

Abstract
AIM: To investigate the impact of dietary copper given at different time points on the onset of fulminant hepatitis.
METHODS: The Long-Evans cinnamon (LEC) rat model of Wilson’s disease (WD) was used to study the impact of high dietary copper (hCu) on the induction of fulminant hepatitis at early or late time points of life. High Cu diet was started in rat pups or in adults (month 5) for three months. Animals that received reduced dietary copper (rCu) throughout their lifetime served as a control. Hepatitis-associated serum markers (alanine aminotransferase, aspartate transaminase, bilirubin) were analyzed in animal groups receiving hCu or rCu. Liver copper content and liver histology were revealed at sacrifice. A set of 5 marker genes previously found to be affected in injured liver and which are related to angiogenesis (Vegfa), fat metabolism (Srebf1), extracellular matrix (Timp1), oxidative stress (Hmox1), and the cell cycle (Cdkn1a) were analyzed by real-time polymerase chain reaction.
RESULTS: Regardless of the time point when hCu was started, LEC rats (35/36) developed fulminant hepatitis and died. Animals receiving rCu (36/36) remained healthy, did not develop hepatitis, and survived long term without symptoms of overt disease, although liver copper accumulated in adult animals (477 ± 75 μg/g). With regard to start of hCu, onset of fulminant hepatitis was significantly (P < 0.001) earlier in adults (35 ± 9 d) that showed pre-accumulation of liver copper as compared to the pup group (77 ± 15 d). Hepatitis-associated serum markers, liver copper and liver histology, as well as gene expression, were affected in LEC rats receiving hCu. However, except for early and rapid onset of hepatitis, biochemical and molecular markers were similar at the early and late time points of disease.
CONCLUSION: Rapid onset of fulminant hepatitis in asymptomatic LEC rats with elevated liver copper suggests that there is a critical threshold of liver copper which is important to trigger the course of WD.
Keywords: Wilson’s disease, Fulminant hepatitis, Acute liver failure, Dietary copper, Long-Evans cinnamon rat, ATP7B
INTRODUCTION
Wilson’s disease (WD) is a genetic disorder transmitted by a recessive gene located on chromosome 13[1,2]. Diagnosis of WD is mostly established on the basis of combined biochemical and clinical parameters, and lately by specific genetic analysis[3-6]. Presentation of WD includes hepatic insufficiency, acute and chronic active hepatitis, fulminant hepatitis and/or extra hepatic manifestations. Although it is a rare disease, fulminant hepatitis is frequently observed in WD patients and is followed by high mortality that accounts for approximately 5% of acute liver failure (ALF) observed worldwide. The disease is linked to an imbalance of copper homeostasis that is due to mutation of the ATP7B copper transporter gene expressed primarily in the liver[7,8]. More than 600 mutations have been reported in human ATP7B. Effective drug treatment of WD has been established which aims at chelation or reduction of copper[1]. However, compliance to take lifelong treatment or to reduce dietary copper intake is low in patients[9]. Liver transplantation remains the only treatment option of WD patients encountering ALF.
In healthy individuals copper is absorbed via the intestine and delivered to the liver, where it is either transferred by ATP7B into ceruloplasmin that is secreted into blood or exported via bile[2]. Malfunction of the ATP7B gene in WD patients leads to large quantities of toxic copper in the liver and other organs. However, manifestation and onset of the disease induced by toxic copper can vary to a great extent between WD patients[10-12]. Individual mutations of the ATP7B gene as well as mutations outside of this locus have been implicated to be important for the course of the disease as suggested, e.g., by rare studies of twins having the same mutation of the ATP7B gene but presenting different courses of WD[13,14]. Besides genetic factors, environmental factors, such as the amount of dietary copper intake, may also have an impact on WD[15,16]. However, studies of such factors are difficult in humans.
Much has been learned from animal models of WD. The Long-Evans cinnamon (LEC) rat has a large deletion of the ATP7B gene[17] and has many characteristics that are also observed in patients, e.g., low ceruloplasmin, high liver copper levels, and sensitivity to anti-copper therapy. The LEC rat strain can be housed on a commercial diet containing standard copper concentrations (about 7-15 mg/kg). Using this standard chow, 40%-60% of LEC rats spontaneously encounter fulminant hepatitis at the age of 80-120 d[18]. The LEC rat is therefore a highly attractive model to analyze pathogenesis of the liver, and is also used to study the effect of novel therapeutic approaches for liver disease, e.g., hepatocyte transplantation or gene transfer[19,20].
In the present study, the role of low and high dietary copper given at different time points was investigated in male and female LEC rats. We hypothesized that preloading of copper may affect the course and onset of disease. A high copper (hCu) diet was given to juvenile and adult rats that could pre-accumulate liver copper. The impact on hepatitis, liver copper, histology, gene expression, and survival was examined.
MATERIALS AND METHODS
Animals
LEC rats that lack functional ATP7B[17] and Long-Evans agouti (LEA) rats expressing wild-type ATP7B were a kind gift of S. Gupta (Albert Einstein College of Medicine, New York, United States). Rats were genotyped for ATP7B by polymerase chain reaction (PCR) analysis using primers essentially as described previously[21]. For the hCu regimen, animals received a solid diet that contains a standard concentration (13 mg/kg) of copper (1324, Altromin, Germany). Animals of the hCu groups additionally received tap water containing 20 mg copper/L [copper (II) chloride, Sigma-Aldrich]. For the reduced copper (rCu) regimen, rats received a specialized solid diet (C1041, Altromin) with a copper content of 0.3 mg/kg and distilled tap water. Female rats (n = 12) which were reported to have higher sensitivity to copper[18,22] and male rats (n = 6) were analyzed for each group. LEA rats, heterozygotes and LEC rats housed on standard diet were used as controls. The incidence of severe jaundice with strong disturbance of common as well as spontaneous behaviour, and weight loss (> 20%) which was accompanied by coma, led to sacrifice of the animals. The protocol for animal use was approved by local authorities.
Analysis of serum samples
Blood was taken under retrobulbar anesthesia with isoflurane (1-chloro-2,2,2-trifluoroethyl difluoromethyl ether, Abbott). Activities of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) and total concentration of bilirubin in serum were analyzed photometrically using a Cobas Modular System (Roche Diagnostics, Germany). Ceruloplasmin oxidase activity was determined using the modified protocol of Schosinsky et al[23] adapted to a 96-well plate.
Liver histology
For histological evaluation, liver specimens were fixed by immersion for at least 24 h in 4% formaldehyde solution, and were subsequently dehydrated and embedded in paraffin wax in order to cut serial sections at a thickness of 5 μm. Morphologic parameters were determined after hematoxylin and eosin staining using standard protocols, and scored by a blinded observer, e.g., for polyploidy, steatosis, apoptosis, and proliferation.
Copper determination
For determination of copper content, liver samples were dried for 72 h at 70 °C. Dry weight was determined using the analytical balance ME235S (Sartorius, Germany). Liver was redissolved in 65% nitric acid (Merck, Germany) and copper concentration was determined by flame atomic absorption spectroscopy (Shimadzu AA6300, Japan).
Real-time PCR analysis
Randomly chosen liver samples were immediately frozen after resection. Total RNA was extracted after disruption of tissue using a homogenizer and RNeasy kit (Qiagen, Hilden, Germany). Reverse transcription-PCR was carried out as described[24]. Briefly, RNA was reverse transcribed using SuperScript II according to the instructions of the manufacturer (Invitrogen). Primers for PCR (Table 1) were synthesized by MWG Biotech (Germany). For quantitative real-time PCR, a SYBR® Green kit (Eurogentec, Belgium) was used. PCR was performed on the ABI PRISMTM 7900HT sequence detector (PE Applied Biosystems, United States). Each sample was tested in three independent experiments. The ct value was normalized to the expression of the house-keeping gene HPRT (∆ct method). Relative expression level of gene normalized to house-keeping gene was determined by the equation 2∆ct and fold-change was calculated thereafter.
Table 1.
Genes analyzed by real-time polymerase chain reaction in liver
| Gene symbol | Synonym | Accession number | Primer (forward/reverse) |
| Vegfa | Vascular endothelial growth factor A | NM031836 | CAGTTCGAGGAAAGGGAAAGGCAAATGCTTTCTCCGCTCTG |
| Hmox1 | Heme oxygenase (decycling) 1 | NM012580 | AGAGGCTAAGACCGCCTTCC AGGCCTCTGGCGAAGAAAC |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A | U24174 | CTTGCACTCTGGTGTCTCACG ATCGGCGCTTGGAGTGATAG |
| Timp1 | TIMP metallopeptidase inhibitor 1 | BC099821 | CCTGGTTCCCTGGCATAATC TTGCAAGGGATGGCTGAAC |
| Srebf1 | Sterol regulatory element binding protein-1 | AF286470 | AGAAGGCCAGTGGGTACCTG TGCGGGCCACAAGAAGTAG |
Statistical analysis
Statistical analysis was performed using SPSS 17.0 software. Data are given as mean ± SE and median. Data were analyzed by the Student’s two tailed t test using Bonferroni correction for post-hoc pairwise comparisons or Kruskal-Wallis test. Kaplan-Meier survival test and logrank test were performed for time to event data.
RESULTS
Onset of fulminant hepatitis
The role of low and high dietary copper in the induction of severe liver disease was studied with respect to the start of a copper diet in LEC rats of different age (Figure 1). A hCu was started early in pups or late in adults at month 5. The latter animals received a rCu up to month 5. The time point of hCu in adults was chosen to encompass the phase of fulminant hepatitis that occurs in LEC rats at the age of 80-120 d when using commercial chow[18]. LEC rats subjected to hCu displayed significantly elevated hepatitis-associated markers (Figure 2A), and developed severe jaundice (bilirubin > 2.0 mg/dL). With regard to start of hCu, levels of serum markers increased significantly earlier (P < 0.001) after the late regimen as compared with the early regimen (Figure 2B). Both genders displayed similar onset of significantly elevated serum markers (Table 2). LEC rats receiving a rCu diet or animals of the control groups did not develop hepatitis.
Figure 1.
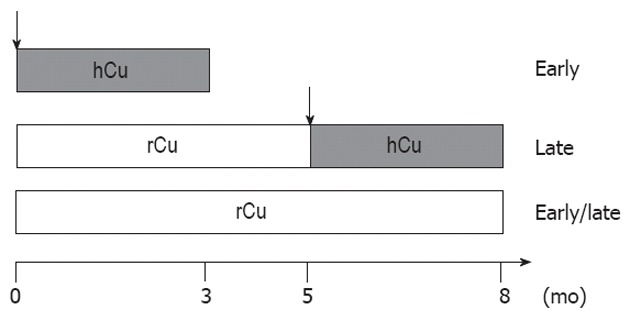
Schematic representation of study design. For the early copper regimen, Long-Evans cinnamon (LEC) rat pups received a high copper (hCu) diet up to month 3. For the late copper regimen, LEC pups were first housed on a reduced copper (rCu) diet until adult (month 5). Thereafter, rats received hCu for 3 mo up to the end of observation time (month 8). LEC rats received rCu diet throughout the observation period (early/late). ATP7B heterozygotes as well as Long-Evans agouti rats were also studied using an early hCu regimen. Arrow indicates start of hCu regimen.
Figure 2.

High copper regimen induces fulminant hepatitis. A: Bilirubin, aspartate transaminase (AST) and alanine aminotransferase (ALT) were determined in Long-Evans cinnamon (LEC) rats (-/-), heterozygotes (+/-) or Long-Evans agouti (LEA) rats (wt). Animals received a high copper (hCu) or reduced copper (rCu) diet as an early or late regimen. Median ± SE of maximal values obtained from animals are shown; B: Time course of serum markers in LEC rats that received early (top) or late (bottom) hCu. X-axis represents day after start of hCu. Animals of other groups showed normal levels. Of note, the onset of increased values following late hCu was at day 189 ± 42 after birth. Each line represents one animal. Logrank test using threshold (mean ± SE of LEA rats ×2) was used; C: Kaplan-Meier survival curve of LEC rats receiving an early and late hCu regimen are shown. Survival is represented as day after start of hCu. Of note, age of animals at death in late group was 190 ± 9 d.
Table 2.
Male and female Long-Evans cinnamon rats after copper diet
| Early hCu | Late hCu | rCu | Long-Evans agouti | |||||
| Male | Female | Male | Female | Male | Female | Female | ||
| Bilirubin | > 0.4 mg/dL1 (d) | 55 (67 ± 5) | 63 (60 ± 1) | 28 (30 ± 1) | 28 (32 ± 1) | 0.32 (0.3 ± 0) | 0.22 (0.2 ± 0) | 0.22 (0.2 ± 0) |
| AST | > 240 U/L1 (d) | 74 (75 ± 3) | 68 (68 ± 1) | 28 (34 ± 3) | 30 (33 ± 1) | 1152 (117 ± 4) | 1032 (103 ± 3) | 1142 (114 ± 5) |
| ALT | > 110 U/L1 (d) | 44 (50 ± 2) | 54 (54 ± 1) | 28 (30 ± 2) | 28 (31 ± 1) | 692 (69 ± 6) | 752 (75 ± 6) | 542 (54 ± 1) |
| Liver Cu | (μg/g) | 868 (880 ± 93) | 1036 (1058 ± 59) | 1494 (1452 ± 161) | 1235 (1178 ± 59) | 503 (495 ± 75) | 309 (326 ± 29) | 46 (44 ± 9) |
| Survival | (day after hCu) | 85 (81 ± 4) | 81 (75 ± 9) | 37 (37 ± 6) | 35 (35 ± 2) | NA | NA | NA |
NA: Not available; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; hCu: High copper; rCu: Reduced copper.
Sera up to sacrifice were analyzed by logrank test using thresholds calculated from Long-Evans agouti rat (mean ± SE) × 2;
Max values up to sacrifice are given. Data of male and females did not differ (logrank, t test, Kruskal-Wallis, Kaplan-Meier).
LEC rats (36/36) receiving rCu survived the observation period (8 mo), and no signs of overt morbidity were recorded in individual rats (18/18) as compared to controls, even when monitored beyond the observation period (up to month 20). In contrast, survival was significantly impaired (P < 0.001) in LEC rats (35/36) that received hCu (Figure 2C). LEC rats subjected to an early hCu regimen showed a mean survival of 77 ± 15 d that was only moderately shorter when compared to control LEC rats that were housed on a commonly used standard diet (84 ± 5 d). In contrast, LEC rats that received a late hCu regimen had a significantly reduced survival (35 ± 9 d) with regard to start of hCu. Survival of male and female LEC rats did not differ (Table 2).
Impact of dietary copper on liver histology
Nuclei of hepatocytes were found to be highly enlarged in LEC rats receiving early and late hCu regimens (Figure 3). Pronounced fatty acid changes, inflammation and, to a lesser extent, necrosis were observed in LEC rats receiving hCu. No marked changes in liver histology, including enlargements, fatty metamorphosis and inflammation, or cholangiofibrosis, that are routinely found in adults[18] were observed in LEC rats that received a rCu diet. The liver histology in the control groups was found to be normal regardless of the copper regimen used.
Figure 3.
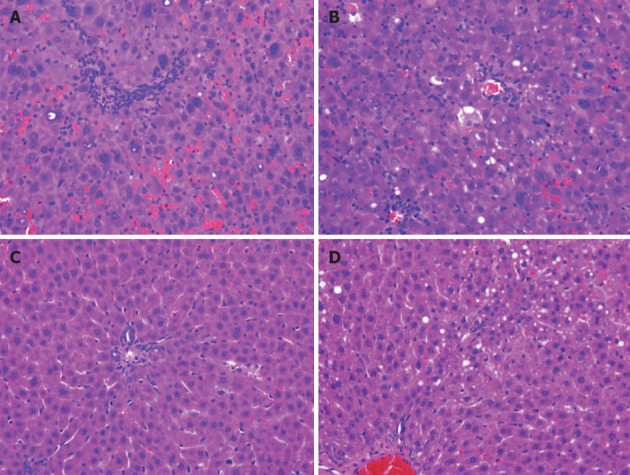
Liver of Long-Evans cinnamon rats is significantly affected by high copper regimen. Hematoxylin and eosin staining of liver. Livers were obtained from Long-Evans cinnamon rats that received high copper at an early or late time point (A, B) or after a reduced copper diet (C: late regimen). As a control, liver of Long-Evans agouti rat is depicted (D). Note highly irregular architecture of liver parenchyma, polyploidy, enlarged nuclei and inflammation (A, B) as compared to C and D. Liver histology of other groups was normal. One representative staining (40× magnification) of female rats is shown.
Accumulation of liver copper
High concentrations of copper were determined in the livers of LEC rats (Figure 4) following hCu, resembling WD[25]. LEC rats receiving rCu exhibited significant liver copper after the early (422 ± 62 μg/g) and late regimen (477 ± 75 μg/g) as compared to heterozygotes or wild type rats. Male and female LEC rats did not significantly differ with respect to liver copper (Table 2). Ceruloplasmin activity, which is absent in the LEC rat strain[20], was not affected by a copper diet (Figure 5).
Figure 4.
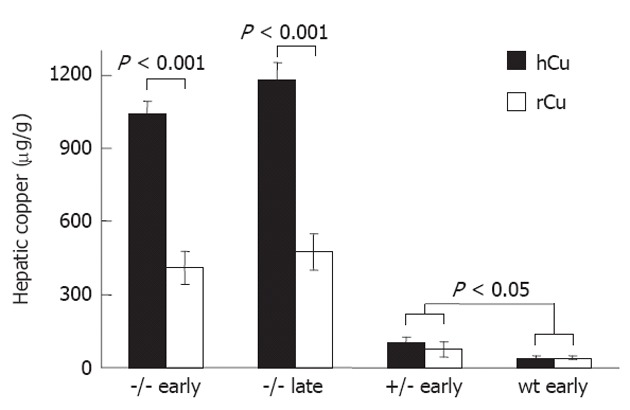
Increased liver copper is found in Long-Evans cinnamon rats irrespective of copper diet. Copper was determined in the liver of Long-Evans cinnamon rats (-/-; n = 9), heterozygotes (+/-; n = 5) and Long-Evans agouti rats (wt; n = 3) after receiving high copper (hCu) or reduced copper (rCu) diet according to an early or late regimen, respectively. Copper concentration is given as mean ± SE of liver dry weight.
Figure 5.
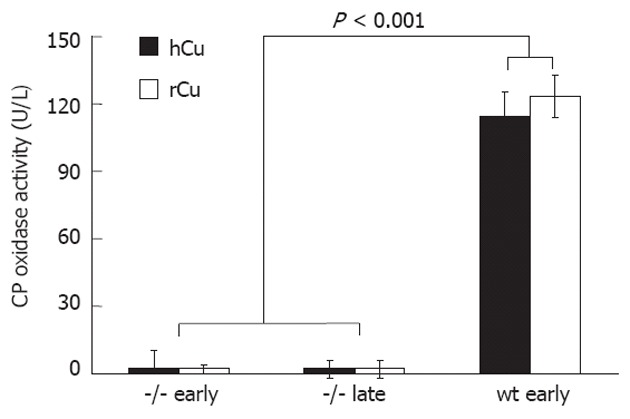
Ceruloplasmin oxidase is independent of high copper treatment. Ceruloplasmin (CP) oxidase was determined in Long-Evans cinnamon rats (-/-) that received high copper (hCu) or reduced copper (rCu). Sera (n = 6) were obtained from animals after an early or late hCu regimen. As control, values obtained from Long-Evans agouti rats (wt) (n = 3) are shown. Data are expressed as mean ± SE.
Gene expression in liver
In order to investigate whether the different copper diet regimens also have an impact on major biochemical pathways, gene expression in the liver was determined in animals at sacrifice. A small set of five marker genes was analyzed that relate to different areas of metabolism previously found to be affected in injured liver, including angiogenesis (Vegfa), fat metabolism (Srebf1), extracellular matrix (Timp1), oxidative stress (Hmox1), and the cell cycle (Cdkn1a). A highly different (fold-change > 8.5) expression of marker genes was observed when LEC rats receiving an early hCu regimen were compared to LEC rats receiving rCu (Figure 6). mRNA levels of Vegfa (8.5) and Srebf1 (13.9) were downregulated at the time points of fulminant hepatitis in LEC rats receiving hCu, whereas Timp1 (9.8), Cdkn1 (10.1) and Hmox1 (10.6) were upregulated at this time point. Marker gene expression was not affected in LEC rats receiving rCu, in LEA rats or in ATP7B heterozygotes (data not shown). Of note, none of the five genes displayed a significantly different (fold-change > 2) level of expression when LEC rats receiving early and late hCu regimen were compared.
Figure 6.

Liver gene expression of Long-Evans cinnamon rat is significantly altered after high copper regimen. Relative expression of individual mRNA in Long-Evans cinnamon (LEC) rats (n = 7) that were subjected to early high copper (hCu) regimen (left boxes) and in age-matched LEC rats (n = 5) receiving reduced copper (rCu) (right boxes). Animals were analyzed at fulminant hepatitis. Fold-change was calculated within groups by the Δct method using the HPRT gene for normalization. Data are shown as a box plot representation as calculated from three independent experiments. Numbers refer to difference of fold-change with regard to the median of each group.
DISCUSSION
The early hCu regimen used here for juvenile LEC rats closely resembles previous protocols. Onset of fulminant hepatitis was observed by us at about day 80, which is of a similar range as compared to others[18,25,26]. The high dietary copper regimen was achieved by copper enriched tap water (20 mg copper/L) and a standard solid diet (13 mg copper/kg). As compared to previous studies that employed a standard solid diet, only the overall dietary copper burden given to the LEC rats was moderately increased[18,25,26]. Much higher dietary copper concentrations have been reported in LEC rats[26-28], and intraperitoneal injection of 3 mg/kg copper for 3 d was shown to result in jaundice and fulminant hepatitis within 2 d after the last injection, suggesting that besides the absolute amount of copper the route of administration is also important[29]. As one result of our high dietary copper regimen, all LEC rats (with the exception of one animal used for the early regimen) encountered fulminant hepatitis and death. This is in contrast to previous studies using a standard solid diet only, where 40%-60% of LEC rats survived[18,25,26]. Since the high dietary copper regimen can induce fulminant hepatitis in nearly all LEC rats, such protocols may now allow a more quantitative analysis of therapy, e.g., by cell-based approaches[30].
Prevention of hepatitis and almost disease-free long term survival were observed in LEC rats that received a low copper diet throughout their lifetime. Hepatitis in LEC rats is closely associated with copper intake[18,31], and it was therefore noted early that a copper deficient diet can reduce serum levels of AST and ALT in addition to diminution of copper concentration in the liver[27]. In the latter study of male LEC rats, animals at the age of 30 d were monitored for 35 d or 15 wk[18,32]. A rCu diet that was enriched for L-proline could also improve survival (up to month 5) in male LEC rats[26]. We could confirm previous results regarding the impact of reduced dietary copper intake in a long-term study (up to 20 mo). Since male LEC rats seem to be more resistant to copper[33,34], we also investigated female LEC rats; however, a significant difference between genders was not observed for onset of hepatitis, liver copper, and survival.
Onset of hepatitis as determined by serum markers ALT, AST, and bilirubin was significant earlier (about 2 wk) in LEC rats that received a late hCu regimen. The finding of a significantly earlier onset of hepatitis and death in animals having elevated liver copper at start of hCu suggests that copper preloading of the liver above a critical threshold may accelerate the onset of disease. In contrast, LEC rats remained apparently asymptomatic showing liver copper levels that were about 10-fold higher as compared to LEA rats, suggesting that a high liver copper level can be tolerated for an extended time but induces a rapid onset of disease when dietary copper intake is increased. It should be stated that the effect of copper preloading could not be demonstrated by inspection of liver tissue since the stains obtained at early and late time points after hCu treatment did not significantly differ. Inflammation and necrosis were almost absent in stains of liver tissue at early and late time points when a rCu diet was used, suggesting that preloading with significant but subcritical levels of copper does not result in gross histological alterations of liver tissue and adverse immune responses.
The liver of LEC rats was characterized by determination of mRNA levels after a hCu regimen to assess the impact of dietary copper on gene expression. Notably, an almost identical gene expression was observed in juvenile and adult LEC rats after the hCu regimen, indicating that pre-accumulated copper levels do not affect gene expression at least at the time point of fulminant hepatitis. Marked differences were however observed between LEC rats at fulminant hepatitis and age-matched asymptomatic LEC rats that received a rCu diet. Inhibitory molecule Cdkn1a (p21) was found to be markedly upregulated suggesting that the cell cycle is blocked at G0 in the phase of fulminant hepatitis, confirming previous results obtained in LEC rats as well as in rats after hepatectomy[35,36]. It is conceivable that arrest of the cell cycle within the liver is a common molecular determinant of fulminant hepatitis. Timp1, an important mediator of extracellular matrix remodeling during toxic liver injury[37], was highly upregulated in LEC rats after hCu corroborating our observations of significant changes in liver architecture. The role (if any) of Timp-1 mRNA regulation by a copper diet with regard to the activity of metalloproteinase (MMP), e.g., MMP-9, which was shown to be highly induced in fulminant hepatic failure[38], remains to be studied. Expression of Srebf1 was down-regulated in LEC rats after the hCu regimen, suggesting that copper overload may be followed by reduction of lipid biosynthesis. Of note, Srebf1 activity was found to be increased in copper-deficient rats[39]. Hmox1, which is related to oxidative stress induced by iron deposition that is also commonly observed in LEC rats, was found to be highly upregulated after the hCu regimen, as reported before[36]. In contrast, Vegfa, a strong mediator of angiogenesis that was recently observed to be induced in hepatoma cells by hCu via a ceruloplasmin promotor, was down-regulated after hCu regimen. LEC rats which have a low ceruloplasmin expression may differ in this respect from wild type[40]. Although our analysis of gene expression in the liver of LEC rats after hCu regimen showed a significant modulation of important markers of copper metabolism, the study is limited since only one time point was examined. Clearly, further studies are needed that are beyond the scope of this article to explore the molecular events following toxic copper exposure of the liver.
Accumulation of dietary copper in the liver over a lifetime may play a role in the course of WD even when the genetic constitution of patients is similar[13-16]. While WD is frequently observed in the 2nd and 3rd decade of life, presentation of typical WD symptoms at the preschool age is relative rare and onset of disease above the age of 40 has been noted[12]. The finding that LEC rats have moderate liver copper but no overt disease may possibly resemble the observation of rare asymptomatic WD patients[41,42]. Conversion from a state of low to high dietary copper is followed by rapid development of severe disease in LEC rats, a situation that may also apply to WD patients after discontinuation of therapy[9,43]. Our findings indicate the importance of a stringent maintenance of anti-copper therapy for management of WD patients in order to balance liver copper level below a critical threshold, and reinforce the lifetime risk of WD patients of developing liver failure when dietary copper intake is increased or anti-copper therapy has failed.
Taken together our results demonstrate that asymptomatic LEC rats having significantly elevated liver copper either rapidly develop fulminant hepatitis when dietary copper is increased or survive long term on a rCu diet, suggesting that there may exist a critical threshold of liver copper important to trigger onset of WD. A stringent diet of low copper throughout the lifetime is followed in LEC rats by escape from hepatitis, moderately low liver copper, and long term survival without overt disease. The data underline the importance of environmental factors, such as dietary copper, with respect to the phenotype and course of WD, and suggest that similar molecular and physiologic mechanisms are initiated when tolerable copper thresholds are exceeded during the lifetime.
ACKNOWLEDGMENTS
We thank Gerβ J for statistical analysis and Cebulla K for technical support.
COMMENTS
Background
Fulminant hepatitis is a life-threatening disorder that is also observed in Wilson’s disease (WD), an inherited disease that is caused by mutation of the ATP7B gene encoding a crucial copper transport protein in the liver. The dietary intake of copper during a lifetime can affect the course of WD. Analysis of dietary copper intake was assessed in a rat model of WD. The study is related to the understanding of copper toxicity and the molecular events of pathogenesis.
Research frontiers
High copper is toxic to cells. In humans, the liver has the role of excreting excessive copper and preventing toxicity. The molecular events following copper toxicity are not understood. The relationship between liver copper levels and onset of disease is important to understand for improving current therapy approaches.
Innovations and breakthroughs
Accumulation of liver copper in individuals carrying two allelic mutations of ATP7B is ultimately followed by development of WD; however, rate, onset, course of disease, including acute liver failure (ALF), varies greatly between patients. Studies of monozygotic twins having identical mutations of ATP7B are most informative to decipher the role of genetic and environmental factors for the course of WD. However, these studies are casual and rare. The Long-Evans cinnamon rat carrying a natural mutation of ATP7B has been established as a model to study the pathophysiology of WD. This study illustrates for the first time that a stringent copper-restricted diet can completely prevent development of fulminant hepatitis in the long-term (up to 20 mo). Reduced dietary copper intake is associated with no overt disease symptoms, although elevated levels of liver copper were observed. This state of disease rapidly shifts to severe disease, liver failure, and death if dietary copper intake is increased. The molecular events following induction of ALF seem to be identical in WD regardless of the time point of disease onset.
Applications
The observation of different degrees of disease progression with regard to liver copper levels underlines the importance of a stringent maintenance of drugs during anti-copper therapy of WD and provides a novel basis for efforts to achieve compliance. Counselling and monitoring of dietary aspects in WD could be reinforced for effective management of patients.
Terminology
Fulminant hepatitis is the appearance of severe, life-threatening complications that occur rapidly after the first signs of liver disease (such as jaundice). The damage to the liver is broad, leading to the death of most (80%-90%) of the liver cells.
Peer review
This article is about the correlation between dietary copper level and the onset of fulminant hepatitis in the rat model of WD, especially at different time points. The article presents an interesting topic of WD to simulate the liver injury and onset of fulminant hepatitis. Although the model has been previously described the current study provides additional new value to the field.
Footnotes
Supported by Deutsche Forschungsgemeinschaft, SCHM 964/10-1; Innovative Medizinische Forschung, Münster
Peer reviewer: Yuan-Ping Han, Assistant Professor, Surgery and Pathology, University of Southern California, 2011 Zonal Ave HMR 813, Los Angeles, CA 91780, United States
S- Editor Gou SX L- Editor Logan S E- Editor Li JY
References
- 1.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 2.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 3.Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S, Parano E, Pavone L, Evgrafov O, Ivanova-Smolenskaya IA, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet. 1997;61:317–328. doi: 10.1086/514864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, Sewell RB. Diagnosis of Wilson’s disease: an experience over three decades. Gut. 2000;46:415–419. doi: 10.1136/gut.46.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut. 2007;56:115–120. doi: 10.1136/gut.2005.087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt HH. Role of genotyping in Wilson’s disease. J Hepatol. 2009;50:449–452. doi: 10.1016/j.jhep.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 8.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 9.Scheinberg IH, Jaffe ME, Sternlieb I. The use of trientine in preventing the effects of interrupting penicillamine therapy in Wilson’s disease. N Engl J Med. 1987;317:209–213. doi: 10.1056/NEJM198707233170405. [DOI] [PubMed] [Google Scholar]
- 10.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- 11.Nazer H, Ede RJ, Mowat AP, Williams R. Wilson’s disease: clinical presentation and use of prognostic index. Gut. 1986;27:1377–1381. doi: 10.1136/gut.27.11.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferenci P, Członkowska A, Merle U, Ferenc S, Gromadzka G, Yurdaydin C, Vogel W, Bruha R, Schmidt HT, Stremmel W. Late-onset Wilson’s disease. Gastroenterology. 2007;132:1294–1298. doi: 10.1053/j.gastro.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 13.Senzolo M, Loreno M, Fagiuoli S, Zanus G, Canova D, Masier A, Russo FP, Sturniolo GC, Burra P. Different neurological outcome of liver transplantation for Wilson’s disease in two homozygotic twins. Clin Neurol Neurosurg. 2007;109:71–75. doi: 10.1016/j.clineuro.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Członkowska A, Gromadzka G, Chabik G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov Disord. 2009;24:1066–1069. doi: 10.1002/mds.22474. [DOI] [PubMed] [Google Scholar]
- 15.Brewer GJ, Yuzbasiyan-Gurkan V, Dick R, Wang Y, Johnson V. Does a vegetarian diet control Wilson’s disease? J Am Coll Nutr. 1993;12:527–530. doi: 10.1080/07315724.1993.10718347. [DOI] [PubMed] [Google Scholar]
- 16.Kegley KM, Sellers MA, Ferber MJ, Johnson MW, Joelson DW, Shrestha R. Fulminant Wilson’s disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant. 2010;10:1325–1329. doi: 10.1111/j.1600-6143.2010.03071.x. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, Forbes JR, Chen HS, Cox DW. The LEC rat has a deletion in the copper transporting ATPase gene homologous to the Wilson disease gene. Nat Genet. 1994;7:541–545. doi: 10.1038/ng0894-541. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Togashi Y, Sato S, Emoto T, Kang JH, Takeichi N, Kobayashi H, Kojima Y, Une Y, Uchino J. Spontaneous hepatic copper accumulation in Long-Evans Cinnamon rats with hereditary hepatitis. A model of Wilson’s disease. J Clin Invest. 1991;87:1858–1861. doi: 10.1172/JCI115208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida Y, Tokusashi Y, Lee GH, Ogawa K. Intrahepatic transplantation of normal hepatocytes prevents Wilson’s disease in Long-Evans cinnamon rats. Gastroenterology. 1996;111:1654–1660. doi: 10.1016/s0016-5085(96)70029-x. [DOI] [PubMed] [Google Scholar]
- 20.Terada K, Nakako T, Yang XL, Iida M, Aiba N, Minamiya Y, Nakai M, Sakaki T, Miura N, Sugiyama T. Restoration of holoceruloplasmin synthesis in LEC rat after infusion of recombinant adenovirus bearing WND cDNA. J Biol Chem. 1998;273:1815–1820. doi: 10.1074/jbc.273.3.1815. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed S, Deng J, Borjigin J. A new strain of rat for functional analysis of PINA. Brain Res Mol Brain Res. 2005;137:63–69. doi: 10.1016/j.molbrainres.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 22.Du C, Fujii Y, Ito M, Harada M, Moriyama E, Shimada R, Ikemoto A, Okuyama H. Dietary polyunsaturated fatty acids suppress acute hepatitis, alter gene expression and prolong survival of female Long-Evans Cinnamon rats, a model of Wilson disease. J Nutr Biochem. 2004;15:273–280. doi: 10.1016/j.jnutbio.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Schosinsky KH, Lehmann HP, Beeler MF. Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin Chem. 1974;20:1556–1563. [PubMed] [Google Scholar]
- 24.Sauer V, Siaj R, Todorov T, Zibert A, Schmidt HH. Overexpressed ATP7B protects mesenchymal stem cells from toxic copper. Biochem Biophys Res Commun. 2010;395:307–311. doi: 10.1016/j.bbrc.2010.03.158. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida MC, Masuda R, Sasaki M, Takeichi N, Kobayashi H, Dempo K, Mori M. New mutation causing hereditary hepatitis in the laboratory rat. J Hered. 1987;78:361–365. doi: 10.1093/oxfordjournals.jhered.a110416. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins RL, Mori M, Inoue M, Torii K. Proline, ascorbic acid, or thioredoxin affect jaundice and mortality in Long Evans cinnamon rats. Pharmacol Biochem Behav. 1995;52:509–515. doi: 10.1016/0091-3057(95)00118-g. [DOI] [PubMed] [Google Scholar]
- 27.Sugawara N, Sugawara C. A copper deficient diet prevents hepatic copper accumulation and dysfunction in Long-Evans Cinnamon (LEC) rats with an abnormal copper metabolism and hereditary hepatitis. Arch Toxicol. 1994;69:137–140. doi: 10.1007/s002040050149. [DOI] [PubMed] [Google Scholar]
- 28.Cheah DM, Deal YJ, Wright PF, Buck NE, Chow CW, Mercer JF, Allen KJ. Heterozygous tx mice have an increased sensitivity to copper loading: implications for Wilson’s disease carriers. Biometals. 2007;20:751–757. doi: 10.1007/s10534-006-9038-7. [DOI] [PubMed] [Google Scholar]
- 29.Sugawara N, Sugawara C, Katakura M, Takahashi H, Mori M. Harmful effect of administration of copper on LEC rats. Res Commun Chem Pathol Pharmacol. 1991;73:289–297. [PubMed] [Google Scholar]
- 30.Sauer V, Siaj R, Stöppeler S, Bahde R, Spiegel HU, Köhler G, Zibert A, Schmidt HH. Repeated transplantation of hepatocytes prevents fulminant hepatitis in a rat model of Wilson’s disease. Liver Transpl. 2012;18:248–259. doi: 10.1002/lt.22466. [DOI] [PubMed] [Google Scholar]
- 31.Okayasu T, Tochimaru H, Hyuga T, Takahashi T, Takekoshi Y, Li Y, Togashi Y, Takeichi N, Kasai N, Arashima S. Inherited copper toxicity in Long-Evans cinnamon rats exhibiting spontaneous hepatitis: a model of Wilson’s disease. Pediatr Res. 1992;31:253–257. doi: 10.1203/00006450-199203000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi M, Kuge T, Endoh D, Nakayama K, Arikawa J, Takazawa A, Okui T. Hepatic copper accumulation induces DNA strand breaks in the liver cells of Long-Evans Cinnamon strain rats. Biochem Biophys Res Commun. 2000;276:174–178. doi: 10.1006/bbrc.2000.3454. [DOI] [PubMed] [Google Scholar]
- 33.Kasai N, Miyoshi I, Osanai T, Yamashita T, Kamimura E, Yoshida MC. Effects of sex hormones on fulminant hepatitis in LEC rats: a model of Wilson’s disease. Lab Anim Sci. 1992;42:363–368. [PubMed] [Google Scholar]
- 34.Togashi Y, Li Y, Kang JH, Takeichi N, Fujioka Y, Nagashima K, Kobayashi H. D-penicillamine prevents the development of hepatitis in Long-Evans Cinnamon rats with abnormal copper metabolism. Hepatology. 1992;15:82–87. doi: 10.1002/hep.1840150116. [DOI] [PubMed] [Google Scholar]
- 35.Hui TT, Mizuguchi T, Sugiyama N, Avital I, Rozga J, Demetriou AA. Immediate early genes and p21 regulation in liver of rats with acute hepatic failure. Am J Surg. 2002;183:457–463. doi: 10.1016/s0002-9610(02)00822-x. [DOI] [PubMed] [Google Scholar]
- 36.Klein D, Lichtmannegger J, Finckh M, Summer KH. Gene expression in the liver of Long-Evans cinnamon rats during the development of hepatitis. Arch Toxicol. 2003;77:568–575. doi: 10.1007/s00204-003-0493-4. [DOI] [PubMed] [Google Scholar]
- 37.Herbst H, Wege T, Milani S, Pellegrini G, Orzechowski HD, Bechstein WO, Neuhaus P, Gressner AM, Schuppan D. Tissue inhibitor of metalloproteinase-1 and -2 RNA expression in rat and human liver fibrosis. Am J Pathol. 1997;150:1647–1659. [PMC free article] [PubMed] [Google Scholar]
- 38.Chen F, Hori T, Ohashi N, Baine AM, Eckman CB, Nguyen JH. Occludin is regulated by epidermal growth factor receptor activation in brain endothelial cells and brains of mice with acute liver failure. Hepatology. 2011;53:1294–1305. doi: 10.1002/hep.24161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang Z, Gasperkova D, Xu J, Baillie R, Lee JH, Clarke SD. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J Nutr. 2000;130:2915–2921. doi: 10.1093/jn/130.12.2915. [DOI] [PubMed] [Google Scholar]
- 40.Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, Eckhardt K, Tröger J, Barth S, Camenisch G, et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: implications for ceruloplasmin regulation. Blood. 2005;105:4613–4619. doi: 10.1182/blood-2004-10-3980. [DOI] [PubMed] [Google Scholar]
- 41.Hefter H, Weiss P, Wesch H, Stremmel W, Feist D, Freund HJ. Late diagnosis of Wilson’s disease in a case without onset of symptoms. Acta Neurol Scand. 1995;91:302–305. doi: 10.1111/j.1600-0404.1995.tb07010.x. [DOI] [PubMed] [Google Scholar]
- 42.Perri RE, Hahn SH, Ferber MJ, Kamath PS. Wilson Disease--keeping the bar for diagnosis raised. Hepatology. 2005;42:974. doi: 10.1002/hep.20893. [DOI] [PubMed] [Google Scholar]
- 43.Walshe JM, Dixon AK. Dangers of non-compliance in Wilson’s disease. Lancet. 1986;1:845–847. doi: 10.1016/s0140-6736(86)90949-9. [DOI] [PubMed] [Google Scholar]