

Abstract
In the last few years, the advent of next generation sequencing (NGS) has revolutionized the approach to genetic studies, making whole-genome sequencing a possible way of obtaining global genomic information. NGS has very recently been shown to be successful in identifying novel causative mutations of rare or common Mendelian disorders. At the present time, it is expected that NGS will be increasingly important in the study of inherited and complex cardiovascular diseases (CVDs). However, the NGS approach to the genetics of CVDs represents a territory which has not been widely investigated. The identification of rare and frequent genetic variants can be very important in clinical practice to detect pathogenic mutations or to establish a profile of risk for the development of pathology. The purpose of this paper is to discuss the recent application of NGS in the study of several CVDs such as inherited cardiomyopathies, channelopathies, coronary artery disease and aortic aneurysm. We also discuss the future utility and challenges related to NGS in studying the genetic basis of CVDs in order to improve diagnosis, prevention, and treatment.
Keywords: Next generation sequencing, Genetics of cardiovascular diseases, Cardiomyopathies, Coronary artery disease, Complex disease
INTRODUCTION
In the years after 2000, the completion of the Human Genome Project (HGP) has completely changed the approach to many genetic research studies.
Indeed, the knowledge of the genome sequence has been increasingly important in order to define the basis of human biology and medicine, providing a single, essential reference for all genetic information. Currently, ten years after the HGP, a new technology, next-generation sequencing (NGS), has revolutionized the genomic and transcriptomic approaches to biology reducing the sequencing cost and significantly increasing the throughput[1]. Whole-genome sequencing has become a possible and efficient way to obtain global genomic information[2].
At present, Roche/454 (Roche), Solexa (Illumina) and AB SOLiD (Applied Biosystem) are the NGS technologies predominantly used in genetics. In all NGS platforms, a whole genome, or targeted regions of the genome, are randomly digested into small fragments (or short reads) which are sequenced and are then, either aligned to a reference genome or assembled[3] (Figure 1). The unique combination of specific protocols distinguishes the NGS technology determining limits or advantages. This new strategy of sequencing producing many short reads (tens or hundreds of Gbp for each run) has made necessary the development of several bioinformatics tools to perform the correct alignment/assembly or to analyze large amounts of data. To date, many bioinformatics programs have been created for the different platforms of NGS[4].
Figure 1.
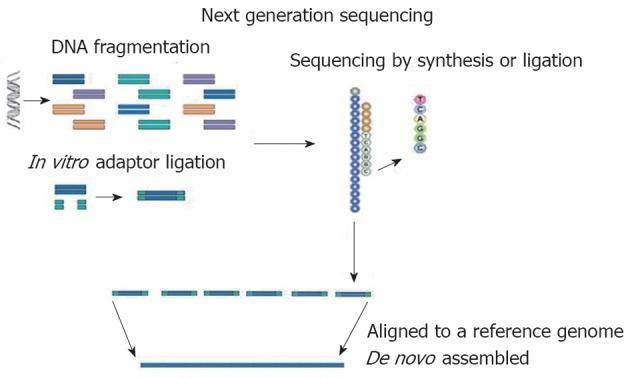
Basic principles of next generation sequencing. A whole genome or a targeted region of the genome are randomly digested into small fragments and then sequenced. The sequence obtained is subsequently aligned to a reference genome or de novo assembled.
For instance, Mapping and Assembly with Quality (MAQ) is a very popular NGS software program developed to efficiently map short reads to a reference genome and derive genotype calls to the consensus sequence with quality scores[5]. MAQ is one of the first reference guided assembly programs. It is accurate, efficient, versatile and has been successfully applied to several NGS projects[6]. Efficient Large-Scale Alignment of Nucleotide Databases (ELAND) is a different NGS program designed to search DNA files for short DNA reads allowing up to 2 errors per match[7,8]. Benchmarks comparing ELAND with other popular NGS software, such as MAQ, Basic Local Alignment Search Tool, Short Oligonucleotide Alignment Program[9] or SeqMap[7] generally place ELAND as one of the fastest available programs. Since many of the programs are open source, additional programming may be needed to modify the program to the needs of a specific NGS project. Finally, some online utility programs, such as EagleView[10] or LookSeq[11], provide some additional assistance on NGS data analysis and interpretation.
However, data management remains the biggest challenge in NGS and the major limiting factor in moving the sequencing to the clinical setting.
Indeed, the production of large numbers of low-cost reads could make the NGS platforms useful for many applications including variant discovery by re-sequencing targeted regions of interest or whole genomes, cataloguing the transcriptomes of cells, and genome-wide profiling of epigenetic marks[12].
NGS has very recently been shown to be successful in identifying novel causative mutations of rare or common Mendelian disorders, also from a very small number of affected individuals[1,12].
Furthermore, it is expected that NGS will be increasingly important in the studies of complex diseases, such as common cardiovascular diseases in which one or more variants in a single gene or more variants in different genes are involved. Currently, the NGS approach to the genetics of cardiovascular diseases (CVDs) represents a territory which has not been widely investigated.
The purpose of this paper is to discuss the recent results of NGS in monogenic classic and in complex genetic cardiovascular disorders, such as inherited cardiomyopathy, channelopathies and coronary artery disease (CAD). We also discuss the potential contribution of future NGS applications in order to significantly improve our understanding of the genetics of CVDs.
NGS AND CVDs
In the etiology of the most CVDs a clear hereditary component has been demonstrated.
The CVDs can be divided in two major categories: the monogenic (more rare) and the polygenic/multifactorial forms (Figure 2).
Figure 2.
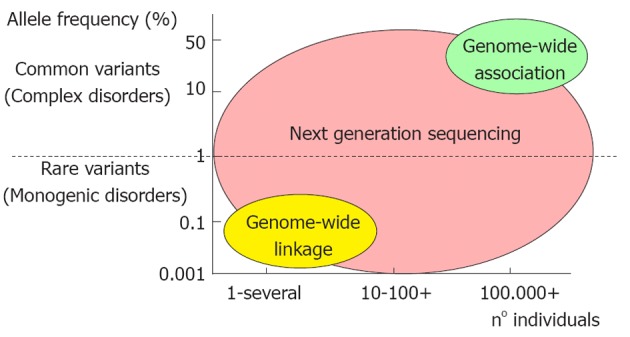
Genetic contribution to monogenic and multigenic cardiovascular diseases and their study approach. The monogenic diseases are caused by a rare mutation in a specific gene and are very rare (< 10 individuals). In the complex diseases, many common genetic variants occur and have a major frequency in the population (> 100 000). The study approach for monogenic diseases is a genome wide linkage study in which one single mutation is identified. Conversely, for complex diseases the genome wide association study is very important to identify a series of common variants contributing to the etiology of the disease. Target re-sequencing by next generation sequencing is an approach which permits the study of both monogenic and complex diseases.
In the monogenic diseases, the mutation of a single gene causes the pathology. These diseases are rare Mendelian traits that show the classical inheritance patterns: autosomal dominant, autosomal recessive, X-linked, or mitochondrial (maternally inherited). Examples of these traits in cardiovascular medicine include structural cardiomyopathies (i.e., hypertrophic or dilated cardiomyopathy) and channelopathies (i.e., Brugada and long QT syndrome) as well as familial dyslipidemias[13].
In clinical practice, the most common CVDs (i.e., CAD) are complex traits that arise from elaborate gene-gene and gene-environmental interactions that confer risk for disease in a probabilistic manner[14]. In these cases, a series of polymorphic variants in several genes increases the risk of developing the disease.
NGS AND INHERITED CARDIOMYOPATHIES
The inherited cardiomyopathies are heterogeneous diseases caused by functional abnormality of cardiac muscle[15]. Hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) are two major clinical forms of inherited cardiomyopathy. HCM, the major cause of sudden death in young people and of heart failure, is characterized by left ventricular hypertrophy, often asymmetric, accompanied by myofibrillar disarrays and reduced compliance (diastolic dysfunction) of cardiac ventricles. Conversely, DCM is characterized by dilated ventricular cavity with systolic dysfunction. The clinical symptom of DCM is heart failure, often associated with sudden death[16]. More than half of HCM patients have a family history of the disease consistent with an autosomal dominant genetic trait[17]. In the case of DCM, about 20%-35% of patients show a family history of the disease (consistent with autosomal dominant inheritance) although some familial cases can be explained by an autosomal recessive or X-linked recessive trait[18-22].
The inherited forms of cardiomyopathies can be caused by mutations in at least 30 different genes. A specific genetic test in patients with cardiomyopathy is of immense clinical importance since some genetic forms of heart muscle diseases are associated with disease manifestation at an early age, an overall poor prognosis, or a high incidence of sudden cardiac death.
Characterizing the genetics of DCM has been a challenging task due to incomplete knowledge of the genes involved in the disease. Unlike HCM, which is largely a disease of the sarcomere (more than 450 mutations in 16 genes have been identified codifying for the sarcomere proteins)[23], the pathways leading to DCM are considerably more diverse, involving genes encoding components of the sarcomere, Z-disk, nuclear lamina proteins, intermediate filaments, and the dystrophin-associated glycoprotein complex[22].
NGS offers a new approach in the diagnosis of cardiomyopathies, and it has recently been used to characterize both HCM and DCM patients. At the present time, despite its good cost-efficiency ratio, NGS is not suitable for clinical practice because of a lack of efficient reduction in genomic complexity and established protocols[24].
Using HCM as a diagnostic model, Dames et al[25] developed a NGS-based approach for multi-gene re-sequencing in a clinical laboratory. Sixteen genes implicated in HCM have been sequenced using an uncharacterized human DNA sample. A Long-Range polymerase chain reaction (PCR) (a PCR method used to amplify a long region of genome) was used for gene enrichment, followed by comparative sequencing on the Illumina Genome Analyzer and Roche 454 GS FLX platforms[25]. Both platforms detected several different variants, of which only 27 were common and have been confirmed by Sanger classical sequencing. According to these results the authors proposed a targeted re-sequencing by combining Long-Range PCR and NGS as a new approach for multigene analysis.
Conversely, Meder et al[24] established a microarray-based target enrichment followed by SOLiD NGS for a comprehensive and cost-efficient genetic diagnosis of cardiomyopathies. This approach increased the mean depth of coverage of cardiomyopathy genes analyzing 1092 disease exons and adjacent intronic regions in only one NGS run and identified 1891 sequence variants within these regions (of which 349 were nonsynonymous)[24].
NGS has also been used to study the maternally-inherited cardiomyopathy caused by mutations in mitochondrial DNA. Zaragoza et al[26] studied 18 patients with mitochondrial cardiomyopathy and two patients with suspected mitochondrial disease. Sequencing PCR-amplified mtDNA with a single run on Roche’s 454 Genome Sequencer identified 427 variants.
Recently, Herman et al[27] analyzed the gene encoding the sarcomere protein, titin (TTN), in subjects with DCM, subjects with HCM, and in controls using NGS, and evaluated the deleterious variants for cosegregation in families and assessed clinical characteristics. Using this approach they found that TTN truncating mutations are a common cause of DCM, occurring in approximately 25% of familial cases of idiopathic DCM and in 18% of sporadic cases. The authors concluded that the incorporation of sequencing approaches which detect TTN truncations in genetic testing for DCM should substantially increase test sensitivity, thereby allowing earlier diagnosis and therapeutic intervention for many patients with this pathology[27].
Finally, the NGS tools may be used to develop RNA sequencing methodologies for high-throughput comprehensive analysis of individual transcriptomic profiles. In Gαq transgenic mice, a well-characterized model of cardiac hypertrophy/cardiomyopathy[28], the results of sequencing through NGS (Illumina platform) have been compared with an array-based transcriptional profiling.
The results of this study revealed superior dynamic range for mRNA expression and enhanced specificity for reporting low-abundance transcripts by RNA sequencing.
Together these studies suggest that the application of NGS tools in the inherited cardiomyopathies will be increasingly important to define the genetic component of these disorders and to detect cardiomyopathy-causing mutations with high accuracy in a fast and cost-efficient manner which will be suitable for daily clinical practice of genetic testing[24].
NGS AND CHANNELOPATHIES
A different group of CVDs included in inherited cardiomyopathies is the primary electrical diseases such as Brugada syndrome (BrS) and long QT syndrome (LQTS). Each of these cardiac channelopathies is characterized by a unique genetic profile and clinical features[29]. Advances in molecular biology have allowed the identification of many disorders linked to specific genes previously ascribed as idiopathic. Genetic studies in families with LQTS have associated this disorder with gene mutations affecting cardiac ion channels - specifically the sodium and potassium channels. To date, hundreds of variants have been identified in 13 LQTS genes[30].
Conversely, BrS has been associated with more than 100 mutations in 7 genes. Loss-of-function mutations in the SCN5A gene, which encodes the alpha-subunit of the sodium ion channel, causes 18%-30% of BrS cases[31].
To date, NGS methodologies have not been applied to these disorders.
In addition, since more genes are involved in these diseases, the application of NGS will provide important advantages for identifying pathogenetic mutations in a fast and cost-efficient manner.
NGS AND CAD
CAD remains the leading cause of death in industrialized and developing countries.
It has been estimated that heritable factors contribute 30%-60% of the inter-individual variation in the risk of CAD[32].
Mendelian disorders such as familial dyslipidemia which lead to alterations in the lipid profile are heritable risk factors for CAD[33]. While these rare mutations are well-recognized and well-characterized, the identification of common genetic variants associated with CAD is more difficult despite strong evidence that disease susceptibility is heritable.
Recently, genome-wide association studies (GWAS) have identified several common variants (single nucleotide polymorphisms, SNPs) associated with the risk of CAD. Notably, these SNPs are not inherited independently, but as “bins” or “blocks”[34]. Furthermore, the genotype of 1 SNP may be sufficient to affect the genotype of all other SNPs within a given linkage disequilibrium block (haplotype), thereby “tagging” an entire region of interest[35].
GWAS are performed using the array technologies that in parallel captured hundreds of thousands and now over a million SNPs in independent haplotype blocks in large case/control samples[14,36-44].
Next generation sequencing has enabled targeted re-sequencing of genomic regions found to be involved in the disease. In particular, the re-sequencing of genomic regions identified by GWAS in healthy and diseased populations represents a powerful strategy for assessing the contribution of rare variants to disease etiology[45]. This is because NGS is able to identify rare genetic variants with a minor allele frequency (MAF) of < 5%, which complement the common susceptibility SNPs (MAF > 5%) established through the GWAS[46].
For instance, after GWAS several other studies confirmed the exceptional role of the chromosome 9p21.3 region on the risk of CAD[36,42].
Interestingly, a very recent study analyzed sequence data from a 240-kilobase (kb) region on chromosome 9p21 in 47 individuals using the Illumina GA platform[47]. The authors compared the results of targeted sequencing with NGS to pilot data from the 1000 Genomes Project[48] (characterized by a description of the location, allele frequency and local haplotype structure of approximately 15 million SNPs). The findings showed that the targeted sequencing provides high sensitivity for lower-frequency variants despite several gaps in sequence coverage which existed after the alignment to a reference genome.
Furthermore, NGS has been used to detect rare variants in the gene encoding adiponectin. In this study, a combination of family-based linkage, whole-exome sequencing (by NGS), direct sequencing and association methods has been developed in order to efficiently identify rare variants associated with large effects in families from the Insulin Resistance Atherosclerosis Family Study[49]. These results suggest that this approach could be advantageous in discovering novel genes influencing complex traits in a wide range of family studies.
NGS AND OTHER CVDs
The NGS technologies have also been applied in other cardiovascular diseases with complex traits, such as aortic aneurysm. Harakalova et al[50] performed a pilot experiment designed to find an efficient method for the detection of rare genetic variants in regions of interest in large sample groups with aortic aneurysm using SOLiD platform. They discussed the challenges and limitations connected with this approach and showed that the high number of novel variants detected per pool can be limiting factors for successful variant prioritization and confirmation. Indeed, they discovered 681 coding variants, however, the majority of the detected candidate novel variants were false positives.
Moreover, a very recent study using exome sequencing of 2 distantly affected relatives, efficiently and successfully identified a frameshift mutation in the SMAD3 gene as the cause of vascular disease in a family with arterial aneurysms and dissections inherited in an autosomal dominant pattern. Subsequent sequencing of families involving multiple members with thoracic aortic aneurysms and acute aortic dissections identified SMAD3 mutations in 2% of familial thoracic aortic aneurysms and dissections[51].
Additionally, Sakai et al[52] used two different technologies (re-sequencing array technology and NGS) to analyze eight genes associated with syndromic aortic aneurysms and/or dissections. They identified eighteen variants with both technologies and concluded that NGS was able to detect almost all types of mutations, but it requires improved informatics methods.
Finally, NGS with the Illumina platform has been used to sequence genomic DNA in a nuclear family with a history of thrombophilia[53]. Two hundred variants have been identified and compared with different groups of HapMap populations. This method has allowed the identification of multigenic risk for inherited thrombophilia and appropriate pharmacological therapy.
FUTURE PERSPECTIVE
Table 1 summarizes the current application of NGS in cardiovascular disorders. NGS technology promises to improve our understanding of the genetic architecture and the missing heritability of CVDs.
Table 1.
Next generation sequencing approaches in cardiovascular diseases
| Disease | NGS approach | Ref. | |
| Monogenic diseases | DCM | Target re-sequencing in 47 associated genes | Meder et al[24] |
| Sequencing of TTN gene | Herman et al[27] | ||
| HCM | Target re-sequencing in 47 associated genes | Meder et al[24] | |
| Target re-sequencing in 16 associated genes | Dames et al[25] | ||
| RNA sequencing | Matkovich et al[28] | ||
| Mitochondrial cardiomyopathies | Mitochondrial DNA sequencing | Zaragoza et al[26] | |
| Channelopathies | No approach | ||
| Complex diseases | CAD | Target re-sequencing of 9p21.3 region | Shea et al[47] |
| Sequencing of ADIPOQ gene | Bowden et al[49] | ||
| AA | Target sequencing in associated genes | Harakalova et al[50], Sakai et al[52] | |
| Thrombophilia | Target sequencing in gene associated | Dewey et al[53] |
DCM: Dilated Cardiomyopathies; HCM: Hypertrophic cardiomyopathies; CAD: Coronary artery disease; AA: Aortic aneurysm; NGS: Next generation sequencing; TTN: Titin; ADIPOQ: Adiponectin.
In the near future, NGS will revolutionize the genetic study of cardiovascular disease allowing unprecedented opportunities to detect mutations in disease-genes with high accuracy in a fast and cost-efficient manner in daily clinical practice.
In particular, the targeted re-sequencing of the region of interest selected by GWAS, using NGS technologies, will allow identification of rare SNPs involved in the risk of CVD.
To date, data on the reproducibility of NGS results in the cardiovascular setting are limited by too few available studies. In addition, results in other more explored fields such as cancer showed that two independent groups can simultaneously arrive at different sets of gene alterations, without overlap between the two sets of mutations identified[54,55]. This finding suggests that the reproducibility of data could be one major limitation of these advanced techniques. Furthermore, as previously cited, Dames et al[25] demonstrated that different variants were found using two different NGS tools of which only a few were successively confirmed by the conventional Sanger approach of sequencing. Thus, new efforts are needed to improve sequencing accuracy and streamline technical processes as the next steps toward transitioning NGS into the clinical laboratory.
The major challenge in NGS is that although it produces an enormous volume of data cheaply, in most cases, the millions of reads generated do not cover the coding regions of disease genes[56]. Indeed, NGS provides only 50-500 continuous base-pair reads[4], making it difficult for both the assembly and the data analysis. Therefore, new methods should be developed to selectively capture DNA from the region of interest in order to sequence only targeted regions.
In addition to short DNA sequence reads, these technologies can generate terabyte-sized data files for each instrument run, greatly increasing the computer resource requirements. Given the vast amount of data produced by NGS, the creation of informatics tools for the storage and analysis of data will be essential to the successful application of NGS[4].
In the future, with the advent of NGS and the progressive increase in data sequences of the human genome from projects such as HapMap and the 1000 Genomes Project, investigators will have to choose between the multiple strategies to test a reference panel of polymorphic sites.
Moreover, parallel genome-wide studies are characterizing a large number of genes affecting the risk factors for CAD including dyslipidemia, hypertension, diabetes mellitus, and obesity. These findings are to be integrated with loci directly associated with CAD to obtain the fullest picture. Thus, in the next few years, the main focus of these studies will be to define a risk prediction as well as a preventive and individual therapy for CAD.
In the last few years, a technological revolution has taken place in the field of epigenomics.
The development of NGS devices are now providing researchers with tools to draw high-resolution maps of DNA methylation and histone modifications in normal tissues and diseases[57]. NGS technologies may be used to profile epigenetic alterations that influence gene expression and to study the genome-wide epigenetic changes that occur in the genome in cardiovascular disease.
Footnotes
Peer reviewers: Cristina Vassalle, PhD, G. Monasterio Foundation and Institute of Clinical Physiology, Via Moruzzi 1, I-56124 Pisa, Italy; Yuri V Bobryshev, PhD, Associate Professor, School of Medical Sciences, Faculty of Medicine, University of New South Wales, Kensington, NSW 2052, Australia; Mohamed Chahine, PhD, Professeur Titulaire, Le Centre de Recherche Université Laval Robert-Giffard, Local F-6539, 2601 chemin de la Canardière, Québec G1J 2G3, Canada
S- Editor Cheng JX L- Editor Webster JR E- Editor Li JY
References
- 1.Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- 2.Su Z, Ning B, Fang H, Hong H, Perkins R, Tong W, Shi L. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev Mol Diagn. 2011;11:333–343. doi: 10.1586/erm.11.3. [DOI] [PubMed] [Google Scholar]
- 3.Flicek P, Birney E. Sense from sequence reads: methods for alignment and assembly. Nat Methods. 2009;6:S6–S12. doi: 10.1038/nmeth.1376. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Chiodini R, Badr A, Zhang G. The impact of next-generation sequencing on genomics. J Genet Genomics. 2011;38:95–109. doi: 10.1016/j.jgg.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang H, Wong WH. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng J, Moorhead M, Weng L, Siddiqui F, Carlton VE, Ireland JS, Lee L, Peterson J, Wilkins J, Lin S, et al. High-throughput, high-accuracy array-based resequencing. Proc Natl Acad Sci USA. 2009;106:6712–6717. doi: 10.1073/pnas.0901902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 10.Huang W, Marth G. EagleView: a genome assembly viewer for next-generation sequencing technologies. Genome Res. 2008;18:1538–1543. doi: 10.1101/gr.076067.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manske HM, Kwiatkowski DP. LookSeq: a browser-based viewer for deep sequencing data. Genome Res. 2009;19:2125–2132. doi: 10.1101/gr.093443.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 13.Damani SB, Topol EJ. Future use of genomics in coronary artery disease. J Am Coll Cardiol. 2007;50:1933–1940. doi: 10.1016/j.jacc.2007.07.062. [DOI] [PubMed] [Google Scholar]
- 14.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 15.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 16.Kimura A. Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. J Hum Genet. 2010;55:81–90. doi: 10.1038/jhg.2009.138. [DOI] [PubMed] [Google Scholar]
- 17.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104:557–567. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 18.Colombo MG, Botto N, Vittorini S, Paradossi U, Andreassi MG. Clinical utility of genetic tests for inherited hypertrophic and dilated cardiomyopathies. Cardiovasc Ultrasound. 2008;6:62. doi: 10.1186/1476-7120-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, Vatta M, Pinamonti B, Muntoni F, Caforio AL, et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol. 1999;34:181–190. doi: 10.1016/s0735-1097(99)00172-2. [DOI] [PubMed] [Google Scholar]
- 20.Kärkkäinen S, Peuhkurinen K. Genetics of dilated cardiomyopathy. Ann Med. 2007;39:91–107. doi: 10.1080/07853890601145821. [DOI] [PubMed] [Google Scholar]
- 21.Kimura A. Contribution of genetic factors to the pathogenesis of dilated cardiomyopathy: the cause of dilated cardiomyopathy: genetic or acquired? (genetic-side) Circ J. 2011;75:1756–1765; discussion 1765. doi: 10.1253/circj.cj-11-0368. [DOI] [PubMed] [Google Scholar]
- 22.Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010;25:198–204. doi: 10.1097/HCO.0b013e328337ba52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voelkerding KV, Dames S, Durtschi JD. Next generation sequencing for clinical diagnostics-principles and application to targeted resequencing for hypertrophic cardiomyopathy: a paper from the 2009 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2010;12:539–551. doi: 10.2353/jmoldx.2010.100043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meder B, Haas J, Keller A, Heid C, Just S, Borries A, Boisguerin V, Scharfenberger-Schmeer M, Stähler P, Beier M, et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ Cardiovasc Genet. 2011;4:110–122. doi: 10.1161/CIRCGENETICS.110.958322. [DOI] [PubMed] [Google Scholar]
- 25.Dames S, Durtschi J, Geiersbach K, Stephens J, Voelkerding KV. Comparison of the Illumina Genome Analyzer and Roche 454 GS FLX for resequencing of hypertrophic cardiomyopathy-associated genes. J Biomol Tech. 2010;21:73–80. [PMC free article] [PubMed] [Google Scholar]
- 26.Zaragoza MV, Fass J, Diegoli M, Lin D, Arbustini E. Mitochondrial DNA variant discovery and evaluation in human Cardiomyopathies through next-generation sequencing. PLoS One. 2010;5:e12295. doi: 10.1371/journal.pone.0012295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matkovich SJ, Zhang Y, Van Booven DJ, Dorn GW. Deep mRNA sequencing for in vivo functional analysis of cardiac transcriptional regulators: application to Galphaq. Circ Res. 2010;106:1459–1467. doi: 10.1161/CIRCRESAHA.110.217513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerrone M, Priori SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J. 2011;32:2109–2118. doi: 10.1093/eurheartj/ehr082. [DOI] [PubMed] [Google Scholar]
- 30.Refsgaard L, Holst AG, Sadjadieh G, Haunsø S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur J Hum Genet. 2012;20:905–908. doi: 10.1038/ejhg.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedley PL, Jørgensen P, Schlamowitz S, Moolman-Smook J, Kanters JK, Corfield VA, Christiansen M. The genetic basis of Brugada syndrome: a mutation update. Hum Mutat. 2009;30:1256–1266. doi: 10.1002/humu.21066. [DOI] [PubMed] [Google Scholar]
- 32.Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. doi: 10.1056/NEJM199404143301503. [DOI] [PubMed] [Google Scholar]
- 33.Miller NE, Miller GJ. Letter: High-density lipoprotein and atherosclerosis. Lancet. 1975;1:1033. doi: 10.1016/s0140-6736(75)91977-7. [DOI] [PubMed] [Google Scholar]
- 34.Damani SB, Topol EJ. Emerging genomic applications in coronary artery disease. JACC Cardiovasc Interv. 2011;4:473–482. doi: 10.1016/j.jcin.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 36.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trégouët DA, König IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel-Nitschke P, Perret C, DeSuremain M, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 41.Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helgadottir A, Thorleifsson G, Magnusson KP, Grétarsdottir S, Steinthorsdottir V, Manolescu A, Jones GT, Rinkel GJ, Blankensteijn JD, Ronkainen A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 43.Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, Thorleifsson G, Helgadottir H, Steinthorsdottir V, Stefansson H, Williams C, Hui J, Beilby J, Warrington NM, James A, Palmer LJ, Koppelman GH, Heinzmann A, Krueger M, Boezen HM, Wheatley A, Altmuller J, Shin HD, Uh ST, Cheong HS, Jonsdottir B, Gislason D, Park CS, Rasmussen LM, Porsbjerg C, Hansen JW, Backer V, Werge T, Janson C, Jönsson UB, Ng MC, Chan J, So WY, Ma R, Shah SH, Granger CB, Quyyumi AA, Levey AI, Vaccarino V, Reilly MP, Rader DJ, Williams MJ, van Rij AM, Jones GT, Trabetti E, Malerba G, Pignatti PF, Boner A, Pescollderungg L, Girelli D, Olivieri O, Martinelli N, Ludviksson BR, Ludviksdottir D, Eyjolfsson GI, Arnar D, Thorgeirsson G, Deichmann K, Thompson PJ, Wjst M, Hall IP, Postma DS, Gislason T, Gulcher J, Kong A, Jonsdottir I, Thorsteinsdottir U, Stefansson K. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. 2009;41:342–347. doi: 10.1038/ng.323. [DOI] [PubMed] [Google Scholar]
- 44.Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bansal V, Harismendy O, Tewhey R, Murray SS, Schork NJ, Topol EJ, Frazer KA. Accurate detection and genotyping of SNPs utilizing population sequencing data. Genome Res. 2010;20:537–545. doi: 10.1101/gr.100040.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topol EJ, Frazer KA. The resequencing imperative. Nat Genet. 2007;39:439–440. doi: 10.1038/ng0407-439. [DOI] [PubMed] [Google Scholar]
- 47.Shea J, Agarwala V, Philippakis AA, Maguire J, Banks E, Depristo M, Thomson B, Guiducci C, Onofrio RC, Kathiresan S, et al. Comparing strategies to fine-map the association of common SNPs at chromosome 9p21 with type 2 diabetes and myocardial infarction. Nat Genet. 2011;43:801–805. doi: 10.1038/ng.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowden DW, An SS, Palmer ND, Brown WM, Norris JM, Haffner SM, Hawkins GA, Guo X, Rotter JI, Chen YD, et al. Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum Mol Genet. 2010;19:4112–4120. doi: 10.1093/hmg/ddq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harakalova M, Nijman IJ, Medic J, Mokry M, Renkens I, Blankensteijn JD, Kloosterman W, Baas AF, Cuppen E. Genomic DNA pooling strategy for next-generation sequencing-based rare variant discovery in abdominal aortic aneurysm regions of interest-challenges and limitations. J Cardiovasc Transl Res. 2011;4:271–280. doi: 10.1007/s12265-011-9263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, Clarke L, Bernier F, Santos-Cortez RL, Leal SM, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakai H, Suzuki S, Mizuguchi T, Imoto K, Yamashita Y, Doi H, Kikuchi M, Tsurusaki Y, Saitsu H, Miyake N, et al. Rapid detection of gene mutations responsible for non-syndromic aortic aneurysm and dissection using two different methods: resequencing microarray technology and next-generation sequencing. Hum Genet. 2012;131:591–599. doi: 10.1007/s00439-011-1105-7. [DOI] [PubMed] [Google Scholar]
- 53.Dewey FE, Chen R, Cordero SP, Ormond KE, Caleshu C, Karczewski KJ, Whirl-Carrillo M, Wheeler MT, Dudley JT, Byrnes JK, et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011;7:e1002280. doi: 10.1371/journal.pgen.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009;27:522–530. doi: 10.1016/j.tibtech.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 57.Martín-Subero JI, Esteller M. Profiling epigenetic alterations in disease. Adv Exp Med Biol. 2011;711:162–177. doi: 10.1007/978-1-4419-8216-2_12. [DOI] [PubMed] [Google Scholar]