

ABSTRACT
Fecal microbiome transplantation by low-volume enema is an effective, safe, and inexpensive alternative to antibiotic therapy for patients with chronic relapsing Clostridium difficile infection (CDI). We explored the microbial diversity of pre- and posttransplant stool specimens from CDI patients (n = 6) using deep sequencing of the 16S rRNA gene. While interindividual variability in microbiota change occurs with fecal transplantation and vancomycin exposure, in this pilot study we note that clinical cure of CDI is associated with an increase in diversity and richness. Genus- and species-level analysis may reveal a cocktail of microorganisms or products thereof that will ultimately be used as a probiotic to treat CDI.
IMPORTANCE
Antibiotic-associated diarrhea (AAD) due to Clostridium difficile is a widespread phenomenon in hospitals today. Despite the use of antibiotics, up to 30% of patients are unable to clear the infection and suffer recurrent bouts of diarrheal disease. As a result, clinicians have resorted to fecal microbiome transplantation (FT). Donor stool for this type of therapy is typically obtained from a spouse or close relative and thoroughly tested for various pathogenic microorganisms prior to infusion. Anecdotal reports suggest a very high success rate of FT in patients who fail antibiotic treatment (>90%). We used deep-sequencing technology to explore the human microbial diversity in patients with Clostridium difficile infection (CDI) disease after FT. Genus- and species-level analysis revealed a cocktail of microorganisms in the Bacteroidetes and Firmicutes phyla that may ultimately be used as a probiotic to treat CDI.
Introduction
Antibiotic-associated diarrhea (AAD) is a widespread phenomenon in hospitals today. The most common opportunistic pathogen in the setting of AAD is the bacterium Clostridium difficile. The incidence of Clostridium difficile infection (CDI) has been increasing over the last decade, fueled in large part by a strain identified as North American pulsotype (NAP) 1 or ribotype 027 (1–3). This strain drives outbreaks in institutional settings and is associated with increased morbidity and mortality in elderly patients (4, 5), with the most severe infections leading to colectomy (surgical removal of the colon) or death. In addition, up to 30% of patients are unable to clear the infection and suffer recurrent bouts of diarrheal disease.
Current guidelines from the Infectious Diseases Society of America suggest that the first-line therapy for CDI is metronidazole, which nonspecifically targets anaerobic bacteria. The second-line therapy for patients with refractory or severe CDI is vancomycin, an antibiotic with broad activity against Gram-positive bacteria. In certain instances, tapering regimens of vancomycin have been recommended (6), and newer drugs for CDI have recently been developed (7). Nevertheless, owing to significant failure rates with all conventional antibiotic regimens for recurrent CDI, clinicians have for several decades resorted to fecal bacteriotherapy or microbiome “transplantation” (FT) (8–14). This type of therapy represents a paradigm shift in treatment of infectious agents, with the objective of restoration of the normal microbiota rather than specific eradication of the pathogen using a conventional antibiotic. Donor stool for this type of therapy is typically obtained from a spouse or close relative and thoroughly tested for various pathogenic microorganisms prior to infusion. Anecdotal reports suggest a very high success rate of FT in patients who fail antibiotic treatment. These clinical results have been lauded in the lay press as a radically alternative approach with great success to antibiotic therapy (15–17).
It has been suggested that the CDI state is marked by reduced microbial diversity and that persistent disruption of commensal microbe-gut interactions may result in interference with the complex interactions between the host and the commensal gut microbiota of healthy individuals (11, 12, 18). We hypothesized that FT successfully restores the gut diversity absent in patients suffering from refractory CDI. In order to test this hypothesis, we used deep-sequencing technology to explore the human microbial diversity in patients with CDI disease after direct instillation of donor stool as a clinical therapeutic intervention. Even though they are very specific, traditional methods such as culture, cloning, or Sanger sequencing of full-length 16S rRNA following PCR are limited in their capacity to elaborate the microbial community present in the gut (19–23). The use of deep-sequencing technology to address this task provides superior depth and breadth of coverage, especially of as-yet-uncultured anaerobic microorganisms in the gut. A previous study has shown that pyrosequencing provided a more precise estimate of relative abundance and an improved confidence of detection in comparison to cloned full 16S rRNA sequence analysis (19).
We targeted the V5-V6 region of bacterial 16S rRNA. The V5-V6 region reads were classified to reference sequence-based operational taxonomic units (refOTUs), which were defined at 97% sequence identity to determine the represented taxon. In the past, unguided probiotic treatments with Lactobacillus casei and Lactobacillus bulgaricus administered empirically have been shown to be inferior to vancomycin and metronidazole therapy (24). Effective probiotic formulations to prevent and treat recurrent CDI will likely require a global understanding of the dynamic changes of the gut microbiome in response to stool transplantation. Here we demonstrate that dramatic shifts in microbial diversity and composition occur with fecal transplantation, resulting in restoration of bacterial diversity and richness. These results have significant implications for tailoring specific biological therapeutics for infections such as CDI in which the microbial community plays a significant, if not primary, role.
(These data were presented in part at the 2011 American Society for Microbiology General Meeting in New Orleans, LA.)
RESULTS
Number of sequence reads and sequences.
Twenty-one stool samples in total were evaluated from patients undergoing FT as described previously (14). Out of the six patients in this sample set, the first FT of patients 5 and 6 failed. The second FT for patient 5 failed, while the second FT for patient 6 was successful. FT was considered successful if the patients did not have recurrent CDI for ≥4 months. From this sample set, we obtained 148,945 V5-V6 pyrosequencing reads with an average length of 210 bp. The number of reads per sample is outlined in Table 1. The sequenced reads were trimmed, filtered, and preprocessed as described in Text S1 in the supplemental material. The distance matrix was used with a 0.03-genetic-distance, furthest-neighbor threshold in mothur to designate the operational taxonomic units (OTUs) (25). For subsequent standardized comparative analyses, the OTUs were classified at the level of 97% sequence homology into 4,029 reference operational taxonomic units (refOTUs). Taxonomic classification was performed using the Ribosome Database Project (RDP) (minimum 80% bootstrap score) (26, 27). Because several OTUs had the same genus-level classification, indicating that these OTUs represent sub-OTU lineages, further classification at the species level was carried out using the metagenomics analysis server (MG-RAST) (28). From these sequence reads, a range of 72 to 154 genera was identified in the stool specimens. The average number of genera for pooled donors was 123, that for recipients pre-FT was 96, and that for recipients post-FT was 120. The two failed FTs resulted in recipients with an average number of genera of 90. Thus, clinically successful FT resulted in an average increase of 25% in genera for recipients post-FT compared to their pre-FT state (extraction of differential gene expression [EDGE] analysis, P < 0.05) (see Table S5 in the supplemental material). The number of species that belonged to the genera with significant changes in abundance post-FT was 90. The successful donor sample sequence classification contained a median of 211 unique species, while the pre-FT and post-FT sample sequence classifications consisted of a median of 164 and 255 unique species, respectively (see Table S4).
TABLE 1.
Summary of patient sample statistics (n = 6) for recipient (R) and donor (D) specimens in this study for both successful (S) and failed (F) FTs
| Patient no. | Transplant outcome(s) | No. of reads |
|---|---|---|
| 1 | Successful | R1 Pre, 15,786; R1 post-S, 13,851; D1-S, 5,346 |
| 2 | Successful | R2 Pre, 6,607; R2 Post-S, 7,339; D2-S, 6,977 |
| 3 | Successful | R3 Pre, 12,971; R3 Post-S, 9,633; D3-S, 8,055 |
| 4 | Successful | R4 Pre, 9,368; R4 Post-S, 9,767; D4-S, 3,982 |
| 5 | First failed; second failed | R5 Pre, 3,975; R5 Post-F (first), 2,959; R5 Post-F (second), 3,727; D5-F (first), 3,131; D5-F (second), 3,408 |
| 6 | First failed; second successful | R6 Pre, 5,017; R6 Post-F, 6,239; R6 Post-S, 4,677; D6-S, 6,130 |
Statistical analyses of changes in microbial diversity after FT.
Rank abundance curves were used for refOTUs obtained from the V5-V6 pyrosequencing read data for all six patients (Fig. 1) to evaluate the distribution and abundance of taxa in rank order. Statistical assessment of variation in the abundance of refOTUs between participants or with respect to the different states was conducted with EDGE software with a false discovery rate of 7% and a P value of 0.05 (29). EDGE analysis of significant relative changes was selected here because it improves the overall performance of multiple significance tests for multivariate data (29, 30). Patients 1 to 3 demonstrated no increase in abundance from the pre-FT to post-FT state, whereas patients 4 to 6 did show an increase for the successful post-FT state. When all samples were pooled, a median of 132 V5-V6 refOTUs was found in the pre-FT state and a median of 157 V5-V6 refOTUs was found in the post-FT state. Rarefaction curves are a measure of the number of refOTUs observed as a function of sampling effort and establish the rapidity with which taxonomic richness increased with each V5-V6 tag sequence. The rarefaction curves for the six patients in the study (Fig. 2) reveal that except for patient 1, the diversity increased after successful FT. When all specimens were pooled, V5-V6 tag abundance ranged between 1,903 and 6,319 unique reads, corresponding to a range of 72 to 154 higher taxonomic classifications (see Table S3 in the supplemental material). In an attempt to catalogue the ecological diversity of each stool specimen, observed taxon richness, the Shannon diversity index, and the Simpson diversity index were established for each patient pre- and post-FT compared to healthy donors (Fig. 3). When the medians for each category were examined, the failed donors had significantly lower richness (P < 0.01) and Shannon diversity (P < 0.0001) than did the majority of successful donors as assessed by a normal distribution nonparametric test.
FIG 1.

Rank abundance curve comparison of refOTUs for all donors and recipients pre- and post-FT. Abundance has been shown in log scale on the y axis, and the rank order of the refOTUs is shown on the x axis. The dashed blue line corresponds (99% detection) to an abundance of 5. Summary statistics are tabulated in Table S1 in the supplemental material. D#-S, donor of successful transplant; D#-F, donor of failed transplant; R# Pre, recipient pre-FT; R# Post-S, recipient post-successful FT.
FIG 2.

Rarefaction curve comparison of refOTUs for all donors and recipients pre- and post-FT. Phylogenetic diversity is plotted against the number of V5-V6 sequence reads for each donor and recipient pre- and post-FT. D-S, donor of successful transplant; D-F, donor of failed transplant; R Pre, recipient pre-FT; R Post-S, recipient post-successful FT; R Post-F, recipient post-failed FT. Each 30th point was plotted with the confidence interval at 95%.
FIG 3.

Diversity statistics observed for all donors and recipients pre- and post-FT. Observed taxon richness, Shannon diversity, and Simpson diversity for each sample are shown. The lines in the plots represent the medians for each group. The asterisks denote the failed-FT samples. R Pre samples are characterized by fewer taxa and lower diversity than those of post-FT and donor samples. R Pre, recipient pre-FT samples; R Post, recipient post-FT samples; D, donor samples.
PCoA.
We used principal coordinate analysis (PCoA) to analyze the variability in refOTUs among donor, pre-FT, and post-FT samples. The first three axes of the PCoA, plotted in Fig. 4A, were sufficient to account for 71.8% of the total variability, and notably, the variability on these axes was higher than interindividual variability between samples. The relative contribution to variability for each successive axis decreases, with the first five axes accounting for all nonrandom variability (Fig. 4B). The Pearson correlation coefficients for using 1, 2, or 3 axes were 0.87, 0.91, and 0.90, respectively. The PCoA plot also revealed a clustering of refOTUs into two groups consisting of (i) pre-FT or failed-FT samples and (ii) donor or successful-FT samples, suggesting that key genera are shared between donors and successful FTs. Clustering may be responsive to both differences in proportions and the presence or absence of sequence data.
FIG 4.
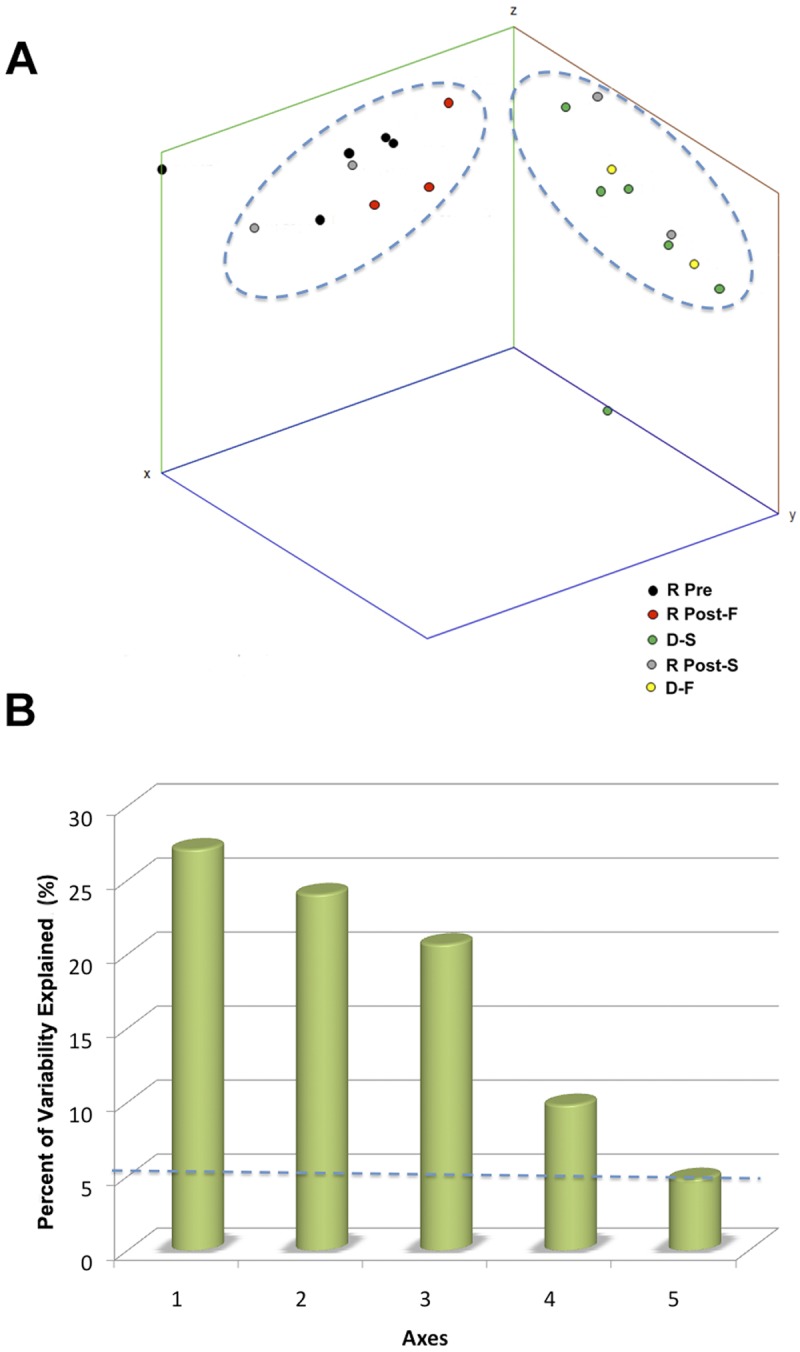
(A) PCoA. PCoA represents multivariate analysis, which explores community similarity by measuring phylogenetic branch length (UniFrac) of 16S rRNA tag sequences. The dots represent the projected shadows of the samples on the respective two-dimensional planes (xy, blue; yz, red; xz, green). The x axis, y axis, and z axis represent 27.1%, 24.0%, and 20.7% of the intersample variability, respectively. R Pre, recipient pre-FT; R Post-F, recipient post-failed FT; D-S, donor of successful transplant; R Post-S, recipient post-successful FT; D-F, donor of failed transplant. (B) PCoA axes in relation to observed variability. The percentage of variability observed is depicted in relation to the first five axes. The dashed line at 5.26% represents the amount of variability expected for each axis based on random events.
Phylum-, genus-, and species-level changes noted after FT.
Examination of changes in the microbiota at the phylum level following FT showed two general trends (Fig. 5). Patients 1, 3, and 5 demonstrated no change in the predominance of Proteobacteria post-FT, whereas patients 2, 4, and 6 revealed a replacement of Proteobacteria with Bacteroidetes post-FT. We examined quantitative changes in specific refOTUs and genera in response to FT. Normalized and classified refOTUs are shown in detail across the samples (Fig. 6A). The greatest number of refOTUs was shared between failed transplants and pretransplant samples (1,435 refOTUs) followed by the refOTUs shared between successful transplants and donor samples (1,189 refOTUs). On the other hand, the lowest number of shared refOTUs was that between successful transplant samples and the pretransplant samples (9 refOTUs). Post-FT, within Proteobacteria significant (P < 0.05, EDGE analysis) decreases in the abundance of Kluyvera, Raoultella, and Parasutterella species were noted. In contrast, for Bacteroidetes a significant increase (P < 0.05, EDGE analysis) in the abundance of Alistipes, Bacteroides, and Parabacteroides was documented post-FT (Fig. 6B). Further significant increases were noted in Firmicutes genera and families such as Lactobacillus, Subdoligranulum, Carnobacterium, Streptococcus, Enterococcus, unclassified Ruminococcaceae, and unclassified Lachnospiraceae. Further dissection of genera that increase post-FT at the species level revealed that the following species contribute significantly to this effect: Alistipes putredinis, Alistipes shahii, Bacteroides intestinalis, Bacteroides caccae, Bacteroides ovatus, Bacteroides sp. strain 2_1_22, and Clostridium lituseburense (Fig. 6C; see also Table S4 in the supplemental material). Pyrosequencing reads corresponding to Clostridium difficile were present pre-FT but not post-FT.
FIG 5.

Phylum-level differences for each FT set. Donor (D) and post-successful (Post-S)-FT microbial populations are distinctive from pre-FT (R Pre) and post-failed (Post-F)-FT samples by higher levels of Bacteroidetes and Firmicutes and lower levels of Proteobacteria. At the phylum level, there is no difference observed between the failed-FT donor samples and the successful-FT donor samples.
FIG 6.

(A) Comparison of refOTU structures across the stool microbial communities. The Venn diagram displays the overlapping refOTUs for pooled samples belonging to each sample type. Each of the communities is characterized by more unique than overlapping refOTUs. The highest overlap is seen between R Pre and R Post-F samples as well as between R Post-S and D-S samples. R Pre, recipient pre-FT samples; R Post, recipient post-FT samples; D-S, donor samples. (B) Relative contributions of significant genera in pooled specimens before and after FT. Genera shown as a percentage of the total genera for each sample type are depicted. Only genera which change significantly (P < 0.05, EDGE analysis) following FT are depicted. R Pre, recipient pre-FT samples; R Post-S, recipient post-FT samples; D-S, donor samples. (C) Relative percentage of species in pooled specimens before and after FT. Species which increased significantly (P < 0.05, EDGE analysis) following FT are shown as absolute counts (number of V5-V6 sequence reads) for each sample type. R Pre, recipient pre-FT samples; R Post, recipient post-FT samples; D, donor samples.
Community balance changes in response to FT.
Changes in taxon abundance after successful FT were calculated based on pooled data. Donor stool samples were characterized by an equal abundance of the taxa Bacteroides, Alistipes, Bifidobacterium, and Enterobacteriaceae, whereas in the pretransplant stool samples, Bacteroides strains were outnumbered by a factor of 10 by unclassified Enterobacteriaceae, Pseudomonadaceae, and Escherichia/Shigella strains. In the post-FT samples, a marked expansion of Bacteroides and Faecalibacterium content and a corresponding decrease in Escherichia/Shigella were noted. The highest fold increases after successful FT (range, 16-fold to 100-fold) were observed in Bacteroides, Lactobacillus, Dorea, Roseburia, and Faecalibacterium, with the corresponding highest fold decreases (range, 4-fold to 20-fold) being observed in Enterobacteriaceae and unclassified Proteobacteria.
DISCUSSION
Refractory CDI can be severely debilitating and life-threatening. Patients often become dependent on chronic metronidazole or vancomycin therapy for symptom alleviation at great financial expense. Discontinuation of these antibiotics often leads to recurrent episodes of diarrhea from CDI. We reported previously on self-administered FT as an effective treatment modality for the cure of patients with refractory CDI (14). FT represents a paradigm shift in the approach to the treatment of CDI, aiming to restore microbial diversity rather than to eradicate the pathogen with antibiotics. While many studies have reported on the efficacy and clinical success of FT, only two have attempted to address the mechanistic basis for this success. The first used a biased approach by amplifying predetermined gut microbes using PCR (8). The second is a single FT case report where the authors used a traditional clone-and-sequence approach to describe changes in the microbiota (12). However, these experimental approaches may underestimate the relative abundance changes that take place in the gut microbiota.
Here we undertook metagenomic analysis of stool specimens from donors and recipients of FT. It has been previously shown that classification accuracy of microbial populations is largely dependent on the choice of the 16S rRNA gene region, sequencing technology, and sequence quality (19, 31). Dethlefsen et al. showed that even though the V3 region provided more unique sequences than did the V6 region, only 85% of V3 reads had exact matches in the database to facilitate classification, relative to 90% for the V6 region (19). In addition, Claesson et al. suggested that the V4 and V5 regions provide representative sequences for the gut microbiota (31). Therefore, we used the long reads of 454 sequencing to combine the V5-V6 regions in order to take advantage of these two important features that these variable regions bring to the CDI patient microbiota analysis. Amplification bias may occur with this method beyond 20 cycles of amplification, but as this is a comparative analysis of specimens, these effects should be minimal.
Statistical analyses of pooled deep-sequencing data from successful FT recipients revealed the dynamic changes in the microbial community that occur pre- and post-FT. At the level of phyla, the healthy gut microbiota consists predominantly of Bacteroidetes, whereas CDI patients in this study demonstrate an overabundance of Proteobacteria. When we measured the ecological diversity of the microbiota, the CDI disease state (refractory cases in this study) is marked by decreased richness and diversity. The failed post-FT samples highlight the importance of increased richness and diversity for a successful FT and eradication of CDI. In the CDI state, fewer taxa are present and they are present in greater abundance (Fig. 6B). Healthy post-FT recipients appear to “adopt” certain elements of the donor microbiota (Fig. 6B). Specifically, the healthy state corresponding to a successful FT is marked by an eradication of key Proteobacteria species and a corresponding restoration of key Firmicutes and Bacteroidetes species, which may well be critical to “outcompeting” or depriving C. difficile of fitness in some way. These data agree with a previous study using a clone-and-sequence strategy (32).
We note in CDI stools the relative paucity of taxa from Lachnospiraceae (formerly Clostridium cluster XIVa), which constitute the majority of butyrate-producing bacteria (19, 33, 34). Lactate breakdown into butyrate by key microorganisms plays a major role in host metabolism, nutrient digestibility, microbiota changes, and epithelial integrity as well as host defense (35–37). In the hindgut, butyrate acts directly on tissue development, repair, and modulation of bacterial virulence gene expression (35–37). Lactate accumulation has been previously associated with disease states such as gut resections and ulcerative colitis both in humans and in horses (34, 38). We speculate that the absence of lactate-utilizing bacteria such as Lachnospiraceae is one of the metabolic reasons leading to disequilibrium and perpetuation of refractory CDI (34). Consistent with this model, successful FT replenishes butyrate-producing bacteria that utilize lactate in the distal gut, along with Roseburia and Bacteroides (33, 34, 39). Roseburia, for example, is a difficult-to-culture, mutualistic, and cellulolytic bacterial genus that is important for plant polysaccharide breakdown. Roseburia spp. such as Roseburia intestinalis and Roseburia inulinivorans are able to use soluble oligosaccharides and polysaccharides to produce butyrate (52). The abundance of these species was on average increased post-FT. One might speculate that butyrate depletion and lactate buildup may be the reason why CDI results in severe disease, particularly in a certain subset of patients being treated with antimicrobials and infected with hypervirulent C. difficile strains. Further studies of the metabolic consequences of changes in microbiota will be required to investigate this possibility.
One limitation of our study is that patients were being treated with vancomycin prior to FT. It has been demonstrated that vancomycin can alter the stool microbiota, and thus, decreases in diversity and richness prior to FT may be in part due to lingering effects of vancomycin as opposed to CDI per se (41). For example, it has been shown that vancomycin has activity against Bacteroidetes (42). Nevertheless, we observe the same paucity of Bacteroidetes and overexpansion of Proteobacteria seen by other studies of the CDI disease state in the absence of prior therapy with vancomycin (18, 43). Furthermore, by use of 16S rRNA gene deep sequencing, our study confirms and extends the depth and breadth of our understanding of the changes in microbiota observed by other groups conducting FT (11, 12). Despite the potential interindividual variability because each FT microbiome was from a unique donor, it was possible to draw predictable patterns of striking similarity in the gut microbial communities among the individuals following a successful FT using a combination of pooled specimen analysis and PCoA. Another concern is spurious PCR amplification from reagents used in the stool extraction. To address this, we noted experimentally that reagents alone can produce bacterial 16S rRNA gene sequence data corresponding to genera such as Leuconostoc and Lactobacillus, but in this comparative analysis, this should not affect interpretation of relative microbial changes. Finally, the use of technical replicates would have increased the confidence of the findings in this study.
Clear separation of pre-FT/failed-FT and donor/successful-FT groups suggests that the presence or absence of key taxa likely outweighs interindividual differences in determining whether or not an FT is successful. Even though variability in microbiota composition depends on historical exposures to microbes and antibiotics, the post-FT samples grouped quite closely with the successful donor samples, suggesting the presence of key genera required for recolonization and bacterial community resilience. Furthermore, FT replenishment of certain specialized bacteria appears to be durable for the most part and results in the restoration of key taxa following FT, as mirrored by an increase in richness and diversity consistent with other previously published reports (11). The apparent success of FT in restoring “normal” microbial diversity raises the question of whether it can be used for other clinical conditions. For example, perturbations of the distal gut microbiota and childhood antibiotic use have been previously implicated in chronic conditions such as asthma, life-threatening pseudomembranous colitis, cancer, and obesity (44, 45). It is possible that the microbial equilibrium is specifically altered by commonly used antibiotics such as the newer-generation fluoroquinolones which have increased activity against healthy Bacteroides, lower activity against Proteobacteria, and no activity against the epidemic North American pulsotype (NAP) 1 strain, which predominates currently (5, 40, 46, 47). The clinical implications of our findings for the development of a rational probiotic drug to treat CDI are significant. It is conceivable that a probiotic containing the species which increase significantly with FT may be sufficient to outcompete CDI and restore gut homeostasis. Future investigation will require a larger analysis of patients pre- and post-FT from randomized controlled trials and animal studies to confirm mechanistic hypotheses raised by our work.
MATERIALS AND METHODS
Participants, fecal transplantation, and specimens.
Patient eligibility criteria, donor screening, and FT protocol were as previously described (14). Briefly, all patients (recipients) underwent a full history and physical examination. The study was approved by the Conjoint Health Research Ethics Board, Faculty of Medicine, University of Calgary (study ID, E-24404). Potential family member fecal donors were selected by the patients and were questioned for any of the following contraindications for donation: (i) history of gastrointestinal illness, including peptic ulcer disease, gastroesophageal reflux, irritable bowel syndrome, inflammatory bowel disease, or polyps; (ii) any malignancy; and (iii) antibiotic use or hospitalization within the past 3 months. Donor screening was conducted as previously described (19). FT was conducted using a home-based enema protocol conducted by a family member with 250 ml of donor stool slurry as previously described (14). Patients discontinued chronic oral vancomycin suppressive therapy 24 to 48 h pre-FT. Pre-FT recipient stool specimens for analysis were obtained after discontinuation of vancomycin, on the morning of the transplantation procedure. For failed transplants, recipient posttransplantation samples were obtained on the second day of diarrhea (>3 watery bowel movements/day) recurrence, prior to the reintroduction of vancomycin. For successful cases, post-FT specimens were obtained 14 days postprocedure. Stool samples were collected using standard methods in sterile containers without preservatives. Samples were frozen at −20°C within 24 h of collection prior to DNA extraction.
DNA extraction.
DNA extraction from 200 mg of each of the 21 stool specimens was performed by following the protocol of isolation of DNA from stool for pathogen detection available with the QIAamp DNA stool minikit (Qiagen, Valencia, CA).
16S rRNA V5-V6 tag deep sequencing.
For the amplification of the 16S rRNA gene from the genomic DNA, amplicon fusion primers were designed for the multiplexing and deep-sequencing procedure. The primers consisted of a directional GS FLX titanium primer (forward and reverse), a four-base library key sequence (5′ TCAG 3′), a multiplex identifier (MID) sequence specific for each stool sample pre- and posttransplant, and the gene complementary primer comprising the V5 and V6 tag region (31, 48). A list of the sequences of these fusion primers is provided in Table S2 in the supplemental material. To amplify the 16S rRNA V5-V6 region sequences, 43 µl of Pfx Accuprime Supermix (Invitrogen, Carlsbad, CA) was mixed with 0.25 µM forward and reverse primers (final concentration) and 5 µl of the stool-extracted DNA. Thermocycling involved a 7-min degeneration step at 95°C and 35 cycles of degeneration (95°C for 30 s), annealing (57°C for 1 min), and elongation (68°C for 1 min) as well as a final extension step of 10 min at 68°C. After amplification, the products were purified using the QIAquick PCR purification columns (Qiagen, Valencia, CA) and were quantified using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA). The amplicons were submitted to Genome Quebec for further quality control and 454 deep sequencing. The MID sequence was used for automated software identification of samples after pooling and deep sequencing. Sequence preparation for alignment and downstream analysis was conducted using the mothur package of algorithms (25).
Reference databases and refOTU definitions.
Operational taxonomic units (OTUs) are units of taxonomy. They were set at 97% sequence identity to resolve the data to the species level. A reference sequence-based operational taxonomic unit (refOTU) was defined as containing all query reads sharing the same reference database sequence (or group of sequences) as their nearest neighbor (19, 32). The data shown represent relative abundance in relation to the total taxa present. To determine genera, the V5 and V6 tag sequences obtained were aligned to the SILVA 16S rRNA sequence database (49) with taxonomic classifications obtained from MG-RAST (29) and the RDP classifier (27, 28) (minimum 95% bootstrap score). To determine species, classifications were obtained from MG-RAST. A preclustering step was implemented to reduce sequencing noise from the deep-sequencing data, as has been previously suggested by Huse et al. (21). A distance matrix of the preclustered aligned sequences was generated in order to assign the sequences to refOTUs using the average neighbor algorithm. The majority of the microbiota sequence reads obtained here have an exact or near match in the reference database. In this study, the diversity of the stool bacteria was measured at the highest taxonomic level available (44, 45, 50, 51).
Statistical calculations.
The comparisons of the nearest database distance to tag abundance, the rank abundance plots, the rarefaction curves, and the diversity and richness summary statistics were calculated using absolute tag counts. All these analyses were performed using mothur (25). Principal coordinate analysis of unweighted refOTUs was also conducted using the PCoA function of the mothur pipeline (25). To determine the statistical significance of variation in the abundance of taxa post-FT, the extraction of differential gene expression (EDGE) software for significance analysis using the optimal discovery procedure (26) was used with a false discovery rate of 7% and a P value of 0.05 (see Table S5 in the supplemental material). Data from SILVA and RDP were used for the EDGE significance analysis. The mothur package and the EDGE optimal discovery procedure were chosen for the comparison of community memberships and structures on the basis of the thoroughness of the results previously published for this type of analysis with pooled data using the refOTU approach (19). Newer pipelines that make use of mothur and updated databases have become available recently, and future studies will have to compare results between these algorithms in order to ensure consistent interpretation of sequence data (25). The OTUs were normalized to relative abundance by dividing read counts relative to sample size. Log2-transformed normalized abundance was used to determine the significance of relative changes pre- and post-FT at the genus level. Significantly increasing or decreasing genera were identified at the species level using MG-RAST (29).
SUPPLEMENTAL MATERIAL
Pyrosequencing read preprocessing steps. Download Text S1, DOCX file, 0.1 MB.
Summary statistics of rank abundance data.
Oligonucleotide primer list used in this study.
Taxonomic analysis master list.
Species-level classification master list.
EDGE significance analysis of significant taxa.
ACKNOWLEDGMENTS
We thank the patients, clinical microbiology staff, and administrative staff who supported this work. We thank Patrick Schloss of the University of Michigan for reading the manuscript and providing valuable comments.
We have no conflict of interest to declare.
Funding for this study was provided by a grant from the OPS Innovation Fund (D.R.P.).
Footnotes
Citation Shahinas D, et al. 2012. Toward an understanding of changes in diversity associated with fecal microbiome transplantation based on 16S rRNA gene deep sequencing. mBio 3(5):e00338-12. doi:10.1128/mBio.00338-12.
REFERENCES
- 1. Lavallée C, et al. 2009. Fatal Clostridium difficile enteritis caused by the BI/NAP1/027 strain: a case series of ileal C. difficile infections. Clin. Microbiol. Infect. 15:1093–1099 [DOI] [PubMed] [Google Scholar]
- 2. O’Connor JR, Johnson S, Gerding DN. 2009. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 136:1913–1924 [DOI] [PubMed] [Google Scholar]
- 3. Quesada-Gómez C, et al. 2010. Emergence of Clostridium difficile NAP1 in Latin America. J. Clin. Microbiol. 48:669–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller M, et al. 2010. Health care-associated Clostridium difficile infection in Canada: patient age and infecting strain type are highly predictive of severe outcome and mortality. Clin. Infect. Dis. 50:194–201 [DOI] [PubMed] [Google Scholar]
- 5. Pillai DR, Longtin J, Low DE. 2010. Surveillance data on outbreaks of Clostridium difficile infection in Ontario, Canada, in 2008–2009. Clin. Infect. Dis. 50:1685–1686 (Reply) doi: 10.1086/653007 [DOI] [PubMed] [Google Scholar]
- 6. Cohen SH, et al. 2010. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect. Control Hosp. Epidemiol. 31:431–455 [DOI] [PubMed] [Google Scholar]
- 7. Louie TJ, et al. 2011. Fidaxomicin versus vancomycin for Clostridium difficile infection. N. Engl. J. Med. 364:422–431 [DOI] [PubMed] [Google Scholar]
- 8. Bakken JS. 2009. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe 15:285–289 [DOI] [PubMed] [Google Scholar]
- 9. Bowden TA, Jr, Mansberger AR, Jr, Lykins LE. 1981. Pseudomembraneous enterocolitis: mechanism for restoring floral homeostasis. Am. Surg. 47:178–183 [PubMed] [Google Scholar]
- 10. Eiseman B, Silen W, Bascom GS, Kauvar AJ. 1958. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery 44:854–859 [PubMed] [Google Scholar]
- 11. Grehan MJ, et al. 2010. Durable alteration of the colonic microbiota by the administration of donor fecal flora. J. Clin. Gastroenterol. 44:551–561 [DOI] [PubMed] [Google Scholar]
- 12. Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. 2010. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 44:354–360 [DOI] [PubMed] [Google Scholar]
- 13. Schwan A, Sjölin S, Trottestam U, Aronsson B. 1983. Relapsing Clostridium difficile enterocolitis cured by rectal infusion of homologous faeces. Lancet ii:845. [DOI] [PubMed] [Google Scholar]
- 14. Silverman MS, Davis I, Pillai DR. 2010. Success of self-administered home fecal transplantation for chronic Clostridium difficile infection. Clin. Gastroenterol. Hepatol. 8:471–473 [DOI] [PubMed] [Google Scholar]
- 15. Zimmer C. 12 July 2010. How microbes defend and define us. N. Y. Times. http://www.nytimes.com/2010/07/13/science/13micro.html?pagewanted=all&_r=0
- 16. Von Blech J. 5 March 2011. Heilsamer Stuhl: Im Kampf gegen chronischen Durchfall erzielen Arzte Erfolge: Sie pflanzen den Patienten die Darm-bakterien gesunder Spender ein. Der Spiegel. http://www.spiegel.de/spiegel/print/d-77299777.html
- 17. Walker EP. 27 January 2011. The enema of your enemy is your friend. Fecal transplants could be a cheap and effective treatment for gastrointestinal disorders. Slate. http://www.slate.com/articles/health_and_science/medical_examiner/2011/01/the_enema_of_your_enemy_is_your_friend.html
- 18. Chang JY, et al. 2008. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197:435–438 [DOI] [PubMed] [Google Scholar]
- 19. Dethlefsen L, Huse S, Sogin ML, Relman DA. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280 http://dx.doi.org/10.1371/journal.pbio.0060280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eckburg PB, et al. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huse SM, et al. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255 http://dx.doi.org/10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sogin ML, et al. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Turnbaugh PJ, et al. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reid G, Jass J, Sebulsky MT, McCormick JK. 2003. Potential uses of probiotics in clinical practice. Clin. Microbiol. Rev. 16:658–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schloss PD, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cole JR, et al. 2007. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 35:D169–D172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cole JR, et al. 2009. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145 10.1093/nar/gkp353 PubMed; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meyer F, et al. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386 http://dx.doi.org/10.1186/1471-2105-9-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Storey JD, Dai JY, Leek JT. 2007. The optimal discovery procedure for large-scale significance testing, with applications to comparative microarray experiments. Biostatistics 8:414–432 [DOI] [PubMed] [Google Scholar]
- 30. Woo S, Leek JT, Storey JD. 2011. A computationally efficient modular optimal discovery procedure. Bioinformatics 27:509–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Claesson MJ, et al. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200 http://dx.doi.org/10.1093/nar/gkq873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Costello EK, et al. 2009. Bacterial community variation in human body habitats across space and time. Science 326:1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duncan SH, et al. 2004. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 91:915–923 [DOI] [PubMed] [Google Scholar]
- 34. Duncan SH, Louis P, Flint HJ. 2004. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 70:5810–5817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guilloteau P, et al. 2010. From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr. Res. Rev. 23:366–384 [DOI] [PubMed] [Google Scholar]
- 36. Guilloteau P, et al. 2010. Dietary sodium butyrate supplementation increases digestibility and pancreatic secretion in young milk-fed calves. J. Dairy Sci. 93:5842–5850 [DOI] [PubMed] [Google Scholar]
- 37. Guilloteau P, et al. 2009. Sodium-butyrate as a growth promoter in milk replacer formula for young calves. J. Dairy Sci. 92:1038–1049 [DOI] [PubMed] [Google Scholar]
- 38. Costa MC, et al. 2012. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS One 7:e41484 http://dx.doi.org/10.1371/journal.pone.0041484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Duncan SH, et al. 2007. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 73:1073–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Woodcock JM, Andrews JM, Boswell FJ, Brenwald NP, Wise R. 1997. In vitro activity of BAY 12-8039, a new fluoroquinolone. Antimicrob. Agents Chemother. 41:101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tannock GW, et al. 2010. A new macrocyclic antibiotic, fidaxomicin (OPT-80), causes less alteration to the bowel microbiota of Clostridium difficile-infected patients than does vancomycin. Microbiology 156:3354–3359 [DOI] [PubMed] [Google Scholar]
- 42. Finegold SM, et al. 2004. In vitro activity of ramoplanin and comparator drugs against anaerobic intestinal bacteria from the perspective of potential utility in pathology involving bowel flora. Anaerobe 10:205–211 [DOI] [PubMed] [Google Scholar]
- 43. Manges AR, et al. 2010. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridium difficile-associated disease. J. Infect. Dis. 202:1877–1884 [DOI] [PubMed] [Google Scholar]
- 44. Bäckhed F, et al. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U. S. A. 101:15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ley RE, et al. 2005. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U. S. A. 102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goldstein EJ, et al. 2006. In vitro activity of moxifloxacin against 923 anaerobes isolated from human intra-abdominal infections. Antimicrob. Agents Chemother. 50:148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McDonald LC, et al. 2005. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 353:2433–2441 [DOI] [PubMed] [Google Scholar]
- 48. Wang Y, Qian PY. 2009. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4:e7401 http://dx.doi.org/10.1371/journal.pone.0007401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pruesse E, et al. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196 10.1093/nar/gkm864. PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915–1920 [DOI] [PubMed] [Google Scholar]
- 51. Ley RE, Peterson DA, Gordon JI. 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837–848 [DOI] [PubMed] [Google Scholar]
- 52. Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA. 2008. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6:121–131 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pyrosequencing read preprocessing steps. Download Text S1, DOCX file, 0.1 MB.
Summary statistics of rank abundance data.
Oligonucleotide primer list used in this study.
Taxonomic analysis master list.
Species-level classification master list.
EDGE significance analysis of significant taxa.