

Abstract
Proliferation of vascular smooth muscle cells is a characteristic of pathological vascular remodeling and represents a significant therapeutic challenge in several cardiovascular diseases. Docosahexaenoic acid (DHA), a member of the n-3 polyunsaturated fatty acids, was shown to inhibit proliferation of numerous cell types, implicating several different mechanisms. In this study we examined the molecular events underlying the inhibitory effects of DHA on proliferation of primary human smooth muscle cells isolated from small pulmonary artery (hPASMCs). DHA concentration-dependently inhibited hPASMC proliferation, induced G1 cell cycle arrest, and decreased cyclin D1 protein expression. DHA activated the unfolded protein response (UPR), evidenced by increased mRNA expression of HSPA5, increased phosphorylation of eukaryotic initiation factor 2α, and splicing of X-box binding protein 1. DHA altered cellular lipid composition and led to increased reactive oxygen species (ROS) production. DHA-induced ROS were dependent on both intracellular Ca2+ release and entry of extracellular Ca2+. Overall cellular ROS and mitochondrial ROS were decreased by RU360, a specific inhibitor of mitochondrial Ca2+ uptake. DHA-induced mitochondrial dysfunction was evidenced by decreased mitochondrial membrane potential and decreased cellular ATP content. DHA triggered apoptosis as found by increased numbers of cleaved caspase-3- and TUNEL-positive cells. The free radical scavenger Tempol counteracted DHA-induced ROS, cell cycle arrest, induction of UPR, and apoptosis. We conclude that Ca2+-dependent oxidative stress is the central and initial event responsible for induction of UPR, cell cycle arrest, and apoptosis in DHA-treated hPASMCs.
Abbreviations: ATF6, activating transcription factor 6; DHA, docosahexaenoic acid; ΔΨm, mitochondrial membrane potential; eIF2α, eukaryotic initiation factor 2α; ER, endoplasmic reticulum; FCS, fetal calf serum; hPASMC, human pulmonary artery smooth muscle cell; HSPA5, heat shock 70-kDa protein 5; IRE1α, inositol-requiring enzyme 1α; n-3 PUFA, n-3 polyunsaturated fatty acid; PERK, protein kinase RNA-like endoplasmic reticulum kinase; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PTP, permeability transition pore; ROS, reactive oxygen species; TG, triglyceride; UPR, unfolded protein response; XBP-1, X-box binding protein 1
Keywords: Oxidative stress, Unfolded protein response, n-3 polyunsaturated fatty acid, Apoptosis, Mitochondria, Cell cycle, Free radicals
Highlights
► DHA induces ROS production, cell cycle arrest, UPR and apoptosis in hPASMC. ► Ca2+ and mitochondria are required for DHA-mediated induction of ROS. ► DHA alters cellular lipid composition and decreases ΔΨm and cellular ATP content. ► Free radical scavenger Tempol counteracts DHA effects in hPASMC.
Vascular remodeling is a pathological process characterized mainly by increased proliferation or decreased apoptosis of vascular smooth muscle cells. It is present in several cardiovascular diseases such as stent restenosis, atherosclerosis [1], and pulmonary hypertension [2] and represents a significant therapeutic challenge.
Clinical studies have shown beneficial cardiovascular effects of n-3 polyunsaturated fatty acid (n-3 PUFA) nutritional supplementation [3,4]. In vitro, n-3 PUFAs have antiproliferative effects on various cell types, including vascular smooth muscle cells. Studies showed that the n-3 PUFAs eicosapentaenoic acid and docosahexaenoic acid (DHA) inhibit proliferation through production of oxidized species at a low concentration [5], block cell cycle progression by inhibiting phosphorylation of Cdk2–cyclin E complex [6], and induce apoptosis partially through the p38 MAP kinase pathway [7,8]. However, the exact mechanism of action still remains unknown.
Two recent studies implicated DHA in the perturbation of calcium homeostasis in the endoplasmic reticulum (ER), resulting in: (i) activation of the unfolded protein response (UPR) and growth arrest in human colon cancer cells [9] as well as (ii) increased mitochondrial ROS production and cell death of murine thymocytes [10].
The UPR comprises upregulation and dissociation of heat shock 70-kDa protein 5 (HSPA5/Bip) from three sensor proteins located in the ER membrane [11], leading to: (i) reduction of protein synthesis due to phosphorylation of the α subunit of eIF2α [12–14], (ii) IRE1α (inositol-requiring enzyme 1α)-mediated splicing of XBP-1 transcription factor [15,16], and (iii) induction of activating transcription factor 6 (ATF6) [17].
In this study we performed a comprehensive analysis of molecular events triggered by DHA in primary human pulmonary artery smooth muscle cells (hPASMCs), known to play a major role in the pathology of pulmonary hypertension. We found that Ca2+-dependent oxidative stress is the central and initial event responsible for induction of the UPR, cell cycle arrest, and apoptosis in DHA-treated hPASMCs.
Materials and methods
Cells
Human pulmonary artery smooth muscle cells were isolated from pulmonary arteries obtained from patients undergoing surgery for lung cancer without a history of pulmonary vascular disease or arterial hypoxemia, as described previously [18]. Briefly, the adventitia from small arteries with diameters of less than 1 mm was carefully removed under microscopic guidance and media pieces less than 1 mm3 were placed into a flask with one drop of culture medium supplemented with 20% fetal calf serum (FCS). The study protocol for tissue donation was approved by the institutional review board of the Medical University of Graz in accordance with national law and with the guidelines on Good Clinical Practice from the International Conference on Harmonization. Written, informed consent was obtained from each patient. Cells were grown in medium containing 5% FCS and growth factors (Lifeline Cell Technology, Frederick, MD, USA) and used between passages 2 and 5. Purity was checked by characteristic appearance under phase-contrast microscopy indirect immunofluorescence staining for α smooth muscle actin, smooth muscle myosin heavy chain, and lack of von Willebrand factor staining.
Chemicals
Antibodies against human phospho-eIF2α, total eIF2α, cyclin D1, p21, α-tubulin, cleaved caspase-3 Alexa 488, and anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody were from Cell Signaling (Beverly, MA, USA). Taq polymerase was from Solis Biodyne (Tartu, Estonia), 10× PCR buffer and dNTP mix were from Promega (Madison, WI, USA). DHA, dimethyl sulfoxide (DMSO), ethanol, CaCl2, KCl, MgCl2, NaCl, Hepes, EGTA, glucose, 4-hydroxy-Tempol (Tempol), bovine serum albumin (BSA), Triton X-100, and propidium iodide were from Sigma (Steinheim, Germany). Tween 20 was from Bio-Rad (Hercules, CA, USA). BAPTA-AM (cell-permeative Ca2+ chelator) and RU360 (mitochondrial calcium uptake inhibitor) were from Merck Biosciences (Nottingham, UK). [methyl-3H]Thymidine was from PerkinElmer Life Sciences (Waltham, MA, USA). DHA was reconstituted in ethanol, aliquotted into glass vials under argon, and stored at − 80 °C until use. Vehicle control contained a maximum of 0.05% ethanol or 0.1% DMSO.
Proliferation
Cells plated in 24-well plates were grown for 3 days with DHA or vehicle control. At the end of the experiment, the cells were harvested by trypsinization, centrifuged, and resuspended in phosphate-buffered saline (PBS). An aliquot of cell suspension was taken for cell number count. The numbers of viable and total cells were determined using a Casy machine (Schärfe System, Reutlingen, Germany), a pulse area-based technology for cell counting and viability testing. Cells were harvested by trypsinization, centrifuged, resuspended in PBS, and measured using the Casy instrument.
Thymidine incorporation
Cells were plated in 96-well plates and incubated with compounds and 2 μCi/well [methyl-3H]thymidine overnight. Cells were harvested (PerkinElmer harvester) and transferred to glass fiber plates (UniFilter-96 GF/C; PerkinElmer) and radioactivity was measured in a scintillation counter (1450 Microbeta Trilux liquid scintillation and luminescence counter; Wallac).
Apoptosis and cell cycle measurements
Cleaved caspase-3 was determined in cells that were incubated for indicated time intervals with DHA in 24-well plates, harvested by trypsinization, washed in PBS, fixed 10 min at 37 °C in 2% formaldehyde, and permeabilized in ice-cold methanol for 30 min. The cells were then washed in PBS with 0.5% FCS, incubated with cleaved caspase-3 antibody at room temperature for 45 min, washed, resuspended in PBS, and measured immediately on a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) flow cytometer.
TUNEL assay for detection of apoptotic cells was performed on cells cultured and treated in glass chamber slides. Cells were washed with PBS, fixed with 4% paraformaldehyde for 1 h at room temperature, and permeabilized on ice with 0.1% Triton X-100 in 0.1% Na-citrate solution. Afterward, the cells were incubated with reagents from the In Situ cell death detection kit according to the manufacturer's instructions (Roche, Mannheim, Germany).
Cell cycle was determined flow cytometrically by staining cells with propidium iodide (PI). Cells were harvested by trypsinization, washed with PBS, incubated at room temperature for 20 min with 50 μg/ml PI in 0.1% Na-acetate with 0.1% Triton X-100, and measured immediately on a FACSCalibur flow cytometer. Acquired data were analyzed with CellQuest Pro software (BD Biosciences).
ROS and mitochondrial membrane potential (MMP) measurements by flow cytometry
ROS production was determined using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) dye (Biotium, Hayward, CA, USA). Cells grown in complete medium with 5% FCS in 24-well plates were loaded with 10 μM H2DCFDA for 30 min. Thereafter, the medium was replaced with fresh medium containing vehicle or DHA, followed by incubation for the indicated time periods and flow cytometric measurements of ROS. For the 24-h time point cells were incubated with vehicle or DHA for 23.5 h followed by labeling with 10 μM H2DCFDA for 30 min before flow cytometric measurements of ROS. In some experiments, 5 μM BAPTA-AM and 10 μM RU360 were added during both labeling with H2DCFDA and incubation with vehicle or DHA. To address the role of the ROS scavenger Tempol, cells were preincubated with Tempol (150 μM) or vehicle (DMSO) for 2 h (90 min before and during the 30-min labeling with H2DCFDA). Thereafter, the cells were exposed to DHA in the presence or absence of Tempol for 3 h. At the end of the experiment, the cells were harvested by trypsinization, washed, and resuspended in PBS, followed by flow cytometry.
For ROS measurements in calcium- and calcium-free buffer cells were first loaded with H2DCFDA in complete medium with 5% FCS for 30 min. Then, the cells were washed with calcium- or calcium-free buffer and incubated with vehicle or DHA in the respective buffer for 1 h, followed by ROS measurement. Calcium-containing buffer contained 2 mM CaCl2, 138 mM NaCl, 1 mM MgCl2, 5 mM KCl, 10 mM Hepes, 10 mM glucose, 5% FCS, pH 7.4. Calcium-free buffer lacked CaCl2 but contained 1 mM EGTA.
Mitochondrial ROS production was measured using MitoSOX red (Invitrogen, Darmstadt, Germany), a fluorogenic dye. Cells were loaded with 1 μM MitoSOX red for 30 min, in the absence or presence of RU360 (10 μM), followed by incubation with vehicle or DHA in the presence or absence of RU360 for 1 h. At the end of the experiment, the cells were harvested by trypsinization, washed, and resuspended in PBS followed by flow cytometry.
ΔΨm was measured using the Mito ID membrane potential detection kit (Enzo Life Sciences, Lorrach, Germany). Cells were incubated for the indicated time with DHA and then harvested by trypsinization, washed, and incubated at room temperature for 15 min with reagent followed by flow cytometry. Loss of MMP was observed as a decrease in orange and increase in green fluorescence. Results are presented as a change in ratio of two fluorescence means that correlates to changes in ΔΨm.
Array confocal laser scanning microscopy
High-resolution imaging of cells loaded with both 1 μM MitoSOX red and 0.5 μM MitoTracker green for 30 min, followed by incubation with vehicle or DHA, was performed using an array confocal laser scanning microscope (ACLSM). The ACLSM was built on an inverse, fully automatic microscope (Axio Observer.Z1 from Zeiss, Göttingen, Germany) that was equipped with a 100× objective (Plan-Fluor 100×/1.45 oil; Zeiss), a Nipkow-based confocal scanner unit (CSU-X1; Yokogawa Electric Corp., Tokyo, Japan), a motorized filter wheel (CSUX1FW; Yokogawa Electric) on the emission side, and an acousto optic tunable filter-based laser merge module for laser lines 405, 445, 473, 488, 515, and 561 nm (Visitron Systems, Pucheim, Germany). Emission was acquired with a charged-coupled device camera (CoolSNAP-HQ; Photometrics, Tucson, AZ, USA). All devices were controlled by VisiView Premier acquisition software (Visitron Systems). MitoSOX red and MitoTracker green were alternatively excited at 561 and 488 nm and fluorescence emission light was collected at 630/75 and 525/40 nm, respectively. Image analysis (colocalization analysis) was performed using MetaMorph software 7.7 (Molecular Devices, Sunnyvale, CA, USA).
Adenine nucleotide analysis
Adenine nucleotides were analyzed as reported previously [19,20] with some modifications. Separation was performed on a Hypersil ODS column (5 μm, 250 × 4 mm i.d.), using a L2200 autosampler, two L-2130 HTA pumps, and an L2450 diode array detector (all from VWR Hitachi). The wavelength for detection of adenine nucleotides was set at 254 nm. EZchrom Elite (VWR) was used for data acquisition and analysis. After trypsinization and mild centrifugation (supernatant discarded) the cellular proteins were precipitated with 250 μl of perchloric acid (0.4 mol/L, 4 °C). After centrifugation (12,000 g), 100 μl of the supernatant was neutralized with 10–12 μl of potassium carbonate (2 mol/L, 4 °C). The supernatant obtained after centrifugation was used for HPLC analysis (injection volume: 40 μl). The pellets of the acid extract were dissolved in 0.5 ml of sodium hydroxide (0.1 mol/L) and used for protein determination (BCA assay; Pierce).
Mass spectrometric determination of phospholipid and triglyceride species
Cells grown in six-well plates were treated with DHA or vehicle for 20 h followed by extraction of cellular lipids according to Bligh and Dyer [21] followed by HPLC and MS as previously described [22,23]. For details please see the supplementary methods.
Measurement of intracellular calcium
Intracellular calcium was measured as described previously [24]. Cells were trypsinized and loaded with 2 μM Fura-2/AM for 45 min. After being washed, the cells were resuspended in either calcium buffer (138 mM NaCl, 1 mM MgCl2, 5 mM KCl, 10 mM Hepes, 10 mM glucose, pH 7.4) or EGTA buffer (1 mM EGTA, 138 mM NaCl, 1 mM MgCl2, 5 mM KCl, 10 mM Hepes, 10 mM glucose, pH 7.4). The ratio of Fura-2 fluorescence intensity at the two excitation wavelengths (340 nm/380 nm ratio) was monitored fluorometrically in a stirring cuvette before and after the injection of vehicle or 10 μM DHA. Values obtained with vehicle were subtracted from those obtained with DHA.
Western blot
Cells were washed once with ice-cold PBS and lysed in M-PER mammalian protein extraction reagent (Thermo Scientific, Rockford, IL, USA) containing protease and phosphatase inhibitors (Roche). Equal amounts of proteins were loaded per lane and analyzed by SDS–PAGE (10%) and subsequent immunoblotting. Nitrocellulose membrane (Bio-Rad) was blocked in 5% milk in Tris-buffered saline with 0.1% Tween 20 and incubated overnight at 4 °C with the indicated primary antibody (1:1000 in 5% BSA). Thereafter, membranes were washed and incubated with HRP-labeled secondary antibody (1:5000 in 1% milk) and developed with SuperSignal West Pico or SuperSignal West Femto Substrate (Thermo Scientific). Membranes were stripped using Restore Plus Western blot stripping buffer (Thermo Scientific).
Quantitative real-time PCR (qRT-PCR)
Isolation of RNA and qRT-PCR were performed as described [23]. mRNA levels of HSPA5 (Primer Assay QT00096404), cyclin D1 (QT00495285), and p21 (QT00062090) were normalized to human β2-microglobulin (QT01665006) and expressed as relative ratio (ΔΔCt). All samples were assayed in duplicate and the average value was used for quantification.
Detection of XBP-1 splice variant
Forward (CCTTGTAGTTGAGAACCAGG) and reverse (GGGGCTTGGTATATATGTGG) primers for XBP-1 PCR were from Invitrogen. The PCR amplification program consisted of 40 cycles each containing three steps: 95, 58, and 72 °C (each 1 min). Electrophoretic separation of the resulting products was performed on 3% agarose gel in 0.5 × TAE buffer at 30 V for 6 h. Unspliced XBP-1 product had a size of 422 bp and the spliced variant had a size of 396 bp.
Statistics
Data are given as means ± SEM. Statistical analyses were performed in GraphPad Prism 5 using the paired t test, for experiments comparing two groups, and ANOVA Kruskal–Wallis with the Dunn multiple comparison posttest, for experiments comparing three or more groups. P < 0.05 was considered statistically significant.
Results
DHA inhibits hPASMC proliferation
We tested the effects of DHA on hPASMC proliferation. After 3 days of incubation with various DHA concentrations ranging from 25 to 150 μM, in fully supplemented medium with 5% FCS, proliferation was determined by cell counting (Fig. 1A) and [3H]thymidine incorporation (Fig. 1B). DHA in both cases dose-dependently inhibited hPASMC proliferation. Based on these results and literature reports on plasma DHA concentration in humans, we chose to use DHA at 100 μM in all subsequent experiments.
Fig. 1.

DHA dose-dependently inhibits hPASMC proliferation. (A) Number of viable cells was determined after 3 days of incubation with indicated concentrations of DHA and results are expressed relative to vehicle control (n = 5). (B) [3H]Thymidine incorporation was measured after overnight incubation with DHA and results are expressed relative to vehicle control (n = 5). (C) Cells were treated with 100 μM DHA for 12 h and analyzed by flow cytometry for DNA content. Cell cycle distribution in G1 phase is expressed as the percentage of total cells (n = 4). (D) Cells were treated with 100 μM DHA and representative time course results of p21 and cyclin D1 Western blots are shown. (E and F) Densitometry evaluation of Western blot data. Signals were normalized to α-tubulin and expressed relative to 4-h vehicle (veh) control (n = 4). Data are given as means ± SEM of at least four independent experiments. *P < 0.05 vs corresponding veh control group.
The observed inhibitory action of DHA on cell proliferation was accompanied by an increased percentage of cells in G1 phase of the cell cycle (Fig. 1C). Protein expression level of cyclin D1, a positive regulator of cell cycle progression, was significantly decreased in DHA-treated cells (Figs. 1D and E). Protein expression level of p21, a negative regulator of cell cycle progression, was unaltered upon DHA treatment (Figs. 1D and F).
DHA induces UPR in hPASMCs
Decreased cyclin D1 protein might be a consequence of DHA-induced ER stress and concomitant activation of UPR. We tested three markers of UPR in DHA-treated cells. The levels of HSPA5 (Bip) mRNA were significantly increased in DHA-treated cells, compared with vehicle-treated control cells (Fig. 2A). Furthermore, the phosphorylation level of eIF2α was significantly increased in DHA-treated cells (Figs. 2B and C). Finally, splicing of XBP-1 mRNA was induced upon incubation with DHA (Fig. 2D).
Fig. 2.

DHA induces UPR in hPASMC. (A) Quantitative RT-PCR measurement of HSPA5 gene expression (n = 4). (B) Representative Western blot detection of phosphorylated eIF2α. Total eIF2α served as loading control. (C) Densitometric evaluation of eIF2α phosphorylation (n = 4). (D) Representative PCR detection of XBP-1 splice variant. Data are given as means ± SEM. *P < 0.05 vs corresponding vehicle (veh) control group.
DHA alters the lipid profile of hPASMCs
Because alterations in cellular lipid composition might be responsible for ER stress and UPR, we performed phospholipid and triglyceride profiling of DHA- and vehicle-treated cells by mass spectrometry. Both phosphatidylcholine (PC) species 38:6, 40:6, and 40:7 (Fig. 3A) and phosphatidylethanolamine (PE) species 40:6 and 40:7 (Fig. 3B) were markedly enriched in DHA-treated cells compared with vehicle-treated control cells. The observed increase in PC and PE species that contain DHA was accompanied by a marked decrease in several phospholipid species that lack DHA (Figs. 3A and B). DHA was also efficiently incorporated into triglycerides (TGs) as reflected by the increase in TG species containing long-chain polyunsaturated fatty acids including DHA (not shown).
Fig. 3.

DHA alters cellular phospholipid composition. hPASMCs were incubated with 100 μM DHA for 20 h followed by lipid extraction and mass spectrometric analysis of phospholipid species. (A) DHA-induced alterations in cellular phosphatidylcholine profile. (B) DHA-induced alterations in cellular phosphatidylethanolamine profile.
DHA induces oxidative stress in hPASMCs: impact on cell proliferation and UPR
To further examine whether DHA-induced oxidative stress is implicated in the promotion of ER stress, we measured intracellular ROS production in DHA-treated cells. We observed a rapid increase in ROS production under DHA treatment, starting as soon as after 10 min incubation, with a maximum after 1 and 3 h, followed by a decreasing trend after 5 h and similar levels in DHA- and vehicle-treated cells after 24 h (Fig. 4A). DHA-induced ROS were significantly decreased by preincubation with the free radical scavenger Tempol (Fig. 4B). Tempol markedly recovered both DHA-mediated inhibition of cell proliferation (Fig. 4C) and cyclin D1 protein expression (Fig. 4D) and diminished DHA-induced phosphorylation of eIF2α (Fig. 4E).
Fig. 4.

Free radical scavenger Tempol attenuates DHA-induced oxidative stress and UPR and improves cell proliferation. (A) Cells were loaded with 10 μM H2DCFDA for 30 min followed by exposure to veh or DHA (100 μM) for indicated time periods up to 5 h, followed by flow cytometry. For the 24-h time point cells were incubated with veh or DHA for 23.5 h and then loaded with H2DCFDA for 30 min in the presence of veh or DHA, followed by flow cytometry. (B) Cells were preincubated with Tempol (150 μM) or veh (DMSO) for 2 h (90 min before loading and during 30-min loading with H2DCFDA). Thereafter, the cells were exposed to DHA in the presence or absence of Tempol for 3 h, followed by ROS measurement. (C) [3H]Thymidine incorporation in veh- or DHA-treated hPASMCs in the absence and presence of Tempol. (D) Representative Western blot of cyclin D1 protein expression in veh- or DHA-treated hPASMCs in the presence and absence of Tempol. (E) Representative Western blot of phosphorylated eIF2α in veh- or DHA-treated hPASMCs in the presence and absence of Tempol. Total eIF2α served as loading control. Data are given as means ± SEM of at least five independent experiments. *P < 0.05.
The role of Ca2+ and mitochondria in DHA-induced ROS generation
To identify the underlying mechanisms responsible for ROS generation upon DHA treatment, we addressed whether Ca2+ triggers ROS generation. For this purpose cells were loaded with the intracellular Ca2+ chelator BAPTA-AM. Cells loaded with BAPTA-AM showed decreased ROS production in response to DHA (Fig. 5A). To examine the role of Ca2+ in DHA-induced ROS production in more detail, we first tested the capacity of DHA to modulate the cytosolic Ca2+ concentration ([Ca2+]i). DHA increased [Ca2+]i in the absence and even more in the presence of extracellular Ca2+ (Fig. 5B). The importance of extracellular Ca2+ for DHA-induced ROS was demonstrated by decreased ROS in cells treated with DHA in the absence of extracellular Ca2+ (EGTA buffer) compared with cells in the presence of extracellular Ca2+ (Ca2+ buffer; Fig. 5C). Overall cellular DHA-induced ROS was decreased in the presence of RU360, a specific inhibitor of Ca2+ uptake into mitochondria (Fig. 5D). Also the DHA-induced increase in mitochondrial ROS, measured in MitoSOX red-loaded cells, was markedly decreased by RU360 (Fig. 5E). The majority of MitoSOX red accumulated in mitochondria as found by its colocalization with a mitochondria-specific marker, MitoTracker green (Supplementary Fig. 1).
Fig. 5.

Ca2+ and mitochondria are involved in DHA-induced ROS production. (A) hPASMCs were loaded with H2DCFDA (10 μM) in the presence or absence of BAPTA-AM (5 μM) for 30 min. Then, the cells were further incubated with vehicle (veh) or 100 μM DHA in the presence or absence of BAPTA-AM for 1 h, followed by ROS measurement by flow cytometry. (B) Fura-2/AM-loaded hPASMCs were resuspended in either Ca2+-containing buffer ([Ca2 +]) or Ca2+-free buffer containing EGTA (EGTA). The ratio of Fura-2 fluorescence intensity at the two excitation wavelengths (340/380 nm ratio) was monitored fluorometrically in a stirring cuvette during exposure to veh or 10 μM DHA. Results are representative single traces of three experiments performed in duplicate. Values for veh were subtracted from those obtained with DHA. (C) Cells were loaded with H2DCFDA in complete medium with 5% FCS for 30 min. Then, the cells were washed with calcium-containing or calcium-free buffer and incubated with vehicle or DHA in the respective buffer for 1 h, followed by ROS measurement. (D) Cells were loaded with H2DCFDA in the absence or presence of RU360 for 30 min. Thereafter, the cells were incubated with veh, RU360 (10 μM), or DHA ± RU360 for 1 h, followed by flow cytometric measurement of ROS. (E) The procedure was exactly as described for (D) except that cells were loaded with MitoSOX red (1 μM). Unless stated otherwise, data are given as means ± SEM of six independent experiments. *P < 0.05.
DHA induces ΔΨm dissipation, ATP depletion, and apoptosis in hPASMCs
Both quantification of cleaved caspase-3-positive cells and TUNEL assay showed significant induction of apoptosis upon prolonged DHA treatment (Figs. 6A and B). Importantly, Tempol markedly diminished DHA-induced apoptosis (Fig. 6C). Furthermore, DHA caused mitochondrial dysfunction as reflected by decreased ΔΨm in DHA-treated cells (Fig. 6D). In line with the decreased ΔΨm, the cellular ATP content (Fig. 6E) and the ATP/ADP ratio (Fig. 6F) were decreased in DHA-treated cells, compared with respective control cells.
Fig. 6.

DHA induces apoptosis in hPASMCs. (A) Flow cytometric determination of cleaved caspase-3-positive cells. (B) Representative immunofluorescence staining for TUNEL-positive cells (green) and DAPI counterstain. Top shows vehicle (veh)-treated cells and bottom shows cells treated with DHA for 24 h. (C) hPASMCs were preincubated with Tempol or veh for 2 h and treated overnight with 100 μM DHA. Percentage of apoptotic cells was determined using flow cytometric assay for cleaved caspase-3. (D) Flow cytometric determination presented as decreased orange/green ratio indicative of a reduced MMP, as described under Materials and methods. Data are given as means ± SEM of six independent experiments. *P < 0.05 vs corresponding veh control group. (E) Cellular ATP content after 6 and 12 h incubation with veh or 100 μM DHA. (F) Cellular ATP/ADP ratio after 6 and 12 h incubation with veh or 100 μM DHA. Results are means ± SEM of six separate incubations for veh and DHA for each indicated time point. *P < 0.05.
Discussion
The major finding of this study is that DHA-induced oxidative stress is the initial and central event responsible for the induction of UPR, inhibition of cell proliferation, and induction of apoptosis in hPASMCs.
These effects of DHA were observed in fully supplemented medium containing serum and growth factors, thus closely resembling the in vivo conditions. Importantly, the applied concentrations of DHA, exhibiting an antiproliferative effect in hPASMCs, were within physiologically/pharmacologically reachable levels in human serum [25–29].
In accordance with the described antiproliferative effect of DHA [30], the proliferation rate of hPASMCs was markedly decreased by DHA. This decrease was accompanied by an increased number of cells in the G1 phase of the cell cycle and a decreased cyclin D1 protein content. Most probably the observed decrease in cyclin D1 reflects a general attenuation of protein synthesis due to activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eIF2α signaling pathway connected with ER stress. Indeed, we found increased levels of phosphorylated eIF2α and HSPA5 as well as the appearance of a spliced variant of XBP-1, indicative of UPR activation upon DHA treatment. DHA was found to induce ER stress and UPR in colon cancer cells [9]. However, a recent publication showed no evidence of ER stress or UPR in DHA-treated rat hepatocytes [31], indicating species- and cell-type-specific cellular responses to DHA. Alternatively, decreased cyclin D1 levels might be due to PERK/eIF2α-mediated increase in cyclin D1 proteasomal degradation, independent of the efficacy of translation [32].
Because protein folding and posttranslational protein modifications are highly sensitive to alterations in the ER luminal environment, we examined the impact of DHA on cellular lipid composition, ROS production, and intracellular Ca2+ homeostasis. In addition to a marked increase in DHA-containing phospholipids, there was a striking decrease in PC and PE species containing mono- (18:1) and di- (18:2) unsaturated fatty acids. This lipid profile is partly similar to that reported for HeLa cells, in which stearoyl-CoA desaturase 1 (SCD1) knockdown decreased membrane phospholipid unsaturation, particularly 18:1 fatty acid, leading to UPR and apoptosis [33]. Although SCD1 mRNA levels were not significantly altered by DHA in our study (not shown), the possibility remains that SCD1 activity is impaired by DHA, leading to the observed lipid profile. Alternatively, DHA might compete with 18:1, 18:2, and other fatty acids for lysophosphatidylcholine acyltransferase-mediated incorporation into cellular phospholipids [34]. Disturbed conformation and consequently dysfunction of ER-membrane-associated chaperones, with concomitant accumulation of unfolded proteins, might therefore be an explanation for the observed induction of ER stress and UPR upon perturbations in phospholipid composition.
The observed DHA-induced ROS production was very rapid in onset, suggesting an acute cellular response to DHA as an underlying mechanism. Indeed, DHA induced a rapid increase in cytosolic calcium (Fig. 5B), a prerequisite for DHA-induced ROS formation (Fig. 5A). In line with the previously described mechanism of the DHA-induced increase in cytosolic calcium [35], both Ca2+ released from ER and Ca2+ entry contributed to DHA-induced increase in cytosolic Ca2+ levels (Fig. 5B), which triggered ROS generation (Figs. 5A and C). Importantly, prolonged exposure of cells to DHA (5 h) did not alter the amount of ionomycin-releasable Ca2+ (not shown), indicating that steady-state ER Ca2+ levels were not altered upon prolonged exposure to DHA. Accordingly, depletion of ER Ca2+ seems not to be the trigger of ER stress and UPR in DHA-treated cells. Rather, the DHA-induced, Ca2+-dependent oxidative stress seems to be responsible for the initiation of UPR and apoptosis. Indeed, Tempol, a strong antioxidant, diminished the amounts of both phosphorylated eIF2α and cleaved caspase-3-positive cells.
It is well established that increased mitochondrial Ca2+ levels promote opening of the permeability transition pores (PTPs) in the mitochondrial inner membrane, leading to a decreased ΔΨm, cytochrome c release, and inhibition of the respiratory chain at complex III, which is accompanied by an increased ROS production [36,37]. Our results showing that the DHA-induced increase in mitochondrial ROS is sensitive to RU360, together with a decreased ΔΨm and ATP content, strongly argue for the role of DHA-induced cytosolic Ca2+ increase in mitochondrial dysfunction. Mitochondria were not the only source of ROS in DHA-treated cells, because overall cellular ROS were significantly decreased but not completely abolished with RU360.
It has been shown that ROS may cause ER stress and that accumulation of misfolded proteins in the ER lumen can initiate ROS production [14,38,39]. Considering a very rapid increase in DHA-induced ROS, preceding the increase in UPR markers, it is possible that the initial increase in ROS, generated outside of the ER, affects the ER folding machinery, leading to UPR and concomitant generation of ER-derived ROS.
Based on the capacity of Tempol to counteract DHA-induced UPR, apoptosis, and cell cycle arrest, the DHA-induced oxidative stress seems to be the initial event triggered by DHA. The failure of RU360 to counteract DHA-mediated attenuation of hPASMC proliferation (Supplementary Fig. 2) clearly demonstrates a rather weak contribution of mitochondrial ROS, suggesting that various other cellular ROS sources are responsible for triggering and mediating DHA effects in hPASMCs. However, the exact intracellular localization and the relative quantitative as well as temporal contributions of the various ROS sources in DHA-treated cells require further investigation.
Supplementary Fig. 2.
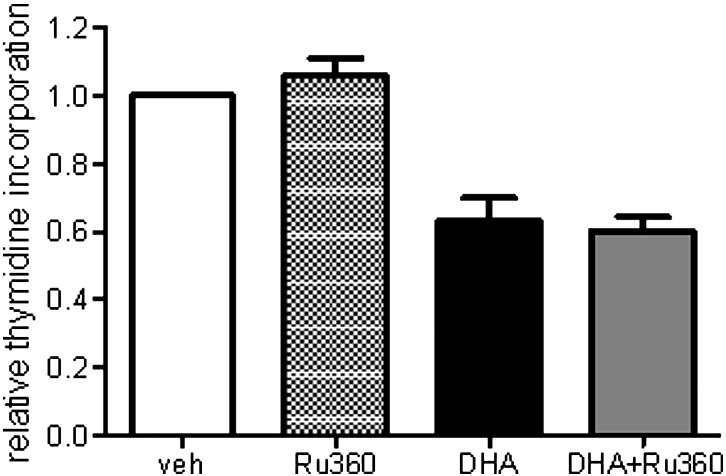
RU360 fails to counteract DHA-mediated attenuation of cell proliferation Cells were plated in 96-well plates and incubated without or with RU360 (10 μM) for 1 h before further incubation with vehicle or DHA (100 μM) along with 2 μCi/well [methyl-3H]thymidine in the absence or presence of RU360 for 24 h. Thereafter, cells were harvested and radioactivity measured in a scintillation counter. Data are mean±SEM of 4 independent experiments. *P<0.05.
In this study DHA induced apoptosis in hPASMCs, evidenced by late apoptotic markers, cleaved caspase-3, and DNA fragmentation. Considering the capacity of Ca2+ influx into mitochondria and concomitantly increased mitochondrial ROS to trigger apoptosis [40], it is conceivable that a similar mechanism underlies DHA-induced apoptosis in our experimental system. We observed a Ca2+-dependent increase in mitochondrial ROS and an attenuating effect of Tempol on caspase-3 cleavage. Furthermore, DHA treatment decreased ΔΨm in hPASMCs, indicating the opening of the mitochondrial inner membrane PTP, an event associated with the release of proapoptotic molecules from mitochondria [41]. Whether and to what extent C/EBP homologous protein, a proapoptotic component of UPR [42,43], contributes to DHA-induced apoptosis remains to be determined.
Conclusion
Based on the results of this comprehensive analysis of molecular events in DHA-treated primary hPASMCs, we conclude that Ca2+-dependent induction of oxidative stress is the central and initial event responsible for induction of UPR, cell cycle arrest, and apoptosis in DHA-treated hPASMCs. The fact that DHA-mediated induction of oxidative stress, UPR, and apoptosis might be beneficial in SMCs undergoing remodeling (pulmonary hypertension or stent restenosis), but detrimental in SMCs constituting the cap of a stable atherosclerotic plaque [44], implies that a systemic application of DHA may simultaneously be both beneficial and harmful. This is of importance, as these pathologic states may coexist in a patient. Therefore, our findings should encourage the development of strategies to achieve targeted, vascular bed-specific utilization of both DHA and appropriate antioxidants capable of modulating DHA effects.
The following are the supplementary materials related to this article.
Supplementary Fig. 1.

MitoSox Red localizes to mitochondria of DHA treated cells. Cells loaded with MitoSox Red (1 μM) and MitoTracker Green (0.5 μM) for 30 min were further incubated with DHA for 60 min and analyzed by an array confocal laser scanning microscope. Image analysis (colocalization analysis) was performed using MetaMorph® Software 7.7; (n=8).
Acknowledgments
We appreciate the excellent technical assistance of Heike Knausz (Medical University of Graz, ZMF Flow Cytometry Core Facility). This work was supported by the Medical University of Graz (Ph.D. Molecular Medicine Program), the Austrian Science Fund FWF (Grant P19473-B05 to S.F.), Jubilee Foundation of the Austrian National Bank Grants 12778 (S.F.) and 12527 (A.O.), and Lanyar Foundation Grant 328 (S.F.). The funding sources had no involvement in study design; collection, analysis, or interpretation of data; writing of the report; or the decision to submit the article for publication.
References
- 1.Dzau V.J., Braun-Dullaeus R.C., Sedding D.G. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat. Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 2.Morrell N.W., Adnot S., Archer S.L., Dupuis J., Jones P.L., MacLean M.R., McMurtry I.F., Stenmark K.R., Thistlethwaite P.A., Weissmann N., Yuan J.X., Weir E.K. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009;54:S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.GISSI-HF Investigators, Tavazzi L., Maggioni A.P., Marchioli R., Barlera S., Franzosi M.G., Latini R., Lucci D., Nicolosi G.L., Porcu M., Tognoni G. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1223–1230. doi: 10.1016/S0140-6736(08)61239-8. [DOI] [PubMed] [Google Scholar]
- 4.Davidson M.H., Stein E.A., Bays H.E., Maki K.C., Doyle R.T., Shalwitz R.A., Ballantyne C.M., Ginsberg H.N. COMBination of prescription Omega-3 with Simvastatin (COMBOS) Investigators. Efficacy and tolerability of adding prescription omega-3 fatty acids 4 g/d to simvastatin 40 mg/d in hypertriglyceridemic patients: an 8-week, randomized, double-blind, placebo-controlled study. Clin. Ther. 2007;29:1354–1367. doi: 10.1016/j.clinthera.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 5.Shiina T., Terano T., Saito J., Tamura Y., Yoshida S. Eicosapentaenoic acid and docosahexaenoic acid suppress the proliferation of vascular smooth muscle cells. Atherosclerosis. 1993;104:95–103. doi: 10.1016/0021-9150(93)90180-3. [DOI] [PubMed] [Google Scholar]
- 6.Terano T., Tanaka T., Tamura Y., Kitagawa M., Higashi H., Saito Y., Hirai A. Eicosapentaenoic acid and docosahexaenoic acid inhibit vascular smooth muscle cell proliferation by inhibiting phosphorylation of Cdk2–cyclin E complex. Biochem. Biophys. Res. Commun. 1999;254:502–506. doi: 10.1006/bbrc.1998.9976. [DOI] [PubMed] [Google Scholar]
- 7.Diep Q.N., Touyz R.M., Schiffrin E.L. Docosahexaenoic acid, a peroxisome proliferator-activated receptor-α ligand, induces apoptosis in vascular smooth muscle cells by stimulation of p38 mitogen-activated protein kinase. Hypertension. 2000;36:851–855. doi: 10.1161/01.hyp.36.5.851. [DOI] [PubMed] [Google Scholar]
- 8.Kim H.J., Vosseler C.A., Weber P.C., Erl W. Docosahexaenoic acid induces apoptosis in proliferating human endothelial cells. J. Cell. Physiol. 2005;204:881–888. doi: 10.1002/jcp.20351. [DOI] [PubMed] [Google Scholar]
- 9.Jakobsen C.H., Storvold G.L., Bremseth H., Follestad T., Sand K., Mack M., Olsen K.S., Lundemo A.G., Iversen J.G., Krokan H.E., Schonberg S.A. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. J. Lipid Res. 2008;49:2089–2100. doi: 10.1194/jlr.M700389-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prasad A., Bloom M.S., Carpenter D.O. Role of calcium and ROS in cell death induced by polyunsaturated fatty acids in murine thymocytes. J. Cell. Physiol. 2010;225:829–836. doi: 10.1002/jcp.22290. [DOI] [PubMed] [Google Scholar]
- 11.Munoz J.P., Zorzano A. Endoplasmic reticulum stress enters a nogo zone. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002708. 88 ps26. [DOI] [PubMed] [Google Scholar]
- 12.Harding H.P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 13.Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R.T., Remotti H., Stevens J.L., Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos C.X., Tanaka L.Y., Wosniak J., Laurindo F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 15.Calfon M., Zeng H., Urano F., Till J.H., Hubbard S.R., Harding H.P., Clark S.G., Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 16.Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H.P., Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 17.Haze K., Yoshida H., Yanagi H., Yura T., Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olschewski A., Li Y., Tang B., Hanze J., Eul B., Bohle R.M., Wilhelm J., Morty R.E., Brau M.E., Weir E.K., Kwapiszewska G., Klepetko W., Seeger W., Olschewski H. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ. Res. 2006;98:1072–1080. doi: 10.1161/01.RES.0000219677.12988.e9. [DOI] [PubMed] [Google Scholar]
- 19.Hallstrom S., Gasser H., Neumayer C., Fugl A., Nanobashvili J., Jakubowski A., Huk I., Schlag G., Malinski T. S-nitroso human serum albumin treatment reduces ischemia/reperfusion injury in skeletal muscle via nitric oxide release. Circulation. 2002;105:3032–3038. doi: 10.1161/01.cir.0000018745.11739.9b. [DOI] [PubMed] [Google Scholar]
- 20.Furst W., Hallstrom S. Simultaneous determination of myocardial nucleotides, nucleosides, purine bases and creatine phosphate by ion-pair high-performance liquid chromatography. J. Chromatogr. 1992;578:39–44. doi: 10.1016/0378-4347(92)80222-c. [DOI] [PubMed] [Google Scholar]
- 21.Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 22.Hartler J., Trotzmuller M., Chitraju C., Spener F., Kofeler H.C., Thallinger G.G. Lipid data analyzer: unattended identification and quantitation of lipids in LC-MS data. Bioinformatics. 2011;27:572–577. doi: 10.1093/bioinformatics/btq699. [DOI] [PubMed] [Google Scholar]
- 23.Riederer M., Ojala P.J., Hrzenjak A., Graier W.F., Malli R., Tritscher M., Hermansson M., Watzer B., Schweer H., Desoye G., Heinemann A., Frank S. Acyl chain-dependent effect of lysophosphatidylcholine on endothelial prostacyclin production. J. Lipid Res. 2010;51:2957–2966. doi: 10.1194/jlr.M006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graier W.F., Simecek S., Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. J. Physiol. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elvevoll E.O., Barstad H., Breimo E.S., Brox J., Eilertsen K.E., Lund T., Olsen J.O., Osterud B. Enhanced incorporation of n-3 fatty acids from fish compared with fish oils. Lipids. 2006;41:1109–1114. doi: 10.1007/s11745-006-5060-3. [DOI] [PubMed] [Google Scholar]
- 26.Rusca A., Di Stefano A.F., Doig M.V., Scarsi C., Perucca E. Relative bioavailability and pharmacokinetics of two oral formulations of docosahexaenoic acid/eicosapentaenoic acid after multiple-dose administration in healthy volunteers. Eur. J. Clin. Pharmacol. 2009;65:503–510. doi: 10.1007/s00228-008-0605-4. [DOI] [PubMed] [Google Scholar]
- 27.Cao J., Schwichtenberg K.A., Hanson N.Q., Tsai M.Y. Incorporation and clearance of omega-3 fatty acids in erythrocyte membranes and plasma phospholipids. Clin. Chem. 2006;52:2265–2272. doi: 10.1373/clinchem.2006.072322. [DOI] [PubMed] [Google Scholar]
- 28.Harper C.R., Edwards M.J., DeFilippis A.P., Jacobson T.A. Flaxseed oil increases the plasma concentrations of cardioprotective (n-3) fatty acids in humans. J. Nutr. 2006;136:83–87. doi: 10.1093/jn/136.1.83. [DOI] [PubMed] [Google Scholar]
- 29.Clore J.N., Allred J., White D., Li J., Stillman J. The role of plasma fatty acid composition in endogenous glucose production in patients with type 2 diabetes mellitus. Metabolism. 2002;51:1471–1477. doi: 10.1053/meta.2002.35202. [DOI] [PubMed] [Google Scholar]
- 30.Bousserouel S., Raymondjean M., Brouillet A., Bereziat G., Andreani M. Modulation of cyclin D1 and early growth response factor-1 gene expression in interleukin-1β-treated rat smooth muscle cells by n-6 and n-3 polyunsaturated fatty acids. Eur. J. Biochem. 2004;271:4462–4473. doi: 10.1111/j.1432-1033.2004.04385.x. [DOI] [PubMed] [Google Scholar]
- 31.Caviglia J.M., Gayet C., Ota T., Hernandez-Ono A., Conlon D.M., Jiang H., Fisher E.A., Ginsberg H.N. Different fatty acids inhibit apolipoprotein B100 secretion by different pathways: unique roles for ER stress, ceramide, and autophagy. J. Lipid Res. 2011;52:1636–1651. doi: 10.1194/jlr.M016931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raven J.F., Baltzis D., Wang S., Mounir Z., Papadakis A.I., Gao H.Q., Koromilas A.E. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2α phosphorylation. J. Biol. Chem. 2008;283:3097–3108. doi: 10.1074/jbc.M709677200. [DOI] [PubMed] [Google Scholar]
- 33.Ariyama H., Kono N., Matsuda S., Inoue T., Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem. 2010;285:22027–22035. doi: 10.1074/jbc.M110.126870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez-Chacon G., Astudillo A.M., Balgoma D., Balboa M.A., Balsinde J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim. Biophys. Acta. 2009;1791:1103–1113. doi: 10.1016/j.bbalip.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Aires V., Hichami A., Filomenko R., Ple A., Rebe C., Bettaieb A., Khan N.A. Docosahexaenoic acid induces increases in [Ca2+]i via inositol 1,4,5-triphosphate production and activates protein kinase Cγ and -δ via phosphatidylserine binding site: implication in apoptosis in U937 cells. Mol. Pharmacol. 2007;72:1545–1556. doi: 10.1124/mol.107.039792. [DOI] [PubMed] [Google Scholar]
- 36.Scorrano L., Penzo D., Petronilli V., Pagano F., Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition: implications for tumor necrosis factor-α apoptotic signaling. J. Biol. Chem. 2001;276:12035–12040. doi: 10.1074/jbc.M010603200. [DOI] [PubMed] [Google Scholar]
- 37.Starkov A.A., Chinopoulos C., Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Malhotra J.D., Kaufman R.J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 39.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W., Kaufman R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chinopoulos C., Adam-Vizi V. Calcium, mitochondria and oxidative stress in neuronal pathology: novel aspects of an enduring theme. FEBS J. 2006;273:433–450. doi: 10.1111/j.1742-4658.2005.05103.x. [DOI] [PubMed] [Google Scholar]
- 41.Szabadkai G., Rizzuto R. Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett. 2004;567:111–115. doi: 10.1016/j.febslet.2004.04.059. [DOI] [PubMed] [Google Scholar]
- 42.Marciniak S.J., Yun C.Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H.P., Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCullough K.D., Martindale J.L., Klotz L.O., Aw T.Y., Holbrook N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]