi: 10.1189/jlb.0910513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aubert D, MacDonald DK, Valvano MA. BcsKC is an essential protein for the type VI secretion system activity in Burkholderia cenocepacia that forms an outer membrane complex with BcsLB. J Biol Chem. 2010;285:35988–35998. doi: 10.1074/jbc.M110.120402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cremer TJ, Shah P, Cormet-Boyaka E, Valvano MA, Butchar JP, Tridandapani S. Akt-mediated proinflammatory response of mononuclear phagocytes infected with Burkholderia cenocepacia occurs by a novel GSK3β-dependent, IκB kinase-independent mechanism. J Immunol. 2011;187:635–643. doi: 10.4049/jimmunol.1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 48.Zakharova E, Grandhi J, Wewers MD, Gavrilin MA. Mycoplasma suppression of THP-1 Cell TLR responses is corrected with antibiotics. PLoS ONE. 2010;5:e9900. doi: 10.1371/journal.pone.0009900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seshadri S, Duncan MD, Hart JM, Gavrilin MA, Wewers MD. Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1beta processing and release. J Immunol. 2007;179:1274–1281. doi: 10.4049/jimmunol.179.2.1274. [DOI] [PubMed] [Google Scholar]
- 50.Wewers MD, Dare HA, Winnard AV, Parker JM, Miller DK. IL-1 beta-converting enzyme (ICE) is present and functional in human alveolar macrophages: macrophage IL-1 beta release limitation is ICE independent. J Immunol. 1997;159:5964–5972. [PubMed] [Google Scholar]
- 51.Abdelaziz DH, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, Abdulrahman BA, Hassan ZA, El-Sharkawi FZ, Bedi SS, et al. Asc-dependent and independent mechanisms contribute to restriction of legionella pneumophila infection in murine macrophages. Front Microbiol. 2011;2:18. doi: 10.3389/fmicb.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gavrilin MA, Bouakl IJ, Knatz NL, Duncan MD, Hall MW, Gunn JS, Wewers MD. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc Natl Acad Sci USA. 2006;103:141–146. doi: 10.1073/pnas.0504271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fahy RJ, Exline MC, Gavrilin MA, Bhatt NY, Besecker BY, Sarkar A, Hollyfield JL, Duncan MD, Nagaraja HN, Knatz NL, et al. Inflammasome mRNA expression in human monocytes during early septic shock. Am J Respir Crit Care Med. 2008;177:983–988. doi: 10.1164/rccm.200703-418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hall MW, Gavrilin MA, Knatz NL, Duncan MD, Fernandez SA, Wewers MD. Monocyte mRNA phenotype and adverse outcomes from pediatric multiple organ dysfunction syndrome. Pediatr Res. 2007;62:597–603. doi: 10.1203/PDR.0b013e3181559774. [DOI] [PubMed] [Google Scholar]
- 55.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 56.Zhang R, LiPuma JJ, Gonzalez CF. Two type IV secretion systems with different functions in Burkholderia cenocepacia K56-2. Microbiology. 2009;155:4005–4013. doi: 10.1099/mic.0.033043-0. [DOI] [PubMed] [Google Scholar]
- 57.Melnikov A, Zaborina O, Dhiman N, Prabhakar BS, Chakrabarty AM, Hendrickson W. Clinical and environmental isolates of Burkholderia cepacia exhibit differential cytotoxicity towards macrophages and mast cells. Mol Microbiol. 2000;36:1481–1493. doi: 10.1046/j.1365-2958.2000.01976.x. [DOI] [PubMed] [Google Scholar]
- 58.Corvol H, Fitting C, Chadelat K, Jacquot J, Tabary O, Boule M, Cavaillon JM, Clement A. Distinct cytokine production by lung and blood neutrophils from children with cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L997–L1003. doi: 10.1152/ajplung.00156.2002. [DOI] [PubMed] [Google Scholar]
- 59.Berger M. Inflammatory mediators in cystic fibrosis lung disease. Allergy Asthma Proc. 2002;23:19–25. [PubMed] [Google Scholar]
- 60.Bamford S, Ryley H, Jackson SK. Highly purified lipopolysaccharides from Burkholderia cepacia complex clinical isolates induce inflammatory cytokine responses via TLR4-mediated MAPK signalling pathways and activation of NFkappaB. Cell Microbiol. 2007;9:532–543. doi: 10.1111/j.1462-5822.2006.00808.x. [DOI] [PubMed] [Google Scholar]
- 61.Shimomura H, Matsuura M, Saito S, Hirai Y, Isshiki Y, Kawahara K. Lipopolysaccharide of Burkholderia cepacia and its unique character to stimulate murine macrophages with relative lack of interleukin-1beta-inducing ability. Infect Immun. 2001;69:3663–3669. doi: 10.1128/IAI.69.6.3663-3669.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nisole S, Stoye JP, Saïb A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3:799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- 63.Gavrilin MA, Wewers MD. Francisella Recognition by Inflammasomes: Differences between Mice and Men. Front Microbiol. 2011;2:11. doi: 10.3389/fmicb.2011.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richards N, Schaner P, Diaz A, Stuckey J, Shelden E, Wadhwa A, Gumucio DL. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem. 2001;276:39320–39329. doi: 10.1074/jbc.M104730200. [DOI] [PubMed] [Google Scholar]
- 65.Waite AL, Schaner P, Hu C, Richards N, Balci-Peynircioglu B, Hong A, Fox M, Gumucio DL. Pyrin and ASC co-localize to cellular sites that are rich in polymerizing actin. Exp Biol Med (Maywood) 2009;234:40–52. doi: 10.3181/0806-RM-184. [DOI] [PubMed] [Google Scholar]
- 66.Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, McCormick M, Zhang Z, Alnemri ES. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell. 2007;28:214–227. doi: 10.1016/j.molcel.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fernandes-Alnemri T, Alnemri ES. Assembly, purification, and assay of the activity of the ASC pyroptosome. Methods Enzymol. 2008;442:251–270. doi: 10.1016/S0076-6879(08)01413-4. [DOI] [PubMed] [Google Scholar]
- 68.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES. The pyroptosome: a supra-molecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–724. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 71.Arlehamn CS, Evans TJ. Pseudomonas aeruginosa pilin activates the inflammasome. Cell Microbiol. 2011;13:388–401. doi: 10.1111/j.1462-5822.2010.01541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Comparison between different stable THP-1 cells, used in the study. THP-YFP-Pyrin show expression of the endogenous pyrin and YFP-pyrin. THP-1 with stable pyrin knock-down (siPyrin) show decrease in the endogenous pyrin expression. All used cells express ASC and show induction of proIL-1β upon stimulation.
Supplementary Figure 2 B. cenocepacia more efficiently activate inflammasome in THP-1 cells expressing YFP-Pyrin. THP-1 and THP-YFP-Pyrin, differing in level of pyrin expression, were infected with B. cenocepacia. Cells over-expressing YFP-Pyrin released more active caspase-1 and processed IL-1β at 6 h post-infection with B. cenocepacia, compared to THP-1 cells.
Supplementary Figure 3. Similar numbers of wild type and T6SS B. cenocepacia are recovered from THP1 cells at early stage of infection. THP1 cells were infected with either WT or T6SS B. cenocepacia at an MOI of 10. Cells were lysed and plated for colony forming units (CFUs) within 1 h of infection.
Concentration of pyrin specks around intracellular B. cenocepacia containing phagosome is prior to the cell pyroptosis. Images have been taken for 3 h at one minute interval using 60X magnification.
ASC speck formation and cell pyroptosis is initiated around intracellular B. cenocepacia containing phagosome. Images have been taken for 2 h at one minute interval using 60X magnification.