

Abstract
Urinary proteins that leak through the abnormal glomerulus in nephrotic syndrome may affect tubular transport by interacting with membrane transporters on the luminal side of tubular epithelial cells. Patients with nephrotic syndrome can develop nephrocalcinosis, which animal models suggest may develop from impaired transcellular Ca2+ reabsorption via TRPV5 in the distal convoluted tubule (DCT). In nephrotic-range proteinuria, filtered plasminogen reaches the luminal side of DCT, where it is cleaved into active plasmin by urokinase. In this study, we found that plasmin purified from the urine of patients with nephrotic-range proteinuria inhibits Ca2+ uptake in TRPV5-expressing human embryonic kidney 293 cells through the activation of protease-activated receptor-1 (PAR-1). Preincubation with a plasmin inhibitor, a PAR-1 antagonist, or a protein kinase C (PKC) inhibitor abolished the effect of plasmin on TRPV5. In addition, ablation of the PKC phosphorylation site S144 rendered TRPV5 resistant to the action of plasmin. Patch-clamp experiments showed that a decreased TRPV5 pore size and a reduced open probability accompany the plasmin-mediated reduction in Ca2+ uptake. Furthermore, high-resolution nuclear magnetic resonance spectroscopy demonstrated specific interactions between calmodulin and residues 133–154 of the N-terminus of TRPV5 for both wild-type and phosphorylated (S144pS) peptides. In summary, PAR-1 activation by plasmin induces PKC-mediated phosphorylation of TRPV5, thereby altering calmodulin-TRPV5 binding, resulting in decreased channel activity. These results indicate that urinary plasmin could contribute to the downstream effects of proteinuria on the tubulointerstitium by negatively modulating TRPV5.
In the kidney, the fine regulation of Ca2+ balance occurs through the activity of the epithelial Ca2+ channel TRPV5.1 TRPV5 is mostly expressed in the distal convoluted tubule (DCT) and connecting tubule of the nephron, where it constitutes the apical entry mechanism for transcellular Ca2+ reabsorption. TRPV5 is a constitutively active ion channel that bears unique electrophysiologic characteristics, including calmodulin (CaM) and Ca2+-dependent inactivation and high selectivity for Ca2+.2,3 The activity of TRPV5 is tightly controlled at multiple levels by an array of different factors, including parathyroid hormone and the serine protease tissue kallikrein. Both parathyroid hormone and tissue kallikrein initiate the phosphorylation of TRPV5 through the cAMP/protein kinase A (PKA) and phospholipase C (PLC)/ diacylglycerol (DAG)/protein kinase C (PKC) signaling cascades, respectively.4,5
Proteinuria is a hallmark of nephrotic syndrome, in which large plasma proteins pass through the disrupted glomerular basement membrane (GBM). Pathologic leakage of glomerular proteins causes multiple tubulointerstitial abnormalities, such as interstitial inflammation and eventually fibrosis, but does not affect tubular structure.6 However, recent data showed that tubular transport processes could be affected by direct effect of urinary protein on membrane transporters at the luminal side of the DCT, such as epithelial sodium channel.7 The observation of nephrocalcinosis in patients with nephrotic-range proteinuria,8,9 associated with impaired transcellular Ca2+ reabsorption via TRPV5 in a mouse model,10 suggests that proteinuria might affect tubular Ca2+ handling.
Patients with nephrotic syndrome (NS) have elevated serum plasminogen levels.11 After leakage into the urine, plasminogen is converted into active plasmin by tubular urokinase-type plasminogen activator and has recently been reported to regulate renal ion transport in nephrotic syndrome by effects on the epithelial sodium channel.7,12 Whether urinary plasmin also can affect tubular Ca2+ handling and, if so, by which mechanisms, has not been established so far. This study aimed to investigate the effects and mechanism of urinary plasmin on TRPV5-mediated Ca2+ reabsorption.
Results
Plasmin in Nephrotic Urine Inhibits TRPV5-Mediated Ca2+ Influx
To investigate the effect of plasmin on TRPV5-mediated Ca2+ transport, human embryonic kidney (HEK) 293 cells were transfected with TRPV5 and treated with 10 nM plasmin for 1 hour before radiotracer 45Ca2+ influx measurements. Plasmin inhibited TRPV5-mediated Ca2+ influx to an extent similar to that seen with ruthenium red, thus indicating completely inhibited TRPV5 activity (Figure 1A). Plasmin inhibits Ca2+ influx with a 50% inhibitory concentration of approximately 3 nM (Figure 1B) after at least 30 minutes of incubation (Supplemental Figure 1). The specific plasmin inhibitor α2-antiplasmin reversed this block (Figure 1A). Purified plasmin from the urine of five nephrotic patients mimicked the inhibitory effect compared with commercial plasmin. This urinary activity could be reversed by heat inactivation and α2-antiplasmin (Figure 1C). The presence of plasmin in urine samples and their activities are depicted in Figure 1D.
Figure 1.

Plasmin in nephrotic urine inhibits TRPV5-mediated Ca2+ influx in HEK293 cells. (A) One-hour incubation of 10 nM commercial plasmin was effective to inhibit Ca2+ influx and was reversed by 50 nM α2-antiplasmin (α2), specific plasmin inhibitor. Ruthenium red (RR), 10 μM, was administered to determine TRPV5-mediated Ca2+ influx. Cells were pretreated with 25 μM BAPTA-AM for 30 minutes. (B) Dose-response curve of Ca2+ uptake. (C) Plasmin purified from nephrotic urine inhibited Ca2+ influx, and this effect was abolished by a heat inactivation (5 minutes at 80°C) and α2. (D) Plasmin expression in urine samples of five nephrotic patients was shown on immunoblot (C, control urine sample; P, plasmin-added sample). Plasmin activities of the commercial plasmin, nephrotic urine–purified, and urine from healthy person are shown in the unit of relative fluorescence unit (RFU) under control (black), heat-inactivated (striped), and α2-antiplasmin (white), respectively. §P<0.05 versus mock. †P<0.05 versus respective control.
Plasmin Does Not Affect TRPV5 Membrane Abundance
Because plasmin has been reported to cleave the membrane-bound epithelial sodium channel,12 we investigated whether plasmin could exert its effects via cleavage of TRPV5 using cell surface biotinylation experiments. Figure 2A shows no change in plasma membrane or total expression of TRPV5 (protein input) after plasmin treatment (immunoblot intensities are shown in Figure 2B). The positive control tissue kallikrein could enhance TRPV5 membrane abundance without affecting the channel total expression5 (Figure 2A).
Figure 2.

Plasmin does not affect surface plasma membrane abundance of TRPV5. (A) TRPV5-transfected HEK293 cells were treated with 10 nM plasmin. Tissue kallikrein (TK), 100 nM, was used as positive control. N-hydroxysuccinimide-long chain-long chain-biotin (NHS-LC-LC-biotin) was added to media for 30 minutes before cells were lysed, and the biotinylated fraction was pulled down with neutravidin-agarose beads. TRPV5 was eluted and analyzed by immunoblot for plasma membrane expression. β-actin was used as an internal control. (B) Immunoblot densities calculated from A. C, control; P, plasmin-treated.
TRPV5 Inhibition by Plasmin Is Mediated by Protease-Activated Receptor-1
Because plasmin failed to cleave TRPV5 at the plasma membrane, we hypothesized that the reduced TRPV5 activity is mediated by a cell surface membrane receptor. Plasmin has been previously shown to bind the protease-activated receptor-1 (PAR-1), prostasin, and megalin.13–16 Therefore, we examined mRNA expressions of F2R, PRSS8, and LRP2, which encode PAR-1, prostasin, and megalin, respectively. Our results showed that only PAR-1 is endogenously expressed in HEK293 cells (Figure 3A). PAR-1 protein expression is shown by immunoblot (Figure 3B). Co-localization of PAR-1 and TRPV5 in late DCT and connecting tubules of mouse kidney cortex is depicted by immunohistochemistry (Figure 3C). Fura-2–based Ca2+ imaging experiments showed that PAR-1 was functional because it stimulated intracellular Ca2+ release (Figure 3D). The PAR-1 inhibitor SCH79797 and PAR-1 antibody prevented TRPV5 inhibition by plasmin, indicating a specific action of plasmin through PAR-1 (Figure 3E).
Figure 3.

PAR-1 is responsible for plasmin-induced reduction in TRPV5-mediated Ca2+ influx. (A) F2R, PRSS8, and LRP2 mRNA expression were examined by RT-PCR. GAPDH was used as a control. Plus and minus signify PCR products of reverse-transcribed and non–reverse-transcribed mRNA, respectively. (B) Immunoblot shows endogenous PAR-1 expression in HEK293 cells in mock (M), TRPV5-transfected (C), and TRPV5-transfected and treated with 10 nM plasmin (P). (C) Immunohistochemistry depicts PAR-1 and TRPV5 co-localization. (D) Fura-2 experiment tracer shows a PAR-1 induction of Ca2+i release in the presence of 10 mM EDTA. (E) Inhibitory action of plasmin (P) on TRPV5-mediated 45Ca2+ influx is reversed by 30-minute pretreatment of 20 μM SCH79797 (S), a PAR-1 antagonist; and 2 μg/mL PAR-1 antibody (Ab). †P<0.05 versus control (C).
Plasmin-Induced Inhibition of TRPV5 Depends on PKC Phosphorylation of Serine-144 Residue
PAR-1 is a member of the G protein–coupled receptor family, which normally exerts its effect through the PKA17 or PLC/PKC pathways.18 We investigated whether these protein kinases are involved in plasmin-activated PAR-1 signaling. TRPV5 possesses three potential phosphorylation sites for PKA: One is on the N-terminus (Nter) and the other two are on the C-terminus (Cter).4 In addition, there are six sites for PKC: three sites on both Nter and Cter.5 HEK293 cells were transfected with Nter-PKA, Cter-PKA, or six-PKC phosphorylation site-mutated TRPV5. All PKA- and PKC-TRPV5 mutants were still able to transport Ca2+ to the same extent as wild-type TRPV5. However, plasmin failed to inhibit Ca2+ uptake in the six-PKC-phosphorylation site mutant, suggesting that at least one of these sites is responsible for plasmin action (Figure 4A). This finding prompted us to find the PKC phosphorylation site responsible for the inactivation of TRPV5. In combination with the Ca2+ uptake assay, serine (S) residue located in the ankyrin repeat domain 3 of the Nter mutated to alanine (A), S144A,5 was found to be unresponsive to plasmin action (Figure 4C). The involvement of the PLC/PKC signaling pathway was further confirmed by establishing the reversal effect of PLC and PKC antagonists (U73122 and chelerythrine, respectively) on the inhibitory action of plasmin (Figure 4B).
Figure 4.

Inhibition of TRPV5 activity by plasmin requires the presence of S144 residue on amino-terminus. (A) Effects of plasmin on Ca2+ uptake were measured in wild-type (WT) TRPV5, amino- and carboxy-terminal PKA-site mutants (N-PKA and C-PKA, respectively), and the six-site PKC TRPV5 mutant (PKC). (B) Incubation of 10 μM U73122 (U), a PLC inhibitor, and 10 μM chelerythrine (Ch), a cell-permeable PKC inhibitor, for 30 minutes reversed the inhibitory effect of plasmin (P) to control (C) level. (C) Effect of plasmin on Ca2+ uptake when TRPV5 contains single-point mutations of six PKC phosphorylation sites. *P<0.05 versus a nonplasmin condition (-) and †P<0.05 versus control (C).
Plasmin Specifically Decreases TRPV5-Mediated Ca2+ Current by Reducing Its Pore Size and Open Probability
The functional effect of plasmin on TRPV5-mediated Ca2+ current measured by patch-clamp technique was studied in HEK293 cells expressing TRPV5, by incubating cells for 1 hour with 10 nM plasmin. Before Ca2+ currents were measured, noninactivating inward-rectifying Na+ currents were recorded in the presence of 50 μM EDTA. Neither the amplitude of the Na+ current nor current-to-voltage relationship was affected by plasmin (Figure 5, A and B). However, in the presence of 10 mM 1,2-bis(o-aminophenoxy)ethane- N,N,N',N'-tetraacetic acid (BAPTA) intracellularly, a significant decrease in the Ca2+ peak current from 1.02±0.07–0.73±0.06 nA/pF was observed in plasmin-treated group without affecting TRPV5 inactivation kinetics (Figure 5, C–E). This effect was not observed in the S144A mutant. Similar results were seen in the presence of 50 μM ethylene glycol tetraacetic acid (Supplemental Figure 2).
Figure 5.

Plasmin-mediated inhibition of TRPV5 Ca2+ currents in HEK293 cells. (A) Representative current traces (current-to-voltage relationship) obtained from TRPV5 incubated with plasmin. Voltage ramps were applied in presence of EDTA (50 μM) until currents reached steady state (left, wild type; right, S144A mutant; solid line, control; dotted line, plasmin-treated). (B) Averaged Na+ current density at −80 mV in the presence of EDTA for TRPV5 wild type and TRPV5-S144A mutant. No differences in the Na+ currents were reported in those conditions. (C) Representative TRPV5 Ca2+ currents evoked by application of a hyperpolarizing voltage step to −100 mV in presence of 10 mM Ca2+ (left, wild type; right, S144A mutant). Plasmin promotes a significant reduction in the peak of Ca2+currents without affecting the inactivation kinetics. (D) Averaged density of the Ca2+-mediated peak currents shows that mutation of the S144 to A abolishes effect of plasmin on Ca2+ currents. (E) Inactivation kinetics analysis from all the Ca2+ recordings in both wild type TRPV5 and S144A mutant in the presence and absence of plasmin. No change was reported at any of the conditions. (F) Relative permeabilities (Px/PNa) of the different organic cations plotted as a function of the cation diameter for TRPV5 in the absence and presence of plasmin (solid line and dotted line, respectively). MA, monomethylammonium; DMA, dimethylammonium; TriMA, trimethylammonium; TetMA, tetramethylammonium. Data points were fitted to a first order exponential decay equation and used to calculate pore diameter (G). *P<0.05 versus a nonplasmin condition (-).
To further substantiate the inhibitory effect of plasmin on the Ca2+-mediated current, we performed sieving experiments after plasmin treatment to estimate changes in TRPV5 effective pore diameter by measuring relative permeability to organic monovalent cations of increasing size. Divalent-free intra- and extracellular solutions were used to avoid Mg2+ and Ca2+ block of monovalent currents. Starting with Na+ as the sole intra- and extracellular cation, TRPV5 currents reversed close to 0 mV. Subsequently, all Na+ in the extracellular solution was replaced by mono-, di-, tri-, or tetra-methyl substituents. Then permeability ratios of the substituents relative to Na+ (PX/PNa) from the bi-ionic reversal potential were determined. Figure 5F shows the permeability ratios of the different cations versus their estimated diameters obtained for TRPV5 treated with plasmin. Data points were fitted using a modified excluded volume equation:
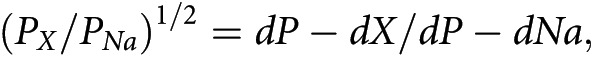 |
where dP, dX, and dNa are diameters of the pore and specific cation X+ and Na+, respectively. Plasmin-treated cells showed a significant decrease in the estimated pore diameter (mean ± SEM, 6.72±0.07 and 6.18±0.04 Å for control and plasmin-treated cells, respectively; P<0.05) (Figure 5G).
In different cell types, PKC activation via PAR-2 affected open probability of TRPV1.19,20 The cell-attached single-channel recordings keep intracellular second messengers and kinases intact, thereby making them suitable for investigating TRPV5 function in the present study. The recordings were made from HEK293 cells expressing TRPV5-wild type (WT) (Figure 6A) or TRPV5-S144A (Figure 6B). The averaged calculated slope conductance was 68±4 picosiemens (pS) for TRPV5-WT and 72±4 pS for TRPV5-WT preincubated with 10 nM plasmin for 1 hour (Figure 6B). Similarly, the averaged calculated slope conductance was 65±3 pS for TRPV5-S144A and 66±2 pS for TRPV5-S144A preincubated with 10 nM plasmin for 1 hour (Figure 6D). In Figure 6, C and F, the averaged open probability upon plasmin incubation was assessed by averaging 10-second intervals for 1 minute using a holding potential of −80 mV (for representative traces, see Supplemental Figure 3). Plasmin significantly decreases the open probability in TRPV5-WT–expressing cells, but does not influence open probability in cells expressing TRPV5-S144A.
Figure 6.

Plasmin inhibits single-channel activity of TRPV5-WT but not TRPV5-S144A. (A and D) Cell-attached single-channel recordings were made from HEK293 cells expressing TRPV5-WT or TRPV5-S144A. Channel activity was elicited by step potentials varying from −100 to 80 mV for TRPV5-WT (A) and TRPV5-S144A (D). Downward currents indicate channel opening state. (B and E) Amplitude histograms were constructed from regions of the single-channel recordings and were fitted by three Gaussian functions corresponding to closed, one open, or two open levels. (C and F) The averaged open probability upon plasmin incubation was assessed by averaging 10-second intervals for 1 minute using a holding potential of −80 mV. *P<0.05 versus nonplasmin condition (n=5–7).
Calmodulin Selectively Binds TRPV5, and This Binding Is Affected by Phosphorylation of S144 in the N-Terminus of TRPV5
The effect of plasmin on TRPV5 has been shown to be highly specific for Ca2+. We decided, therefore, to test whether the ubiquitous Ca2+-sensor CaM was required for plasmin-mediated TRPV5 inhibition. The Calmodulin Target Database (2002 Ikura Lab, Ontario Cancer Institute; http://calcium.uhnres.utoronto.ca) indicated that the specific region surrounding the highly conserved S144 amino acid holds the highest probability for potential CaM binding (RGASVSARA) (Figure 7A). 15N-1H-heteronuclear single quantum coherence (HSQC) spectra of 15N-labeled CaM were measured by high-resolution nuclear magnetic resonance (NMR) spectroscopy under various concentrations of the synthetic peptides corresponding to residues 133–154 of Nter. In the presence of Ca2+, both WT and S144pS mutant peptides specifically bound CaM. As seen from the overlaid 15N-1H-HSQC spectra of CaM in Figure 7, B and C, CaM peaks shift in a similar fashion upon addition of the peptides. This indicates that both peptides interact with CaM in a generally similar way, with some slight differences. Namely, in case of the WT peptide, the intensity of the peaks remains the same in the course of the titration, which implies fast exchange (on the NMR time scale). However, in case of the S144pS mutant, the intensity of the shifted signals significantly decreases (e.g., I27, A57, D64), indicating that the CaM-peptide complex is now in intermediate exchange regime. Thus, phosphorylation of S144 clearly affects CaM-TRPV5 binding and alters its dynamic properties.
Figure 7.

S144 residue located on the third ankyrin repeat domain-3 (ANK3) of the Nter-TRPV5 is conserved among mammalian species. The potential CaM-binding sequence (RGASVSARA) is depicted (A). 15N-1H-HSQC spectra of 15N-calmodulin at various concentrations of the synthetic peptides: (B) WT, (C) S144pS mutant. Calmodulin-to-peptide molar ratios are represented as follows: black, 1:0; red, 1:1; blue, 1:2. Each peak corresponds to a unique proton attached to a 15N nucleus. Spectra were acquired at 308 K (600 MHz Bruker Avance III spectrometer).
Discussion
This study shows that plasmin present in the urine of patients with nephrotic syndrome inhibits TRPV5-mediated Ca2+ transport. This inhibition is mediated by PAR-1 through PKC phosphorylation of the Ca2+ channel at the S144 residue. Our conclusion is based on the following findings: (1) Plasmin, both the commercially available form and that concentrated from the urine of patients with nephrotic syndrome, inhibits TRPV5-mediated Ca2+ transport without affecting TRPV5 surface membrane expression; inhibition of Ca2+ transport could be prevented by the specific plasmin inhibitor α2-antiplasmin and heat inactivation. (2) Plasmin activity requires an intact PAR-1/PLC/PKC signaling pathway, which phosphorylates TRPV5 at S144. (3) Plasmin decreases TRPV5-mediated Ca2+ currents mainly by reducing the open probability of TRPV5 without affecting monovalent currents. (4) Residues 133–154 of TRPV5 selectively bind CaM, and this binding is affected by phosphorylation of S144.
Plasmin is well known for its action in fibrinolytic processes. Our data suggest that plasmin may be involved in modulating Ca2+ reabsorption in the kidney under pathophysiologic conditions, namely glomerular protein leakage. Plasmin has recently been identified as a crucial factor for Ca2+ homeostasis and normal bone maintenance.21 Plasminogen-deficient mice manifest decreased trabecular and cortical bone density because of enhanced osteoclast activity and retarded osteoblast activity. In the kidney, plasmin is known to play a major role in the degradation and turnover of the extracellular matrix and especially the glomerular mesangial matrix.22 In addition, plasmin has long been hypothesized to be involved in renal stone formation because plasmin and urokinase-type plasminogen activator activities are decreased in stone-forming patients.23 In that study, van Aswegen and colleagues reported that urate inhibited urokinase/plasmin activities at a pH of 3.9 (in the presence of acetate) but is minimal under alkaline condition (in the absence of acetate). Therefore, it can be inferred that plasmin is still stable in our patients’ urine, with its pH of 7.5–7.7. Our laboratory previously showed that extracellular alkalinization resulted in a pool of TRPV5-containing vesicles recruited to the cell surface.24 Our findings suggest that, once present at the cell surface, TRPV5 activity is inhibited by plasmin in nephritic urine via its receptor PAR-1. In vivo studies on Ca2+ balance, however, would be required to fully substantiate this assumption. Of note, in line with our data, pharmacologic reduction of proteinuria leads to a corresponding reduction in calciuria in clinical study.25
PAR-1 has been reported to couple to various heterotrimeric G proteins and conveys its signal through several kinases, including Src family tyrosine kinases, JNK, Rho kinases, mitogen-activated protein K, PKA, and PKC.17,19 Protein kinases have been found to differentially regulate ion channel gating and trafficking. Upon stimulation, PAR-1 enhances PLC-β–dependent hydrolysis of PIP2, which results in DAG and inositol trisphosphate production. DAG further stimulates PKC-dependent phosphorylation of TRPV5. In this study we show that S299 and S654 double mutant is still responsive to plasmin effect (Supplemental Figure 4A). On the other hand, the S144 mutant is also stimulated by tissue kallikrein. Therefore, plasmin and tissue kallikrein induce PKC phosphorylation of TRPV5 at independent sites. 12-O-tetradecanoylphorbol-13-acetate has been previously shown to downregulate α, β, and ε.5 Accordingly, the present data showed that 24-hour 12-O-tetradecanoylphorbol-13-acetate treatment prevented the effects of both tissue kallikrein and plasmin (Supplemental Figure 4B). The specific subtype of PKC involved in this process has not yet been characterized. However, PKC-α and PKC-ε have been reported to mediate PAR-1 signaling,26 and we would like to suggest that the Ca2+-independent PKC-ε isoform is more likely to be involved in this pathway because plasmin-mediated PAR-1 activation is sufficient to inhibit TRPV5 in the presence of intracellular Ca2+ chelator BAPTA. The atypical PKC isoforms were probably not involved because the effect of plasmin is DAG dependent (Supplemental Figure 4B).
TRPV5 is the most Ca2+-selective member of the TRP superfamily involved in Ca2+ reabsorption in renal epithelia, as shown by knockout mice studies.27 TRPV5 activity depends on calciotropic hormones to ensure proper Ca2+ reabsorption,28 but other molecules are also involved in the TRPV5 regulation. Activation of PKC by tissue kallikrein via the bradykinin receptor phosphorylates TRPV5 at S299 and S654, resulting in an inhibition of channel internalization.5 This process retains the channel at the plasma membrane, thus increasing Ca2+ reabsorption. A similar effect of plasmin on TRPV5 surface expression was, however, not observed.
Electrophysiologic analysis of HEK293 cells expressing TRPV5 showed that plasmin specifically decreased Ca2+ permeability but had no effect on Na+ permeability. This can be explained by examining the selectivity filter of Ca2+ channels, which have the terminal carboxyl side chains of their aspartates and glutamates in the permeation pathway, directly interacting with passing Ca2+ ions.29,30 This structural organization is thought to be flexible, and, therefore, its configuration can change as a consequence of rearrangements of the tertiary and quaternary structure of the channel. Here, we have shown that phosphorylation of the S144 amino acid in the Nter of TRPV5 is required to observe the inhibitory effect of plasmin. We can, therefore, infer an alteration of pore structure, probably through changes in ion-ion interactions in the pore. This notion is supported by the observation that the effective pore diameter of TRPV5 (6.18 Å) after plasmin treatment is smaller than the hydrated size of Ca2+ ion (6.2 Å).31 At present it is unclear whether this reduction contributed to the diminished TRPV5 activity. Further analysis suggests that our reduction in Ca2+ transport is probably a consequence of a decrease in open probability induced by plasmin. This observation further suggests a role of plasmin in regulating Ca2+ reabsorption in the context of nephrotic syndrome because the glomerular protein leakage allows this enzyme to enter into the pro-urine to reach the DCT.
Because of the specific effect of plasmin on TRPV5 Ca2+ current and open probability, we hypothesized that another player could be involved in the plasmin-activated pathway. CaM has been recently shown to enhance Ca2+-dependent TRPV5 inactivation by binding to the distal region of the Cter domain.32 The phosphorylation of T709 by PTH (via the PKA pathway) impedes CaM binding to the Cter and subsequently increases the open probability of TRPV5.32 CaM can be activated by Ca2+ influx via TRPV532 or the release of Ca2+ from intracellular stores.33 During the resting state, intracellular free Ca2+ ([Ca2+]i) is relatively low and CaM remains inactive. Once [Ca2+]i rises, Ca2+ binds to CaM via Ca2+-binding EF-hand structures,2 which in turn stretches CaM as each pair of EF hands exposes a hydrophobic patch available to bind target amino acid sequences.34 One of the consequences is the inhibition of Ca2+ channel activities.35 In addition to binding of CaM to the Cter domain, several studies have shown that CaM is able to bind the Nter domain of TRPV5 in a Ca2+-dependent manner,32,36,37 although no functional experiments have been performed to assess the effects of Nter binding on channel activity. In fact, CaM has recently been reported to bind in vitro to the Nter (residues 133–154 and 310–330).38 The present study revealed that plasmin-induced S144 phosphorylation modifies CaM binding affinity of the surrounding region of S144, in addition to a decrease of TRPV5 open probability and changing the channel pore diameter (Figure 8).
Figure 8.

Molecular model of plasmin action on TRPV5 in nephrotic syndrome. The basal level of TRPV5-dependent Ca2+ influx is constitutively established by the endogenous CaM binding (A). Plasmin (PL) from nephrotic urine catalyzes PAR-1, promoting S144 phosphorylation by PKC. This results in decreased open probability (NPo) and pore size of TRPV5 and modification of CaM binding to the CaM-binding sequence (B).
In conclusion, this study demonstrates that plasmin inhibits TRPV5 activity through the activation of the PAR-1 receptor via a PLC/PKC-dependent pathway. PKC activation results in phosphorylation of the S144 residue and consequently inhibits TRPV5 activity by decreasing its open probability and pore diameter. Urinary plasmin, by inhibiting TRPV5 activity, could potentially be involved in disturbances in renal Ca2+ handling found in nephrotic patients and may play a role in the tubulo-toxic effects of proteinuric urine.
Concise Methods
Collection of Urine Samples
Twenty-four-hour urine samples were collected from five nephrotic patients (all male; mean age, 53±2.6 years; mean creatinine clearance, 24±9 ml/min) before their visits to the outpatient nephrology clinic. Their underlying diseases proven by biopsies were myeloperoxidase-ANCA–associated GN, IgA nephropathy, membranous nephropathy, FSGS, and diabetic nephropathy. All patients were overtly proteinuric (i.e., the median urinary protein excretion was 6.5 [interquartile range, 5.3–8.2] g/d). Collections were performed at University Medical Centre Groningen, and all patients provided informed consent.
Plasmin Purification and Activity Assay
Urine samples were dialyzed in Spectra/Por dialysis membrane (Spectrum Laboratories) for 12 hours with dialysis buffer (10 mM Tris HCl [pH, 8.1] and 5 mM EDTA). Urine precipitates were purified with high-affinity ion exchange chromatography with mono-Q column based on the theoretical isoelectric point of plasmin (7.08; http://expasy.org/tools/pi_tool.html). Urine samples were injected in the column with buffer A (10 mM Tris [pH, 8.1] and 5 mM EDTA) and eluted with buffer B (10 mM Tris [pH, 8.1], 5 mM EDTA, and 1 M NaCl). Chromatography fractions were concentrated with 50K Amicon Ultra Centrifugal Filter Units (Millipore), and the presence of plasmin was determined by immunoblotting. Plasmin activity was measured with SensoLyte AFC plasmin activity assay kit (AnaSpec) according to a provided protocol.
PCR Analysis
To evaluate mRNA expression of F2R, PRSS8, and LRP2 (encoding PAR-1, prostasin, and megalin, respectively), total RNA was extracted from HEK293 cells using TriZol Total RNA Isolation Reagent (Life Technologies BRL, the Netherlands) according to the manufacturer’s protocol. The obtained RNA was subjected to deoxyribonuclease treatment (Promega) to prevent genomic DNA contamination. Thereafter, 2 µg of RNA was reverse transcribed by Moloney-murine leukemia virus-reverse transcription (Invitrogen, the Netherlands). The cDNA was used to determine mRNA expression levels by PCR of the target genes of interest and of the housekeeping gene GAPDH as an endogenous control. Primers targeting the genes of interest were designed using the computer program Primer3 (version 0.4.0) and are listed in Table 1.
Table 1.
Homo sapiens oligonucleotide sequences used in RT-PCR
| Gene | Forward Primer | Reverse Primer | Product Length (bp) |
|---|---|---|---|
| F2R | CTCTGCCTGCCGCGAAGACC | CCTGCGATGGCCAAAGCCA | 813 |
| PRSS8 | GGCCAGACGGTGCTGGTGAC | GGGTGCTGACCTTGGCGTCC | 582 |
| LRP2 | GGCGGGTCGCTCGTGTCAAA | GTGACCGGGGCTGGACTTGC | 981 |
| GAPDH | GTGAAGGTCGGAGTCAACGG | TCTTCTGGGTGGCAGTGATG | 650 |
F2R, coagulation factor II receptor gene; PRSS8, serine protease 8 gene; LRP2, low-density lipoprotein receptor-related protein 2 gene; GAPDH, glyceraldehyde-3-phosphate dehydrogenase gene.
DNA Constructs and Cell Culture
TRPV5 pCINeo/internal ribosome entry site-green fluorescent protein (IRES-GFP) constructs were generated, as described previously.39 Single and combined PKC mutants were generated by alanine substitution of the six putative phosphorylation sites of TRPV5 (S144A, S299A, S316A, S654A, S664A, S698A) using in vitro mutagenesis (QuikChange Site-Directed Mutagenesis kit, Stratagene).5 HEK293 cells were transfected at 70% of confluency using polyethylenimine (Polysciences, Inc.) or Lipofectamine 2000 (Invitrogen). After 48 hours, cells were used for 45Ca2+ uptake assays, patch-clamp, or biotinylation experiments. Before the assays, cells were incubated for 1 hour in serum-free cell culture medium containing the particular compounds.
45Ca2+ Uptake Assay
HEK293 cells were transfected with TRPV5-HA pCINeo/IRES-GFP constructs. Ca2+ uptake was determined in uptake medium (110 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 0.1 mM CaCl2, 10 mM Na-acetate, 2 mM NaH2PO4, and 20 mM HEPES–Tris [pH, 7.4], supplemented with 10 µM felodipine, 10 µM methoxy-verapamil, and 1 µCi/ml 45CaCl2) for 10 minutes at 37°C. Each well was washed extensively with stop buffer (110 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 0.5 mM CaCl2, 1.5 mM LaCl3, 10 mM Na-acetate, 20 mM HEPES–Tris [pH, 7.4]) at 4°C, incubated with 0.05% wt/vol SDS, and the lysates were counted for radioactivity using liquid scintillation.
Cell Surface Biotinylation and Immunoblotting
HEK293 cells (9☓104 cells/cm2) were plated and transfected with 15 µg TRPV5 pCINeo/IRES-GFP or pCINeo/IRES-GFP in poly-l-lysine (Sigma) coated 10-cm dishes. At 48 hours after transfection, cells were incubated for 1 hour with 10 nM plasmin and 50 nM α2-antiplasmin inhibitor. Cells were homogenized in 1 ml lysis buffer, as described previously4 using the NHS-LC-LC-biotin (Pierce, the Netherlands). Finally, biotinylated proteins were precipitated using NeutrAvidin beads (Pierce). TRPV5 expression was analyzed by immunoblotting for the precipitates (plasma membrane fraction) and for the total cell lysates using the guinea-pig antirabbit TRPV5 antibody.1
Ca2+ Imaging Using Fura-2/Acetoxymethyl
HEK293 cells were seeded on fibronectin-coated coverslips. After 24 hours, cells were loaded with 3 μM fura-2-acetoxymethyl and 0.01% (vol/vol) Pluronic F-129 in DMEM medium at 37°C for 20 minutes. After loading, cells were washed twice with PBS and allowed to equilibrate at 37°C for another 10 minutes in 132 mM NaCl, 4.2 mM KCl, 1.0 mM MgCl2, 5.5 mM d-glucose, 10 mM EDTA, and 10 mM HEPES, and titrated to a pH of 7.4 with Tris. Plasmin with final a concentration of 10 nM was added directly to buffer solution. Changes in intracellular Ca2+ concentration ([Ca2+]i) were monitored with fura-2 excited at 340 and 380 nm using a monochromator (Polychrome IV, Germany). All measurements were performed at room temperature. Details of this experiment were described previously.32
Immunohistochemistry
Mouse kidney sections were incubated for 16 hours at 4°C with rabbit polyclonal antibody against PAR-1 (1:100). To visualize PAR-1, goat antirabbit Alexa Fluor 488-conjugated antibody (1:300) (Molecular Probes) was used. TRPV5 staining has been performed as previously described.40 All negative controls, including sections incubated with preimmune serum or conjugated antibodies solely, were devoid of any staining.
Electrophysiology and Solutions
Patch-clamp experiments were performed under the whole-cell configuration using an EPC-9 patch-clamp amplifier controlled by the Pulse software (HEKA Elektronik, Germany). Borosilicate patch pipettes had a resistance between 3 and 4 MΩ after being filled with the intracellular solutions. Series resistance (3–10 MΩ) was continuously monitored with the automatic capacitance compensation of Pulse software. The extracellular solution consisted of (in mM) 150 NaCl, 6 CsCl, 10 glucose, 10 HEPES, 44 mannitol, and 0.05 EDTA (divalent-free solution with EDTA [pH, 7.4] and NaOH). The intracellular solution included (in mM) 20 CsCl, 100 aspartate, 1 MgCl2, 10 BAPTA, 1 Na2ATP, and 10 HEPES (pH, 7.2 CsOH). For the recording of Ca2+ current, 10 mM CaCl2 was added to the extracellular solution. Cells were exposed for a maximum of 1 minute to a Krebs solution containing 1 mM Ca2+ before sealing the patch pipette to the cell. The analysis and display of patch-clamp data were performed using Igor Pro software (WaveMetrics).
NMR Spectroscopy
Titration of 15N-labeled CaM by nonlabeled peptides was done as previously described.32,38 Briefly, 15N-labeled CaM was titrated by synthetic peptides with CaM-to-peptide molar ratios ranging from 1:0–1:3 in steps of 0.5. Before titrations, both CaM and peptides were dialyzed against the same buffer (1/100 of 50 mM KCl, 10 mM CaCl2, 20 mM Tris [pH, 7.0], HCl), freeze-dried and reconstituted in the same buffer. D2O and NaN3 (both from Sigma) were added to the NMR samples to the final concentrations of 5%–7% (vol/vol) and 0.01% (vol/vol), respectively. The concentration of CaM in the samples was 0.4 mM. Spectra were acquired at 308 K, 600 MHz Bruker Avance III spectrometer.
Compounds
Plasmin, plasminogen, and α2-antiplasmin inhibitor were purchased from Sigma. PAR-1 antagonist (SCH79797) was from Axon Medchem (the Netherlands). A polyclonal PAR-1 antibody (rabbit IgG) was from Santa Cruz Biotechnology. A polyclonal plasminogen antibody (goat IgG) was obtained from Abcam (United Kingdom). Chelerythrine was from Research Biochemicals International (Germany). 15NH4Cl was from Buchem BV (the Netherlands). Synthetic peptides corresponding to the residues 133–154 of hTRPV5 (WT and with phosphorylated S144, S144pS) were purchased from GenicBio Limited (China).
Statistical Analyses
If not specified otherwise, the data are expressed as mean ± SEM. Multiple sets of data were compared by ANOVA. The significant differences between the means of two groups were analyzed by unpaired t test, and multiple comparisons between groups were performed by Tukey post hoc analysis. The level of statistical significance is P<0.05. All data were analyzed by GraphPad Prism (version 4.0c for Mac OS X; GraphPad Software).
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Dr. G.W. Vuister for valuable discussion.
This work was supported by the Netherlands Organization for Scientific Research (grant number ZonMw 9120.6110, ZonMw 9120.8026). J.G.J.H. is supported by a EURYI award from the European Science Foundation, and the Dutch Kidney Foundation (grant number C05.2134).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2011111126/-/DCSupplemental.
References
- 1.Hoenderop JG, van der Kemp AW, Hartog A, van de Graaf SF, van Os CH, Willems PH, Bindels RJ: Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J Biol Chem 274: 8375–8378, 1999 [DOI] [PubMed] [Google Scholar]
- 2.Lambers TT, Weidema AF, Nilius B, Hoenderop JG, Bindels RJ: Regulation of the mouse epithelial Ca2(+) channel TRPV6 by the Ca(2+)-sensor calmodulin. J Biol Chem 279: 28855–28861, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Nilius B, Vennekens R, Prenen J, Hoenderop JG, Droogmans G, Bindels RJ: The single pore residue Asp542 determines Ca2+ permeation and Mg2+ block of the epithelial Ca2+ channel. J Biol Chem 276: 1020–1025, 2001 [DOI] [PubMed] [Google Scholar]
- 4.de Groot T, Lee K, Langeslag M, Xi Q, Jalink K, Bindels RJ, Hoenderop JG: Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol 20: 1693–1704, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gkika D, Topala CN, Chang Q, Picard N, Thébault S, Houillier P, Hoenderop JG, Bindels RJ: Tissue kallikrein stimulates Ca(2+) reabsorption via PKC-dependent plasma membrane accumulation of TRPV5. EMBO J 25: 4707–4716, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuusniemi AM, Lapatto R, Holmberg C, Karikoski R, Rapola J, Jalanko H: Kidneys with heavy proteinuria show fibrosis, inflammation, and oxidative stress, but no tubular phenotypic change. Kidney Int 68: 121–132, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skøtt O: Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 20: 299–310, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burke EC, Holley KE, Stickler GB: Familial nephrotic syndrome with nephrocalcinosis and tubular dysfunction. J Pediatr 82: 202–206, 1973 [DOI] [PubMed] [Google Scholar]
- 9.Mocan H, Yildiran A, Camlibel T, Kuzey GM: Microscopic nephrocalcinosis and hypercalciuria in nephrotic syndrome. Hum Pathol 31: 1363–1367, 2000 [PubMed] [Google Scholar]
- 10.Alexander RT, Woudenberg-Vrenken TE, Buurman J, Dijkman H, van der Eerden BC, van Leeuwen JP, Bindels RJ, Hoenderop JG: Klotho prevents renal calcium loss. J Am Soc Nephrol 20: 2371–2379, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaziri ND, Gonzales EC, Shayestehfar B, Barton CH: Plasma levels and urinary excretion of fibrinolytic and protease inhibitory proteins in nephrotic syndrome. J Lab Clin Med 124: 118–124, 1994 [PubMed] [Google Scholar]
- 12.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR: Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem 283: 36586–36591, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanalas JJ, Makker SP: Identification of the rat Heymann nephritis autoantigen (GP330) as a receptor site for plasminogen. J Biol Chem 266: 10825–10829, 1991 [PubMed] [Google Scholar]
- 14.Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE: Plasmin desensitization of the PAR1 thrombin receptor: Kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 38: 4572–4585, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Majumdar M, Tarui T, Shi B, Akakura N, Ruf W, Takada Y: Plasmin-induced migration requires signaling through protease-activated receptor 1 and integrin alpha(9)beta(1). J Biol Chem 279: 37528–37534, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Svenningsen P, Uhrenholt TR, Palarasah Y, Skjødt K, Jensen BL, Skøtt O: Prostasin-dependent activation of epithelial Na+ channels by low plasmin concentrations. Am J Physiol Regul Integr Comp Physiol 297: R1733–R1741, 2009 [DOI] [PubMed] [Google Scholar]
- 17.Zieger M, Tausch S, Henklein P, Nowak G, Kaufmann R: A novel PAR-1-type thrombin receptor signaling pathway: Cyclic AMP-independent activation of PKA in SNB-19 glioblastoma cells. Biochem Biophys Res Commun 282: 952–957, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Harper MT, Sage SO: PAR-1-dependent pp60src activation is dependent on protein kinase C and increased [Ca2+]: Evidence that pp60src does not regulate PAR-1-dependent Ca2+ entry in human platelets. J Thromb Haemost 4: 2695–2703, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R: Proteinase-activated receptors. Pharmacol Rev 53: 245–282, 2001 [PubMed] [Google Scholar]
- 20.Gu Q, Lee LY: Effect of protease-activated receptor 2 activation on single TRPV1 channel activities in rat vagal pulmonary sensory neurons. Exp Physiol 94: 928–936, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanno Y, Ishisaki A, Kawashita E, Chosa N, Nakajima K, Nishihara T, Toyoshima K, Okada K, Ueshima S, Matsushita K, Matsuo O, Matsuno H: Plasminogen/plasmin modulates bone metabolism by regulating the osteoblast and osteoclast function. J Biol Chem 286: 8952–8960, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto T, Noble NA, Miller DE, Gold LI, Hishida A, Nagase M, Cohen AH, Border WA: Increased levels of transforming growth factor-beta in HIV-associated nephropathy. Kidney Int 55: 579–592, 1999 [DOI] [PubMed] [Google Scholar]
- 23.van Aswegen CH, Neitz AW, Becker PJ, du Plessis DJ: Renal calculi-urate as a urokinase inhibitor. Urol Res 16: 143–148, 1988 [DOI] [PubMed] [Google Scholar]
- 24.Lambers TT, Oancea E, de Groot T, Topala CN, Hoenderop JG, Bindels RJ: Extracellular pH dynamically controls cell surface delivery of functional TRPV5 channels. Mol Cell Biol 27: 1486–1494, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slagman MC, Waanders F, Hemmelder MH, Woittiez AJ, Janssen WM, Lambers Heerspink HJ, Navis G, Laverman GD, HOlland NEphrology STudy Group : Moderate dietary sodium restriction added to angiotensin converting enzyme inhibition compared with dual blockade in lowering proteinuria and blood pressure: randomised controlled trial. BMJ 343: d4366, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otani H, Yoshioka K, Nishikawa H, Inagaki C, Nakamura T: Involvement of protein kinase C and RhoA in protease-activated receptor 1-mediated F-actin reorganization and cell growth in rat cardiomyocytes. J Pharmacol Sci 115: 135–143, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Hoenderop JG, van Leeuwen JP, van der Eerden BC, Kersten FF, van der Kemp AW, Mérillat AM, Waarsing JH, Rossier BC, Vallon V, Hummler E, Bindels RJ: Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112: 1906–1914, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Groot T, Bindels RJ, Hoenderop JG: TRPV5: An ingeniously controlled calcium channel. Kidney Int 74: 1241–1246, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Koch SE, Bodi I, Schwartz A, Varadi G: Architecture of Ca(2+) channel pore-lining segments revealed by covalent modification of substituted cysteines. J Biol Chem 275: 34493–34500, 2000 [DOI] [PubMed] [Google Scholar]
- 30.Wu XS, Edwards HD, Sather WA: Side chain orientation in the selectivity filter of a voltage-gated Ca2+ channel. J Biol Chem 275: 31778–31785, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Tai CY, Smith QR, Rapoport SI: Calcium influxes into brain and cerebrospinal fluid are linearly related to plasma ionized calcium concentration. Brain Res 385: 227–236, 1986 [DOI] [PubMed] [Google Scholar]
- 32.de Groot T, Kovalevskaya NV, Verkaart S, Schilderink N, Felici M, van der Hagen EA, Bindels RJ, Vuister GW, Hoenderop JG: Molecular mechanisms of calmodulin action on TRPV5 and modulation by parathyroid hormone. Mol Cell Biol 31: 2845–2853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersen OH, Gerasimenko OV, Gerasimenko JV: Pathobiology of acute pancreatitis: Focus on intracellular calcium and calmodulin. F1000 Med Rep 3: 15, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babu YS, Sack JS, Greenhough TJ, Bugg CE, Means AR, Cook WJ: Three-dimensional structure of calmodulin. Nature 315: 37–40, 1985 [DOI] [PubMed] [Google Scholar]
- 35.Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H: Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399: 159–162, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Phelps CB, Wang RR, Choo SS, Gaudet R: Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J Biol Chem 285: 731–740, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mercado J, Gordon-Shaag A, Zagotta WN, Gordon SE: Ca2+-dependent desensitization of TRPV2 channels is mediated by hydrolysis of phosphatidylinositol 4,5-bisphosphate. J Neurosci 30: 13338–13347, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovalevskaya NV, Bokhovchuk FM, Vuister GW: The TRPV5/6 calcium channels contain multiple calmodulin binding sites with differential binding properties. J Struct Funct Genomics 13: 91–100, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Graaf SF, Hoenderop JG, Gkika D, Lamers D, Prenen J, Rescher U, Gerke V, Staub O, Nilius B, Bindels RJ: Functional expression of the epithelial Ca(2+) channels (TRPV5 and TRPV6) requires association of the S100A10-annexin 2 complex. EMBO J 22: 1478–1487, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Topala CN, Schoeber JP, Searchfield LE, Riccardi D, Hoenderop JG, Bindels RJ: Activation of the Ca2+-sensing receptor stimulates the activity of the epithelial Ca2+ channel TRPV5. Cell Calcium 45: 331–339, 2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.