

Abstract
There is a substantial body of evidence that spontaneous recurrent seizures occur in a subset of patients with Alzheimer disease (AD), especially the familial forms that have an early onset. In transgenic mice that simulate these genetic forms of AD, seizures or reduced seizure threshold have also been reported. Mechanisms underlying the seizures or reduced seizure threshold in these mice are not yet clear and are likely to be complex, because the synthesis of amyloid β (Aβ) involves many peptides and proteases that influence excitability. Based on transgenic mouse models of AD where Aβ and its precursor are elevated, it has been suggested that seizures are caused by the downregulation of the Nav1.1 sodium channel in a subset of GABAergic interneurons, leading to a reduction in GABAergic inhibition. Another mechanism of hyperexcitability appears to involve tau, because deletion of tau reduces seizures in some of the same transgenic mouse models of AD. Therefore, altered excitability may be as much a characteristic of AD as plaques and tangles—especially for the familial forms of AD.
Highlights
Spontaneous recurrent seizures occur in patients with AD and mouse models of the disease, especially the familial forms.
There are similarities between patients with AD and those with temporal lobe epilepsy (TLE), and between animal models of AD and TLE, but there are also differences.
Almost all peptides and proteases involved in Aβ synthesis influence excitability and do so by diverse mechanisms.
One mechanism for seizures in AD, based on experiments in mouse models, is a reduction in Nav1.1 sodium channel expression in fast-spiking GABAergic interneurons, which is interesting because mutations in the gene for Nav1.1, SCN1A, are responsible for two epilepsy syndromes—generalized epilepsy with febrile seizures plus (GEFS+) and severe myoclonic epilepsy of infancy (SMEI).
Tau plays a critical role in excitability and interacts with Aβ. Reducing tau can decrease seizures in transgenic mice with high levels of Aβ. Tau reduction also decreases the effects of convulsants in normal mice. Therefore, tau may be a therapeutic target for epilepsy.
Coming of Age: Seizures in AD
In 2007, a landmark article was published about transgenic mice that simulated familial AD. This article surprised the AD research community because it showed that epileptiform discharges and spontaneous recurrent seizures occur in some of the most commonly used mouse models of AD (i.e., the mice had bona fide epilepsy) (1).
Notably, seizures were robust: electrographic seizure activity was recorded at many sites in the brain simultaneously. However, electrographic seizures were not associated with convulsive behavior. As a result, it seemed likely that many investigators in AD research were unaware that their experimental animals were experiencing intermittent periods of generalized nonconvulsive seizures (2–4). The implications were potentially important: patients with AD might also have epileptiform discharges or nonconvulsive seizures, and not know.
Although it has been known for some time that seizures occur in some patients with AD (5, 6), and early studies in mouse models of AD had described spontaneous behavioral seizures (7, 8) or decreased seizure threshold (9, 10), there has been a reluctance to accept the idea that seizures are an integral part of AD. The reluctance is probably a result of several factors, such as the clinical definition of AD, which is based on characteristics that differ from epilepsy, rather than emphasizing the characteristics that are shared. The definition of AD emphasizes the neuropathology that was originally used to characterize AD—amyloid-β (Aβ) plaques and neurofibrillary tangles. Notably, plaques do occur in patients with epilepsy; they are more numerous in patients with epilepsy relative to age-matched controls, but they are not as prevalent and do not reach the most severe stages on the standard rating scale, called the Braak scale (11).
The experiments of Palop and colleagues (1) have had a significant impact, which may be related to the fortuitous timing of the published findings. At about the same time as the publication, other investigators studying rodents with epilepsy showed that brief episodes of epileptiform activity or interictal spikes impair fundamental neurobiological phenomena associated with memory formation and consolidation (12, 13). These studies and others provided support for the idea that even brief episodes of epileptiform activity (e.g., interictal spikes) can impair memory.
Notably, additional studies in mouse models of AD have now shown that seizures can be convulsive and severe, reaching stage 5 on the Racine scale (14). These findings are consistent with the observations that familial forms of AD are often associated with convulsive behavior.
On the other hand, there is still reticence to accept the idea that seizures are a characteristic of AD, because many people with AD do not have seizures, and mouse models of AD mostly simulate familial AD. The more common type of AD is sporadic. Regrettably, there are few animal models of sporadic AD. However, clinical data suggest that patients who have sporadic AD have convulsive seizures, although seizures may only occur in a fraction of patients (5, 6, 15). Other patients with sporadic AD appear to have increased excitability (inferred from neuroimaging) (16, 17). Notably, the abnormal excitability can be reduced with anticonvulsants (17). Therefore, hyperexcitability, defined for the present purposes as an abnormally high level of synchronous action potential discharge, typically in a subset of interconnected principal cells located in cortical or limbic regions, seems increasingly relevant to AD. The reader is referred to the accompanying article for a review on clinical science aspects of epilepsy and Alzheimer's Disease.
Of Mice and Men: Pathology in Mouse Models of AD Suggest Similarities to TLE
Palop and colleagues (1) used animal models of familial AD that have been studied by many laboratories interested in mechanisms underlying AD. These transgenic mice overexpress a mutated form of human amyloid precursor protein (APP), the precursor to Aβ. The mutations in APP are relevant to AD because they are found in families with AD. Expression in mouse models is usually driven by a relatively widespread promoter (e.g., platelet-derived growth factor β). The APP mutations cause increased metabolism of APP along the so-called amyloidogenic pathway of APP metabolism, which leads to increased production of Aβ (Figure 1). The other route of APP metabolism, the nonamyloidogenic pathway, does not result in Aβ production (Figure 1). By approximately 6 to 12 months of age (early adulthood to middle age in a mouse), Aβ plaque is present in many areas of the mice that overexpress mutated APP.
FIGURE 1.
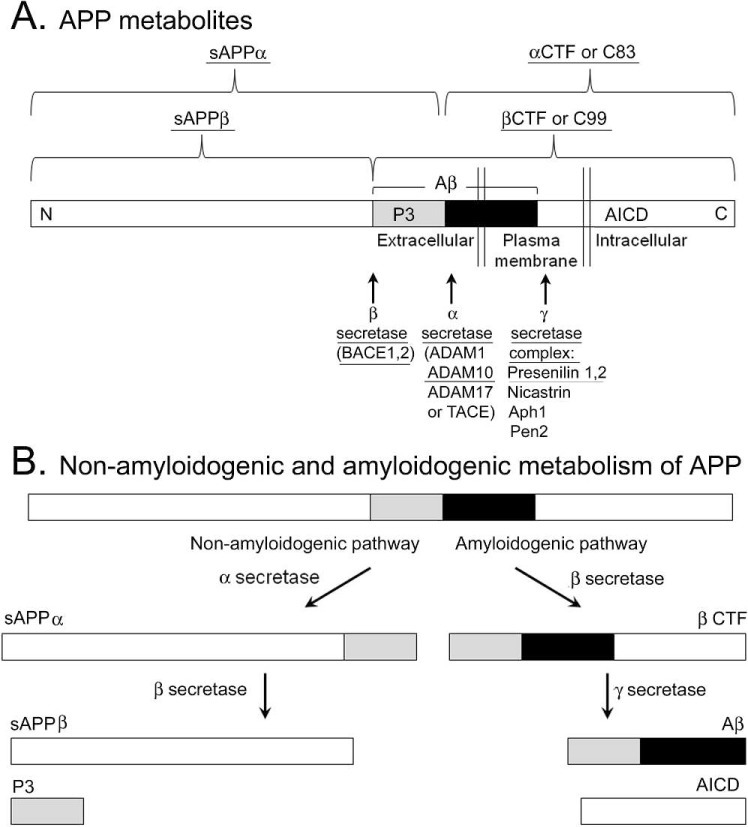
Amyloid precursor protein metabolism produces several peptides that influence excitability. A. A diagram of APP processing is shown. Amyloid β is composed of a peptide fragment called P3 (shaded rectangle) and another fragment (black). The peptide products and associated enzymes (α, β, γ secretases) that lower seizure threshold or appear to induce seizures in transgenic mice or are mutated in a familial form of AD with convulsive seizures are underlined. Peptides that are likely to influence epileptogenesis because of diverse roles in proliferation and outgrowth are also underlined. B. A diagram of non-amyloidogenic and amyloidogenic routes of APP metabolism is shown. For the non-amyloidogenic pathway. α secretase cleavage occurs first, followed by γ secretase. For the amyloidogenic route, β secretase cleavage is followed by γ secretase cleavage, leading to Aβ. CTF, C terminal fragment; sAPP, soluble APP; BACE, β-site APP cleavage enzyme, ADAM, a disintegrin and metalloprotease, TACE, tumor necrosis factor α cleavage enzyme; Aph1, anterior pharynx defective; Pen, presenilin enhancer.
Because of the similarity of the seizures in these “APP mice” to complex partial seizures, which is a common type of seizure in AD patients (4), the hippocampus was examined further, and the mice have been discussed in the context of TLE. The comparison with TLE and the investigation of the hippocampus were logical because TLE is considered to be the type of epilepsy that involves memory centers, and the hippocampus is perhaps the best studied. However, AD and TLE should be compared cautiously, because there are many differences when one ”scratches the surface.“ For example, the type of memory impairment in AD is not necessarily comparable to deficits in TLE (18). The neurons that are most vulnerable in AD and TLE are located in similar areas (e.g., entorhinal cortex, hippocampus) but are not identical. For example, the neurons in the entorhinal cortex that are most vulnerable in AD are the layer II stellate cells, and in TLE the neurons that are most vulnerable are layer III pyramidal cells (19, 20); for review, see Scharfman and Chao (21).
When the hippocampus of mice with APP overexpression/mutation (e.g., J9, J20 mouse lines, termed collectively “APP mice” below) were studied by Palop and colleagues (1), they found several characteristics that have been observed in the so-called status epilepticus (SE) models of TLE. In the SE models, recurrent spontaneous convulsive seizures are induced by an initial period of SE in adult life. Some of the characteristics of the APP mice were ectopic expression of neuropeptide Y (NPY) in the mossy fiber axons of dentate gyrus granule cells (DGCs) (1), which is a characteristic of SE models (22). In addition, there was a loss of calbindin D28K in DGCs in the APP mice, which was also evident in patients with AD (23) and has been reported in patients with TLE and SE models of TLE (24); for review, see Scharfman (25). Notably, mossy fiber expression of NPY and decreased expression of calbindin in granule cells has been found in other APP mouse models (26) and in mice with both APP and additional mutations (14).
What Palop and colleagues (1) did not find was just as interesting as what they did. For example, there was little evidence of widespread neuronal loss. Others have found cell loss in APP mouse models of AD pathology (27) or mice with multiple mutations (28). However, the degree of neuronal damage in mouse models of AD is limited, in general, and is typically less than in patients at the end stage of AD, after progressive hippocampal and entorhinal neurodegeneration. This is an interesting point because a range of neuronal loss also occurs in patients with TLE, despite the fact that hippocampal sclerosis is often considered to define TLE. In TLE animal models (e.g., kindling, febrile seizures, neonatal hypoxia, adult SE), there also appears to be a range of cell loss, and progressive neurodegeneration has been suggested both in animals and patients but is subject to debate (29, 30).
APP mice also lack a common hallmark of TLE, a reorganization of the DGC axons called mossy fiber sprouting. However, some patients with TLE and some animal models of TLE do not exhibit extensive sprouting, and it is not necessarily a cause of seizures (31, 32). Furthermore, sprouting is often robust at temporal regions of the hippocampus (33), which seem to be evaluated rarely in APP mice. Therefore, the lack of mossy fiber sprouting in mouse models of AD needs to be interpreted with caution.
Pass the Salt: Mechanisms of Seizures in Mouse Models of AD May Involve Sodium Channels
Many basic questions are raised by the studies of seizures in patients with AD and in mouse models of AD. Do seizures contribute to the progressive pathology and memory impairment in AD? A very interesting study suggests that this might be the case. Seizure activity in animals, induced by stimulating the perforant path input to the dentate gyrus, led to increased levels of Aβ, detected extracellularly (34). Based on this study and others, it has been suggested that the seizures in AD could initiate a ”vicious cycle” (4). For example, if a seizure increased Aβ levels, the increase in Aβ might increase excitability further, leading to more seizures. Therefore, the emergence of seizures may be a critical point in the progression of AD. After seizures begin, there may be an increased rate of plaque deposition and a more severe phase of the disease. Although there is little direct evidence for this idea in AD patients, it has been reported that seizures begin at about the time of plaque deposition, at least in some of the mouse models of AD (14).
Perhaps the most important question—still a question of considerable debate—is the mechanism of seizure generation in AD. One might predict that the mechanism would involve the increased production of Aβ and the subsequent deposition of plaque. Indeed, some data support the idea that Aβ causes an increase in excitatory synaptic transmission, at least at low concentrations (35). Higher concentrations of Aβ decrease glutamatergic transmission however (33, 35, 36). Therefore, an initial increase in excitability might occur early in AD when Aβ levels are just above normal, but it is harder to understand how high levels of Aβ would lead to seizures. It is important to interpret studies of exogenous application of Aβ with caution, however, because the results could be very different from the effects of endogenous Aβ. It may be more relevant to study the changes in excitability that occur as soluble Aβ initially accumulates and then plaques form. One study that has examined excitability related to endogenous plaques found that excitability was increased in the areas of cortex adjacent to plaques relative to areas further away (37, 38). However, more research is needed before it can be concluded that Aβ plaque is responsible for seizures.
One reason to suggest that more research is needed is that the mouse models of AD have other abnormalities besides elevated Aβ that could alter excitability. APP itself has numerous actions, and the peptides that are by-products of Aβ production also influence excitability (Figure 1). The APP intracellular domain (AICD), a result of Aβ production (Figure 1) is a good example. Animals with AICD overexpression have spontaneous seizures (39), and aged mice with AICD overexpression have ectopic NPY expression in mossy fibers, and mossy fiber sprouting (40). Therefore AICD may have robust proconvulsant effects in mice. The secretases that are responsible for APP proteolysis also influence seizures (41; Figure 1).
The effects of APP and its metabolites are complicated because they are not unidirectional—more of a peptide may lead to greater excitability, but deletion can have similar effects. For example, deleting APP or β-secretase (BACE, a critical step in Aβ synthesis; Figure 1) might seem like a potential therapeutic strategy, but spontaneous seizures or decreased seizure threshold occurs after deletion of APP or BACE1 (9, 42, 43). The results of these studies suggest that there is a delicate balance in the amount of APP and APP metabolism in the normal brain.
Although there is a long list of potential targets of APP and APP-derived peptides, one that has received a great deal of attention recently is the sodium channel Nav1.1. Nav1.1 is interesting in this context, because mutations cause two pediatric epilepsy syndromes: generalized epilepsy with febrile seizures plus (GEFS+), where there are mutations in the gene for Nav1.1, SCN1A, and severe myoclonic epilepsy of infancy (SMEI or Dravet syndrome), where there is a more severe loss-of-function of SCN1A (44).
The idea that Nav1.1 plays a role in the seizures in mouse models of AD has emerged from studies of BACE1 and APP mice. One study showed that BACE1 regulates sodium channel surface expression, causing a downregulation of surface expression (45). Another study found that APP mice had a deficit in Nav1.1 expression primarily in a subset of fast-spiking GABAergic interneurons that express parvalbumin (46). Whole-cell recordings from the interneurons showed that there were defects in action potential firing (46), similar to studies of mice with SCN1A mutations (44). In both the APP and SCN1A mice, seizures can potentially be explained by disinhibition of principal cells, innervated by interneurons that do not release adequate GABA. It has also been suggested that impairments in cognitive function, which occurs in both the APP mice and SMEI, can also be explained by a loss of function in critical fast-spiking interneurons (47).
Another similarity between APP mice, GEFS+, and SMEI is that antiepileptic drugs that block sodium channels make seizures worse rather than better. A potential explanation for this paradox is that sodium channels on interneurons are decreased in APP mice. Therefore, a sodium channel antagonist exacerbates the defect (46, 48, 49). Remarkably, correcting the Nav1.1 defect in the interneurons of APP mice reduced the seizures and the memory impairments (46). This study was a tour de force, but some questions still remain, because the studies of BACE1, which suggest that it regulates sodium channel expression, show that the decrease in sodium channel expression occurs in principal cells rather than interneurons (45). Furthermore, additional studies of sodium channel-blocking AEDs in other mouse models of AD show that phenytoin reduces seizures (50). However, it may be that these seemingly contrasting results are all correct when it comes to the patient with AD. In other words, these studies can potentially explain the heterogeneity of symptoms in AD: when interneurons are primarily affected, seizures result; when principal cells are also affected, seizures may be rare or even absent, and cognitive impairment occurs in isolation.
Although most of the studies of excitability have used mice with disturbed APP expression and metabolism, tau is important to mention because it appears to play a critical role in Aβ-driven pathology (51–53). Normally, tau is an important component of the cytoskeleton and subject to phosphorylation by diverse kinases. In AD, tau becomes hyperphosphorylated and forms neurofibrillary tangles. Although APP mice do not develop tangles, transgenic manipulation of tau in APP mice has shown that it is associated with seizures in the mice. Using APP mice crossed with another mouse line with deletion of tau, seizures were reduced and memory was improved (24). Therefore, tau appears to be critical for seizures in APP mice, although the mechanisms are not clear. Several possibilities exist, such as interactions with Fyn, a kinase that phosphorylates tau (26). Remarkably, tau exerts an anticonvulsant effect even in wild type mice (26).
Conclusions: Multiple Mechanisms, Multiple Targets?
There is increasing evidence that mechanisms of AD and epilepsy overlap. Acknowledging that AD and epilepsy are related is beneficial to both fields, because potential therapeutic advances in one may help translational efforts in the other. However, there also is a challenge—the complexities of the clinical disorders and the limitations of mouse models need to be carefully considered. Nevertheless, this is a time of opportunity. For example, the remarkable anticonvulsant effects of tau suggest that it could be a new target for AED development. There are likely to be many ways to help patients with AD through epilepsy research and vice versa, as difficult as it may be to “untangle” AD and epilepsy.
Acknowledgments
The author thanks Dr. Jorge Palop for comments on the manuscript. This study was supported by a grant from the National Institutes of Health (MH-090606) and the New York Office of Mental Health.
Footnotes
Editor's Note: Authors have a Conflict of Interest disclosure which is posted under the Supplemental Materials (253.5KB, doc) link.
References
- 1.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- 3.Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noebels J. A perfect storm: Converging paths of epilepsy and Alzheimer's dementia intersect in the hippocampal formation. Epilepsia. 2011;52(suppl 1):39–46. doi: 10.1111/j.1528-1167.2010.02909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, Albert M, Amatniek JC, Marder K, Bell K, Hauser WA, Stern Y. Seizures in Alzheimer disease: Who, when, and how common? Arch Neurol. 2009;66:992–997. doi: 10.1001/archneurol.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman D, Honig L, Scarmeas N. Seizures and epilepsy in Alzheimer's disease. CNS Neurosci Ther. 2012;18:285–294. doi: 10.1111/j.1755-5949.2011.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 8.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the α-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 9.Steinbach JP, Muller U, Leist M, Li ZW, Nicotera P, Aguzzi A. Hypersensitivity to seizures in β-amyloid precursor protein deficient mice. Cell Death Differ. 1998;5:858–866. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- 10.Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett. 2004;367:164–167. doi: 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- 11.Thom M, Liu JY, Thompson P, Phadke R, Narkiewicz M, Martinian L, Marsdon D, Koepp M, Caboclo L, Catarino CB, Sisodiya SM. Neurofibrillary tangle pathology and braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: A postmortem study. Brain. 2011;134:2969–2981. doi: 10.1093/brain/awr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleen JK, Scott RC, Holmes GL, Lenck-Santini PP. Hippocampal interictal spikes disrupt cognition in rats. Ann Neurol. 2010;67:250–257. doi: 10.1002/ana.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes GL, Lenck-Santini PP. Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 2006;8:504–515. doi: 10.1016/j.yebeh.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, Penke B, Zilberter Y, Harkany T, Pitkanen A, Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendez M, Lim G. Seizures in elderly patients with dementia: Epidemiology and management. Drugs Aging. 2003;20:791–803. doi: 10.2165/00002512-200320110-00001. [DOI] [PubMed] [Google Scholar]
- 16.Dickerson BC, Sperling RA. Large-scale functional brain network abnormalities in Alzheimer's disease: Insights from functional neuroimaging. Behav Neurol. 2009;21:63–75. doi: 10.3233/BEN-2009-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeman A, Butler C. Transient epileptic amnesia. Curr Opin Neurol. 2010;23:610–616. doi: 10.1097/WCO.0b013e32834027db. [DOI] [PubMed] [Google Scholar]
- 19.Du F, Whetsell WO, Abou-Khalil B, Blumenkopf B, Lothman EW, Schwarcz R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993;16:223–233. doi: 10.1016/0920-1211(93)90083-j. [DOI] [PubMed] [Google Scholar]
- 20.Kordower J, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennet D, Mufson EJ. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49:202–213. [PubMed] [Google Scholar]
- 21.Scharfman HE, Chao MV. The entorhinal cortex in Alzheimer's disease. Eur J Neurodegen Dis. 2012;1:53–65. [Google Scholar]
- 22.Sperk G, Bellmann R, Gruber B, Greber S, Marksteiner J, Roder C, Rupp E. Neuropeptide Y expression in animal models of temporal lobe epilepsy. Epilepsy Res Suppl. 1996;12:197–203. [PubMed] [Google Scholar]
- 23.Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagerl UV, Mody I, Jeub M, Lie AA, Elger CE, Beck H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-d(28k) in temporal lobe epilepsy. J Neurosci. 2000;20:1831–1836. doi: 10.1523/JNEUROSCI.20-05-01831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scharfman HE. Alzheimer's disease and epilepsy: Insight from animal models. Future Neurol. 2012;7:177–192. doi: 10.2217/fnl.12.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi H, Brasnjevic I, Rutten BP, Van Der Kolk N, Perl DP, Bouras C, Steinbusch HW, Schmitz C, Hof PR, Dickstein DL. Hippocampal inter-neuron loss in an APP/PS1 double mutant mouse and in Alzheimer's disease. Brain Struct Funct. 2010;214:145–160. doi: 10.1007/s00429-010-0242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DaRocha-Souto B, Scotton TC, Coma M, Serrano-Pozo A, Hashimoto T, Serenó L, Rodríguez M, Sánchez B, Hyman BT, Gómez-Isla T. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropath Exp Neurol. 2011;70:360–376. doi: 10.1097/NEN.0b013e318217a118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sutula TP, Pitkanen A. Do Seizures Damage the Brain? New York: Elsevier; 2002. [Google Scholar]
- 30.McNamara JO, Scharfman HE. Temporal lobe epilepsy and the BDNF receptor, TrkB. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies, 4th edition. Oxford: Oxford University Press; pp. 514–531. [PubMed] [Google Scholar]
- 31.Nissinen J, Lukasiuk K, Pitkanen A. Is mossy fiber sprouting present at the time of the first spontaneous seizures in rat experimental temporal lobe epilepsy? Hippocampus. 2001;11:299–310. doi: 10.1002/hipo.1044. [DOI] [PubMed] [Google Scholar]
- 32.Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–2347. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scharfman HE, Sollas AL, Smith KL, Jackson MB, Goodman JH. Structural and functional asymmetry in the epileptic rat dentate gyrus. J Comp Neurol. 2002;454:424–439. doi: 10.1002/cne.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 35.Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 37.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 38.Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2009;32:1725–1729. doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosal K, Pimplikar SW. Aging and excitotoxic stress exacerbate neural circuit reorganization in amyloid precursor protein intracellular domain transgenic mice. Neurobiol Aging. 2010;32:2320–2329. doi: 10.1016/j.neurobiolaging.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clement AB, Hanstein R, Schroder A, Nagel H, Endres K, Fahrenholz F, Behl C. Effects of neuron-specific ADAM10 modulation in an in vivo model of acute excitotoxic stress. Neuroscience. 2008;152:459–468. doi: 10.1016/j.neuroscience.2007.10.060. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi D, Zeller M, Cole T, Buttini M, McConlogue L, Sinha S, Freedman S, Morris RG, Chen KS. BACE1 gene deletion: Impact on behavioral function in a model of Alzheimer's disease. Neurobiol Aging. 2008;29:861–873. doi: 10.1016/j.neurobiolaging.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci. 2010;30:8819–8829. doi: 10.1523/JNEUROSCI.1334-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catterall WA, Kalume F, Oakley JC. Nav1.1 channels and epilepsy. J Physiol. 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH, He P, Lee VM, Woolf CJ, Kovacs DM. BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol. 2007;9:755–764. doi: 10.1038/ncb1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneu-ron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini PP. SCN1A mutations in Dravet syndrome: Impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav. 2012;23:177–186. doi: 10.1016/j.yebeh.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerrini R, Belmonte A, Genton P. Antiepileptic drug-induced worsening of seizures in children. Epilepsia. 1998;39(suppl 3):S2–10. doi: 10.1111/j.1528-1157.1998.tb05118.x. [DOI] [PubMed] [Google Scholar]
- 49.Loscher W. Molecular mechanisms of drug resistance in status epilepticus. Epilepsia. 2009;50(suppl 12):19–21. doi: 10.1111/j.1528-1167.2009.02367.x. [DOI] [PubMed] [Google Scholar]
- 50.Ziyatdinova S, Gurevicius K, Kutchiashvili N, Bolkvadze T, Nissinen J, Tanila H, Pitkanen A. Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res. 2011;94:75–85. doi: 10.1016/j.eplepsyres.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 51.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 53.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]