

Abstract
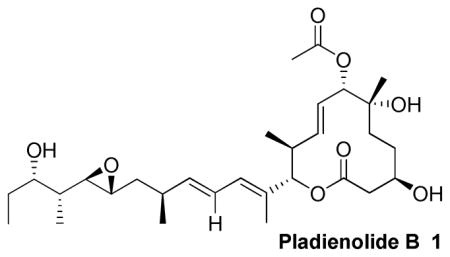
An enantioselective and convergent total synthesis of pladienolide B (1) is described. Pladienolide B binds to the SF3b complex of a spliceosome and inhibits mRNA splicing activity. The synthesis features an epoxide opening reaction, an asymmetric reduction of a β-keto ester, and a cross metathesis strategy for the side chain synthesis.
In 2004, Sakai et al. reported the isolation and structural characterization of seven novel 12-membered macrocyclic compounds named pladienolide A-G from a culture of an engineered strain of Streptomyces platensis, Mer-11107.1 Initial screening studies showed these compounds were capable of inhibiting the proliferation of human cancer cells with low nanomolar IC50 values.2 Interestingly, pladienolides maintained this activity against multiple drug resistant cancer cells. Furthermore, they exhibited a novel mechanism of action by binding to the SF3b subunit of the spliceosome, inhibiting the splicing of pre-mRNA to translatable mRNA. The unspliced pre-mRNA is exported from the nucleus to the cytoplasm resulting in inhibition of cell growth.3 To date, the pladienolides and the structurally distinct FR901464 are the only known molecular scaffolds capable of modulating splicing and generating an antitumor response. Consequently, these compounds have shown tremendous potential to become a new class of anticancer agents. In 2008, clinical trials were initiated using E7107, a pladienolide D analog, for the treatment of various cancers.4 Not surprisingly, the chemistry and biology of the pladienolides attracted immense attention. To date, Kotake and co-workers reported the first total synthesis of the most active compounds, pladienolide B and D.5 Skaanderup et al. published a synthetic pathway to the macrocyclic core of the enantiomer of pladienolide B, while Burkart and co-workers have reported their efforts on constructing and modifying the side chain.6 Recently, both Maier7a and Webb7b have reported progress in the development of pladienolide based analogs albeit with reduced biological activity. Our interest in pladienolide B arose from its novel mechanism of action and its unique structural features. We sought to develop a convergent route to pladienolide B that could facilitate subsequent structure activity relationship studies and synthesis of structural variants. Herein, we report a concise, enantioselective synthesis of pladienolide B.
As shown in Figure 1, we planned to utilize a late stage Julia-Kocienski olefination8 similar to Kotake and co-workers5 to append the epoxide containing side chain 2 to the macrocyclic core 3, providing the trans-trans diene system. The hydroxy groups in both intermediates 2 and 3 would be conveniently protected with silyl ether groups thereby allowing a global deprotection at the final stage of the synthesis. Side chain 2 could be efficiently synthesized by cross metathesis of alcohol 4 and mesylate 5. Shi epoxidation of the resulting olefin would install the desired epoxide moiety.9 Both chiral alcohol 4 and mesylate 5 are readily available in optically active form. We planned to construct the macrocyclic core 3 by ring closing metathesis (RCM) of triene 6 in hopes that the free allylic alcohol would produce an activating effect resulting in higher yields and shorter reaction times.10 Of particular relevance, a related RCM macrocyclization strategy by Kotake and co-workers5 with a protected diol provided low yield and significant amount of isomerized olefin. Triene 6 in turn could be synthesized by Yamaguchi esterification of acid 8 and homoallylic alcohol 7.11 The synthesis of alcohol 7 could be achieved by Brown’s crotylation of the corresponding aldehyde.12 Acid 8 would be synthesized by a ring opening reaction of epoxide 9 with the dianion of tert-butyl acetoacetate 10 followed by asymmetric reduction of the resulting β-keto ester. Epoxide 9 could be synthesized using a desymmetrization strategy via Sharpless epoxidation.13
Figure 1.

Retrosynthetic Analysis of Pladienolide B
The synthesis of the C1–C8 fragment is outlined in Scheme 1. Asymmetric desymmetrization of commercially available divinyl carbinol 11 using Sharpless epoxidation followed by PMB protection of the corresponding alcohol provided epoxide 12 in 43% yield as a single isomer according to literature methods.14 Ring opening of epoxide 12 with the dianion of tert-butyl acetoacetate 10 followed by silyl protection of the resulting alcohol with TESCl afforded acyclic β-keto ester.15 Asymmetric reduction of 13 with NaBH4-L-tartaric acid complex as prepared by Yatagai and Ohnuki16 successfully reduced the ketone with excellent stereoselectivity (> 95% dr). The depicted stereochemistry is based on the observed stereochemical outcome of β-keto esters as reported.16 Silyl protection of the newly formed alcohol provided ester 14 as a single isomer in high yields (61% from 12). Oxidation of 14 with 3.5 eq. of IBX selectively removed the TES ether and oxidized the alcohol to a ketone without migration of the terminal olefin.17 The resulting ketone was treated with MeMgBr in THF at −78 °C providing the desired tertiary alcohol 15 as a single isomer (>95% by NMR) in 85% yield (quant. BRSM). The stereochemistry was assigned based on Cram’s chelation model.18 Treatment of alcohol 15 with an excess of TESOTf in the presence of Et3N in CH2Cl2 protected the tertiary alcohol as a TES ether with concomitant formation of the silyl ester which was hydrolyzed upon aqueous workup providing acid 8. Our asymmetric synthesis of C1–C8 segment is quite efficient and provides convenient access to other possible stereoisomers. Previous syntheses of this segment5,6a,7a relied on terpene (nerol and R-linalool) modification strategies.
Scheme 1.

Synthesis of C1–C8 Intermediate 8
We briefly examined the scope of the NaBH4/tartaric acid reduction of β-keto ester 13. As shown in Table 1, reduction of the β-keto ester in the absence of tartaric acid resulted in only a small degree of selectivity (entry 3). With the TES group removed (entry 1) the selectivity was increased suggesting the free hydroxy group had a beneficial directing effect. When the NaBH4/tartaric acid system was employed, a further improvement in selectivity was observed (entry 2). However, the best results were obtained with NaBH4/tartaric acid with the hydroxy group protected as a bulky silyl ether suggesting that steric effects were the dominating factor (entries 5, 7 and 8) for good diastereoselectivity. The size of the ester group also played an important role with the bulky t-butyl providing the best selectivity (compare entries 4 and 5). Interestingly, using D-tartaric acid (entry 6) gave a similar degree of selectivity for the other diastereomer suggesting the chirality was transferred from the tartaric acid and not induced from the chirality inherent to the substrate.
Table 1.
Asymmetric Reduction of β–keto Ester
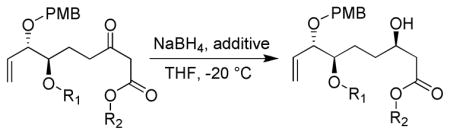
| ||||
|---|---|---|---|---|
| entry | R1 | R2 | additive | S:R |
| 1 | H | t-butyl | none | 37:63 |
| 2 | H | t-butyl | L-tartaric acid | 15:85 |
| 3 | TES | t-butyl | none | 45:55 |
| 4 | TES | t-butyl | L-tartaric acid | 5:95 |
| 5 | TES | methyl | L-tartaric acid | 8:92 |
| 6 | TES | methyl | D-tartaric acid | 94:6 |
| 7 | TBS | methyl | L-tartaric acid | 5:95 |
| 8 | TPS | methyl | L-tartaric acid | 5:95 |
Synthesis of optically active homoallylic alcohol 7 is shown in Scheme 2. Prenyl alcohol 16 was protected as a trityl ether with trityl chloride and Et3N in the presence of a catalytic amount of DMAP. Riley oxidation of the resulting trityl ether provided allylic alcohol 17 in 52% yield along with some aldehyde 18.19 The resulting mixture was exposed to MnO2 oxidation in CH2Cl2 to provide aldehyde 18 in good overall yield. Brown’s asymmetric crotylation12 of aldehyde 18 furnished homoallylic alcohol 7 in > 82% ee and 64% yield over two steps.
Scheme 2.

Synthesis of C9–C13 Intermediate 7
With acid 8 and alcohol 7 in hand we proceeded to form the macrocyclic ring structure. As shown in Scheme 3, esterification of acid 8 with alcohol 7 according to Yamaguchi’s protocol11 followed by DDQ oxidative removal of the PMB ether provided allylic alcohol 19. Ring closing metathesis (RCM) with Grubbs’ 2nd generation catalyst (>95% E by HPLC) followed by treatment with Ac2O in pyridine furnished lactone 20.20 Despite the presence of a free allylic alcohol, the RCM still required elevated temperatures although higher yields were observed compared to Kotake et al.5,10 Interestingly, when the allylic alcohol was protected as a PMB ether, RCM was very sluggish. Treatment of 20 with DDQ for 24 h selectively removed the trityl ether while oxidation of the resulting allylic alcohol with IBX afforded aldehyde 3 in 75% yield over two steps.17 The use of BCl3 at low temperatures was also effective in selectively removing the trityl ether, however, DDQ provided more robust conditions.21
Scheme 3.

Synthesis of Macrocyclic Core 3
With the synthesis of the macrocycle core complete, we then focused on development of a concise route to the epoxide containing side chain C12–C23. As shown in Scheme 4, cross metathesis between homoallylic alcohol 4 and mesylate 5 proceeded in moderate yield with a high degree of selectivity for the trans olefin (5:1 E:Z). Subsequent protection of the homoallylic alcohol as a TES ether enabled the separation of olefin 21 from the other undesired cross metathesis products. Displacement of the mesylate with the anion of 1-phenyl-1H-tetrazole-5-thiol followed by ammonium molybdate catalyzed hydrogen peroxide oxidation gave sulfone 22 in 71% yield over two steps. Shi epoxidation9 of sulfone 22 followed by TES protection of the secondary alcohol afforded the side chain intermediate 2 as a single isomer.
Scheme 4.

Synthesis of Sulfone 2
The final assembly of pladienolide B is shown in Scheme 5. Julia-Kocienski olefination8 between sulfone 2 and aldehyde 3 provided diene 24 which was subsequently deprotected by the addition of TBAF yielding pladienolide B. The 1H and 13C NMR of our synthetic pladienolide B {[α]D 23 = +7.3 (c 0.26, MeOH)} is identical to the reported spectra for the natural pladienolide B {Lit.5 [α]D 27 = +7.9 (c 1.1, MeOH)} thus confirming the absolute configuration of our synthetic material.
Scheme 5.

Synthesis of Pladienolide B
In summary, we have accomplished an enantioselective synthesis of pladienolide B. The synthetic route is convergent and readily scaleable. Overall, our synthesis involved 31 total steps from commercially available material. The longest linear path was 16 steps with an overall yield of 1.4%. This represents a significant improvement over Kotake’s route which required 59 total steps (22 longest linear) from commercially available material. The syntheses featured an effective aymmetric reduction of a β-keto ester, a cross metathesis reaction and Julia-Kocienski olefinations. Other key reactions included a Sharpless asymmetric epoxidation and Brown asymmetric crotylboration reactions. The synthesis will provide convenient access to a variety of derivatives. Further investigations of structural and biological studies are in progress.
Supplementary Material
Acknowledgments
Financial support by the National Institutes of Health is gratefully acknowledged. We also thank the ACS Division of Medicinal Chemistry and the American Foundation for Pharmaceutical Education for predoctoral fellowships to David Anderson. We thank Mr. Khriesto Shurrush (Purdue University) for preliminary experimental assistance.
Footnotes
Supporting Information Available. Experimental procedures and 1H and 13C NMR spectra for all new compounds have been provided. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y. J Antibiot. 2004;57:173–179. doi: 10.7164/antibiotics.57.173. [DOI] [PubMed] [Google Scholar]; (b) Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y. J Antibiot. 2004;57:180–187. doi: 10.7164/antibiotics.57.180. [DOI] [PubMed] [Google Scholar]; (c) Asai N, Kotake Y, Niijima J, Fukuda Y, Uehara T, Sakai T. J Antibiot. 2007;60:364–369. doi: 10.1038/ja.2007.49. [DOI] [PubMed] [Google Scholar]
- 2.Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M. J Antibiot. 2004;57:188–196. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Nat Chem Biol. 2007;3:570–5. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]; (b) Yokoi A, Kotake Y, Takahashi K, Kadowaki T, Matsumoto Y, Minoshima Y, Sugi NH, Sagane K, Hamaguchi M, Iwata M, Mizui Y. FEBS J. 2011;278:4870–80. doi: 10.1111/j.1742-4658.2011.08387.x. [DOI] [PubMed] [Google Scholar]
- 4.Butler MA. In: Natural Product-Derived Compounds in Late Stage Clinical Development at the End of 2008. Buss A, Butler M, editors. The Royal Society of Chemistry; Cambridge, UK: 2009. pp. 321–354. [Google Scholar]
- 5.Kanada RM, Itoh D, Nagai M, Niijima J, Asai N, Mizui Y, Abe S, Kotake Y. Angew Chem Int Ed Engl. 2007;46:4350–5. doi: 10.1002/anie.200604997. [DOI] [PubMed] [Google Scholar]
- 6.(a) Skaanderup PR, Jensen T. Org Lett. 2008;10:2821–2824. doi: 10.1021/ol800946x. [DOI] [PubMed] [Google Scholar]; (b) Mandel AL, Jones BD, La Clair JJ, Burkart MD. Bioorg Med Chem Lett. 2007;17:5159–5164. doi: 10.1016/j.bmcl.2007.06.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Muller S, Mayer T, Sasse F, Maier ME. Org Lett. 2011;13:3940–3943. doi: 10.1021/ol201464m. [DOI] [PubMed] [Google Scholar]; (b) Gundluru MK, Pourpak A, Cui X, Morris SW, Webb TR. MedChemComm. 2011;2:904–908. doi: 10.1039/C1MD00040C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blakemore PR, Cole WJ, Kocienski P, Morley A. Synlett. 1998;1:26–28. [Google Scholar]
- 9.Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J Am Chem Soc. 1997;119:11224–11235. [Google Scholar]
- 10.Hoye T, Zhao H. Org Lett. 1999;1:1123–1125. doi: 10.1021/ol990947+. [DOI] [PubMed] [Google Scholar]
- 11.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Japan. 1979;52:1989–1993. [Google Scholar]
- 12.(a) Brown H, Bhat K. J Am Chem Soc. 1986;108:5919–5923. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]; (b) Brown H, Bhat K. J Org Chem. 1986;108:293–294. [Google Scholar]
- 13.Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless B. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]
- 14.Schreiber S, Smith D. J Org Chem. 1989;54:9–10. [Google Scholar]
- 15.(a) Weiler L. J Am Chem Soc. 1970;92:6702–6704. [Google Scholar]; (b) Lygo B, O’Connor N, Wilson PR. Tetrahedron. 1988;44:6881–6888. [Google Scholar]; (c) Kieczykowski G, Roberts M, Schlessinger R. J Org Chem. 1978;43:788–789. [Google Scholar]
- 16.Yotagai M, Ohnuki T. J Chem Soc, Perkin Trans 1. 1990;6:1826–1828. [Google Scholar]
- 17.(a) Frigerio M, Santagostino M, Sputore S, Palmisano G. J Org Chem. 1995;60:7272–7276. [Google Scholar]; (b) Wu Y, Huang JH, Shen X, Hu Q, Tang CJ, Li L. Org Lett. 2002;4:2141–2144. doi: 10.1021/ol025946n. [DOI] [PubMed] [Google Scholar]
- 18.(a) Cram DJ, Kopecky K. J Am Chem Soc. 1959;81:2748–2755. [Google Scholar]; (b) Reetz M. Angew Chem Int Ed Engl. 2003;23:556–569. [Google Scholar]
- 19.Umbriet M, Sharpless K. J Am Chem Soc. 1977;99:5526–5528. [Google Scholar]
- 20.(a) Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (b) Chatterjee AK, Choi TL, Sanders D, Grubbs R. J Am Chem Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 21.Jones G, Hynd G, Wright J, Sharma A. J Org Chem. 2000;65:263–265. doi: 10.1021/jo9913255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.