

Abstract
Traumatic brain injury (TBI) is one of the leading causes of death and disability in children and adolescents. The neuropathological sequelae that result from TBI are a complex cascade of events including edema formation, which occurs more frequently in the pediatric than the adult population. This developmental difference in the response to injury may be related to higher water content in the young brain and also to molecular mechanisms regulating water homeostasis. Aquaporins (AQPs) provide a unique opportunity to examine the mechanisms underlying water mobility, which remain poorly understood in the juvenile post-traumatic edema process. We examined the spatiotemporal expression pattern of principal brain AQPs (AQP1, 4, and 9) after juvenile TBI (jTBI) related to edema formation and resolution observed using magnetic resonance imaging (MRI).
Using a controlled cortical impact in post-natal 17 day-old rats as a model of jTBI, neuroimaging analysis showed a global decrease in water mobility (apparent diffusion coefficient, ADC) and an increase in edema (T2-values) at 1 day post-injury, which normalized by 3 days. Immunohistochemical analysis of AQP4 in perivascular astrocyte endfeet was increased in the lesion at 3 and 7 days post-injury as edema resolved. In contrast, AQP1 levels distant from the injury site were increased at 7, 30, and 60 days within septal neurons but did not correlate with changes in edema formation. Group differences were not observed for AQP9. Overall, our observations confirm that astrocytic AQP4 plays a more central role than AQP1 or AQP9 during the edema process in the young brain.
Keywords: Edema, Astrocyte, Aquaporin, juvenile traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) has been termed a ‘silent epidemic’ in the United States in recent years because of its increased medical and financial burden. TBI affects about 1.7 million people annually and contributes to 30.5% of all injury-related deaths in the U.S. An important subgroup of this population are children and adolescents ranging in age from 0-14 years, of which half a million visit emergency departments for TBI (Faul M, 2010). Juvenile TBI (jTBI) is the primary cause of death and disability in children and adolescents (Schneier et al., 2006) with long-term impairments in motor and cognitive abilities, including deficits in intellectual functioning, attention, memory, language, sensorimotor, visual-spatial, and executive skills (Adelson and Kochanek, 1998, Adelson et al., 1998). Despite increases in prevalence and resulting catastrophic effects of jTBI, there are no effective pharmacological treatments.
TBI in infants and children is more frequently associated with severe brain swelling than in adults (Lang et al., 1994, Bauer and Fritz, 2004) that may affect the edema process. Two mechanisms may account for these age-related differences: (i) increased post-injury cerebral blood flow in the young, and (ii) developmental and mechanical properties of the brain and skull (Kochanek, 2006). Experimental studies suggest that post-traumatic edema in the immature brain may also be related to enhanced diffusion of excitotoxic neurotransmitters, an intensified inflammatory response (Kochanek, 2006), and higher brain water content in the young rat compared to the adult rat (Dobbing and Sands, 1981).
Developmental differences in water homeostasis between children and adults may also account for the observed greater risk of post-traumatic edema in the younger population. Aquaporins (AQPs), a family of water channel proteins, are recognized to have an important role in brain water regulation (Badaut et al., 2011b). In rodents and humans, AQP4 and AQP1 show increased expression during brain development (Wen et al., 1999, Gomori et al., 2006, Hsu et al., 2011). To date, three different AQPs (AQP1, AQP4, and AQP9) have been identified in vivo in the brain and each are hypothesized to play different roles during normal physiological and neuropathological states (Badaut et al., 2007). For example, AQP1 in epithelial cells of the choroid plexus appears to contribute primarily to cerebrospinal fluid formation whereas the neuronal AQP1 may play a role in pain processing (Oshio et al., 2006). AQP9 in astrocytes and catecholaminergic neurons may contribute to the regulation of brain energy metabolism (Badaut, 2010). In contrast, AQP4 appears to have multiple roles including: (i) water homeostasis and edema formation (Papadopoulos et al., 2004, Badaut et al., 2011a, Badaut et al., 2011b, Lee et al., 2011); (ii) regulation of synaptic plasticity with regulation of p75NTR (Skucas et al., 2011); and (iii) modulation of neurogenesis (Zheng et al., 2010).
The intricate role of brain AQPs after injury may depend on underlying pathology and type of edema (Badaut et al., 2011b), and AQP findings in several TBI studies using adult rodents have provided conflicting results. In some reports, AQP4 expression is increased (Sun et al., 2003, Guo et al., 2006, Ding et al., 2009, Higashida et al., 2011, Tomura et al., 2011) whereas other investigators have reported decreased expression (Ke et al., 2001, Kiening et al., 2002, Zhao et al., 2005). Interestingly, more recent studies have shown both AQP1 (Tran et al., 2010, Oliva et al., 2011) and AQP9 (Ding et al., 2009, Oliva et al., 2011) increasing after injury. Notably, these previous studies were undertaken in adult rats and the majority only examined AQP4 expression for up to 48 hours, without correlations of AQP levels with edema formation and resolution via neuroimaging. In other injury models, AQP expression is observed acutely and in the long-term, such as in spinal cord injury where AQP1 (Nesic et al., 2008) and AQP4 (Nesic et al., 2010) are increased 5 and 11 months after injury.
Developmental differences in water homeostasis and AQP expression between children and adults raises the question whether AQPs play a critical role in regulating brain water content in the developing brain after TBI. To address this question, we examined edema formation and resolution using magnetic resonance imaging (MRI) in conjunction with protein levels of AQP1, 4, and 9 by immunohistochemistry and western blot in a model of jTBI.
Material and Methods
Animals
All protocols and procedures were in compliance with the U.S. Department of Health and Human Services Guide and were approved by the Institutional Animal Care and Use Committee of Loma Linda University. Briefly, juvenile male Sprague-Dawley rats (P10, Harlan, Indianapolis, IN) were housed with their dams on a 12-hour light-dark cycle schedule, at constant temperature and humidity, for seven days prior to surgery at P17. Upon weaning at seven days after surgery, the rats were housed two per cage. Animals were fed with standard lab chow and water ad libitum.
Juvenile traumatic brain injury model
Controlled cortical impact (CCI) was induced in 17 day old rat pups as previously described (Ajao et al., in press). Briefly, P17 juvenile rats were anesthetized with isoflurane (Webster Veterinary Supply, Inc., Sterling, MA) and placed into a stereotaxic frame (David Kopf Instruments, USA). Following a midline skin incision over the skull, a 5mm craniotomy was performed over the right frontal-parietal cortex (Bregma: -1mm anterior-posterior and 2mm medial-lateral). CCI was induced in the jTBI group using a 3mm rounded-tip metal impactor fixed to an electromechanical actuator and centered over the exposed dura at a 20° angle to the cortical surface (Leica Microsystems Company, Richmond, IL). The CCI was delivered at a 1.5 mm depth with impact duration of 200 ms at a velocity of 6 m/s. The surgical site was sutured after recording any bleeding or herniation of cortical tissues (Ajao et al., in press). After the craniotomy, the dura was intact in all of the animals in both groups (sham and jTBI). After induction of jTBI none of the animals had bleeding and all of the animals had a similar minimal damage on the dura. Body temperature was maintained at 37°C during surgery. Following surgery, animals received one subcutaneous injection of buprenorphine (0.01mg/kg; dilution: 0.01mg/ml) for pain relief before the animals were returned to their cages.
Magnetic Resonance Imaging and Analysis
Magnetic Resonance Imaging (MRI) was performed at 1, 3, 7, and 30 days post-injury (d). Rats were lightly anesthetized using isoflurane (1.0%) and then imaged on a Bruker Avance 11.7 T MRI (Bruker Biospin, Billerica MA, 8.9 cm bore) for the 1, 3, and 7d timepoints, or on a larger bore (40 cm) 4.7T MRI (Bruker Biospin, Billerca, MA) for the 30d scan, based on the size of the animals (Badaut et al., 2011a; Ajao et al., in press). Two imaging data sets were acquired: 1) a 10 echo T2 weighted imaging (T2WI), and 2) a diffusion weighted imaging (DWI) sequence where each sequence collected twenty coronal slices (1 mm thickness and interleaved by 1 mm). The T2 sequence had the following parameters: TR/TE = 4600 ms/10.2 ms, matrix = 128 × 128, field of view (FOV) = 3 cm, NEX = 2 and acquisition time = 20 min. The spin echo diffusion sequence parameters were TR/TE = 3000 ms/25 ms, b-values = 0.72, 1855.64 s/mm2, matrix = 128 × 128, FOV = 3 cm, NEX = 2 and acquisition time = 25 min.
Spin-spin relaxation time (T2) and apparent diffusion coefficient (ADC) values were quantified using standardized protocols (Badaut et al., 2007). T2 relaxation rates were determined for each pixel and T2 maps generated. ADC maps were calculated using a linear two point fit. Four primary regions of interest (ROIs) within ipsi- and contralateral hemispheres (cortex and striatum) were delineated on T2WI. These ROIs were overlaid onto corresponding T2 and ADC maps and the mean, standard deviation, number of pixels, and area for each ROI were extracted. MRI analysis was performed blinded without knowledge of experimental group. Data were represented as percentage of sham values to facilitate comparison to AQP changes.
Tissue Processing
Rats were transcardially perfused with 4% paraformaldehyde (PFA) prepared in phosphate buffered saline (PBS) at 1, 3, 7, 30, and 60 d. Brains were immersed in 30% sucrose at 4°C for 48 hours and then frozen on dry ice and stored at -20°C (Badaut et al., 2004). Free-floating coronal sections were cut on a cryostat (Leica CM1850, Leica Microsystems GmbH, Wetzlar, Germany) at 15μm thickness for 1, 3, 7, and 30 d tissue, and at 50μm thickness for 60d tissue.
Immunohistochemistry
All antibody incubations were carried out in PBS (Fisher Scientific, Pittsburgh, PA) containing 0.25% Triton X-100 and 0.25% bovine serum albumin (BSA) (both from Sigma-Aldrich Co., St. Louis, MO). Briefly, after washes in PBS, sections were pre-incubated for 90 minutes in PBS with 1% BSA, and then incubated overnight at 4°C with the various primary antibodies and their respective dilutions as described in Table 1. After rinsing, sections were incubated for 90 minutes at room temperature with the secondary antibodies (Table 1). After subsequent washes in PBS for 3×10min, sections on glass slides were cover-slipped with anti-fading medium VectaShield containing DAPI (Vector, Vector laboratories, Burlingame, CA). Negative control staining where the primary antibody was omitted showed no detectable labelling, and depletion of the AQP4 antibody by an excess of the specific peptide (Chemicon International, Temecula, CA) was also carried out and gave negative results as previously observed (Ribeiro Mde et al., 2006).
Table 1.
Antibodies and dilutions used in this study
| Antibody Name | Company | Dilution |
|---|---|---|
| Anti-AQP1 (rabbit) | Millipore | 1:300 |
| Anti-AQP4 (rabbit) | Millipore | 1:300 |
| Anti-AQP4 (rabbit) | Alpha Diagnostics | 1:300 |
| Anti-AQP9 (rabbit) | Alpha Diagnostics | 1:100 |
| Anti-GFAP (chicken) | Millipore | 1:1000 |
| Alexa 594 goat anti-rabbit | Invitrogen | 1:1000 |
| Alexa 488 goat anti-rabbit | Invitrogen | 1:1000 |
| Alexa 568 goat anti-chicken | Invitrogen | 1:1000 |
| Alexa 488 goat anti-chicken | Invitrogen | 1:1000 |
| Alexa 488 goat anti-mouse | Invitrogen | 1:1000 |
Immunohistochemistry analysis
Intensity of the AQP4 immunoreactivity was quantified using the LI-COR-Odyssey analysis software as previously described (Badaut et al., 2011a). Briefly, the level of fluorescence was quantified in ROIs similar to that of MRI analysis: ipsilateral hemisphere (lesion and perilesion) and contralateral hemisphere (cortex and striatum). Values obtained for the jTBI were also normalized to the sham values of the corresponding ROIs.
Non-infrared stained tissues were observed under an epifluorescent light microscope (Olympus, BX41, Center Valley, PA USA) and pictures were obtained using Fluo-Up (Explora-Nova, La Rochelle, France) and confocal microscope (Zeiss). AQP1 and 4 immunostaining were scored using a relative scale including a combination of the intensity and pattern of staining. AQP1 was scored in the following brain structures: choroid plexus, ependymal cells of the lateral and third ventricle, and lateral septal nucleus (n=5-8 animals/region/timepoint). Scoring was given a value from 0-4 with (0) no staining seen; (1) faint staining in a few positive cells, (2) bright staining in a small area, or low intensity staining in a larger area (more positive cells), (3) bright staining in a larger area, or (4) very intense staining over a large area. AQP4 staining was scored on the tissue collected at 60d with a scale representing the distribution patterns of the protein on astrocyte endfeet along brain vessels in the ipsilateral and contralateral sides of the lesion at multiple coronal bregma levels, from 4.2mm to -5.2mm throughout the longitudinal brain axis (demonstrated in Figure 5). The regions scored for AQP4 were: dorsal frontal-parietal cortex and lateral parietal temporal cortex in the sham and jTBI (n=5-8 animals/region). The AQP4 scale was from 0-4 with (0) no perivascular staining, (1) few scattered areas of perivascular staining at moderate brightness, (2) many areas of perivascular staining at moderate brightness, (3) several high density areas of perivascular staining at moderate brightness (4) entire region has extensive and very bright staining of AQP4 on perivascular astrocyte endfeet. No analyzed regions received a score of 0, and category 4 was most often observed in anterior sections far from the injury site.
Figure 5.
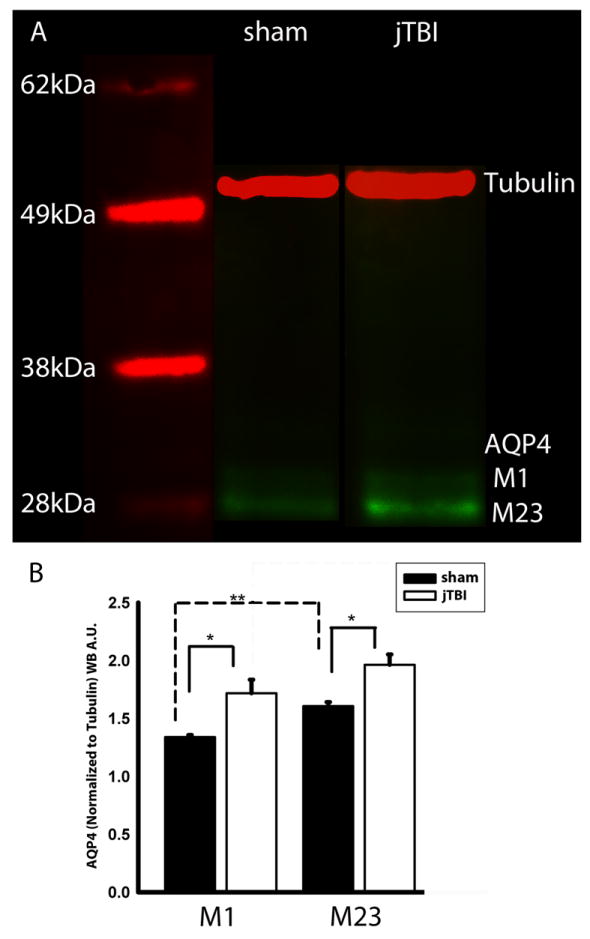
(A) Western blot of AQP4 at 7d shows two distinct bands of AQP4 at around 30kDa, corresponding to the M1 and M23 isoforms of AQP4. (B) jTBI shows a significantly higher expression of both M1 and M23 compared to sham. AQP4-M23 was significantly higher than the M1 in sham. (AQP4, aquaporin 4; kDa, kilo Dalton; jTBI, juvenile traumatic brain injury; *p<0.05, **p<0.01)
AQP4 western blot analysis
After magnetic resonance imaging at 7d, brains from 3 animals per group were freshly dissected with collection of cortex adjacent to the site of the impact. Tissues were prepared in RIPA buffer with protease inhibitor cocktail (PIC, Roche, Basel, Switzerland) and sonicated for 30 seconds. One μg of protein was then subjected to SDS polyacrylamide gel electrophoresis on a 4-12% gel (Nupage, Invitrogen, Carlsbad, CA). Proteins were then transferred to a polyvinylidene fluoride membrane (PerkinElmer, Germany). The blot was incubated with a polyclonal antibody against AQP4 (Millipore, California, 1:2000) and a monoclonal antibody against tubulin (Sigma, Switzerland, 1:25,000) in Odyssey blocking buffer (LI-COR, Bioscience, Germany) for 2 hrs at room temperature. After washing in PBS, the filter was incubated with two fluorescence-coupled secondary antibodies (1:10,000, anti-rabbit Alexa-Fluor-680nm, Molecular Probes, Oregon and anti-mouse infra-red-Dye-800-nm, Roche, Germany) for 2 hours at room temperature. After washing in PBS, the degree of fluorescence was measured using an infrared scanner (Odyssey, LI-COR, Germany).
Statistical Analysis
For MRI, infrared ROI, and western blot analysis, a student’s t-test was performed between groups. Categorical data were assessed using a non-parametric Mann-Whitney U (M-W) test to assess group differences.
RESULTS
Apparent diffusion coefficient (ADC) and T2 values
To address the time course of edema following TBI in our juvenile rat model, ADC and T2 values were analysed in the ipsilateral cortex including the lesion and the perilesion regions. Analysis was also performed distant from the injury site, for the contralateral cortex, ipsilateral striatum and contralateral striatum (Fig. 1). ADC and T2 values were reported relative to sham values. At 1d, ADC was significantly decreased within the lesion (39.5 ± 2.4%) and perilesion (45.0 ± 2.7%) regions compared to shams. A significant decrease in ADC values was also observed in the contralateral cortex (41.1 ± 9.4%) and striatum (41.3 ± 9.4%) (Fig 1A, B, C). ADC values normalized at 3 and 7d in all regions (Figure 1A, B, C). However, ADC became significantly elevated in the jTBI animals at 30 d within the lesion.
Figure 1.
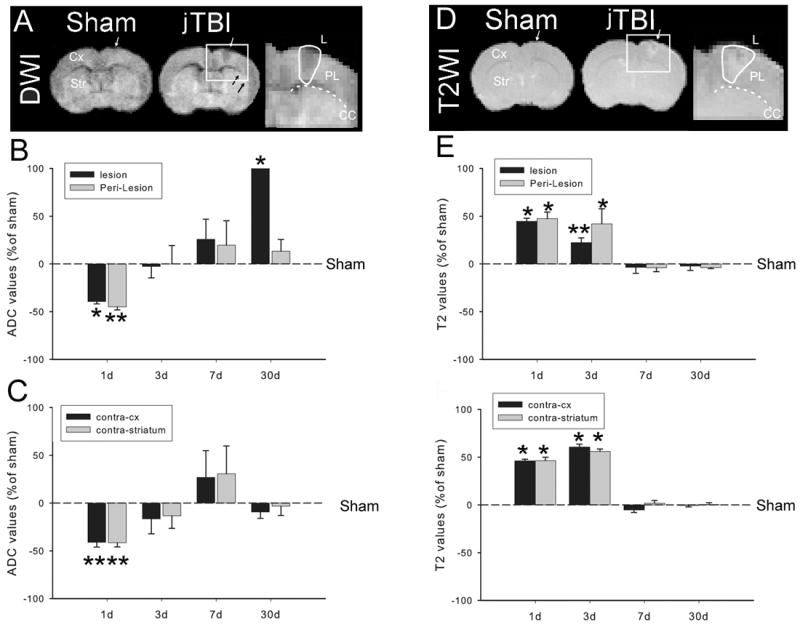
(A) DWI at 24hours after jTBI shows an increase in the size of the lesion and the perilesion compared to the sham group (B, C) Relative changes of the ADC values in the lesion and perilesion regions (B) and contralateral cortex (contra-cx) and contra-striatum show a decrease in the ADC values in all ROIs at 1d then increase at 30d in the lesion (*p<0.01). (D) T2 weighted images (T2) at 24h after jTBI show the increase of T2 signal in different brain regions compared to the sham. (E, F) Relative changes of the T2 values in the lesion and perilesion (B) and contralateral cortex (contra-cx) and contra-striatum show a increase of the T2 values in all ROIs at 1and 3d before to be normalized in all ROIs
(DWI, diffusion weighted images; jTBI, juvenile traumatic brain injury; ADC, apparent diffusion coefficient; ROI, region of interest; d, days post injury; *p<0.01).
Parallel to the ADC changes, T2 values were significantly higher in the jTBI animals compared to shams at 1d (44.4 ± 3.3% increase in the lesion, 47.5 ± 6.9% in the perilesion, 46.1 ± 1.6% in contralateral cortex and 46.3 ± 3.5% in the striatum). Edema remained elevated at 3d for all the regions assessed but returned to sham values at 7 and 30d (Figure 1 D, E, F).
Increase of AQP1 expression in neurons after jTBI
To address the contribution of AQPs following jTBI, we first evaluated AQP1 levels by immunohistochemistry. AQP1 staining was predominantly observed in the apical membrane of the choroid plexus at similar levels between jTBI and sham groups (Figure 2 A and B). The absence of differences between groups may be due to the relatively large distance between the choroid plexus and the site of the impact, which is approximately 2mm.
Figure 2.
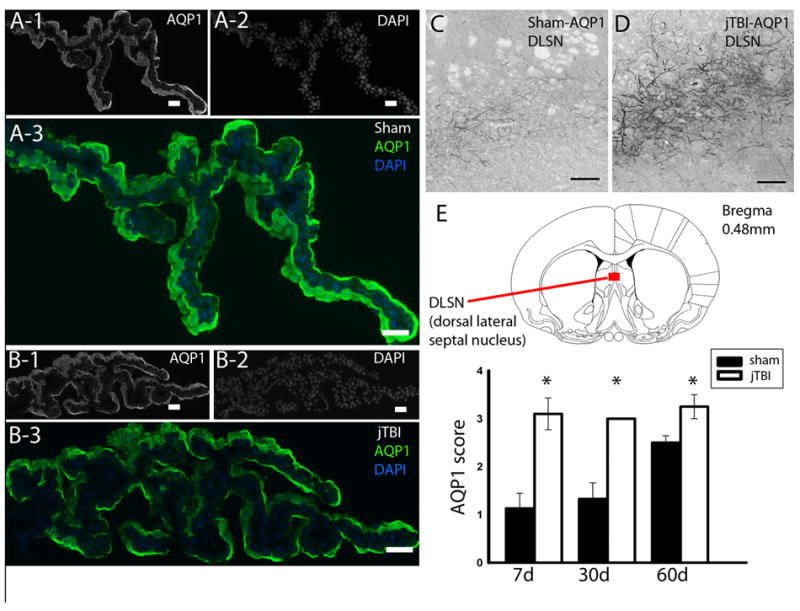
(A) AQP1 staining in the choroid plexus of sham and (B) jTBI shows no difference in intensity or pattern of staining. (C) AQP1 staining in the dorso-lateral septal nucleus of sham compared to that of (D) jTBI shows a (E) significant increase in staining in the jTBI animals at 7d, 28d, and 60d.
(AQP1,aquaporin1; jTBI, juvenile traumatic brain injury; DLSN, dorsal lateral sepal nucleus; d,days post injury; scale bar = 100 μm, *p<0.01).
A few AQP1 positive neurons were observed in the ipsilateral and contralateral parietal cortex in sham and jTBI rat pups at each time point as reported previously (Ribeiro Mde et al., 2006). No overt differences of the AQP1 staining were observed in the cortical and striatal regions where ADC and T2 values were changed. However, a striking increase in AQP1 staining was observed in jTBI rat pups after 7d, specifically in the neuronal dendrites in the dorso-lateral septal nucleus, just below the medial corpus callosum (Figure 2C-E). While quantification based on the intensity and pattern of the AQP1 staining at 1 and 3 d showed no significant differences, jTBI animals exhibited a significant increase at 7, 30, and 60 d (p<0.001) with greater staining intensity on several neuronal processes compared to sham (Figure 2). The changes in AQP1 expression starting at 7d strongly suggested that the changes in neuronal AQP1 expression may not be directly related to edema formation and resolution as observed in MRI.
AQP4 expression in astrocytes
We determined the evolution of AQP4 immunoreactivity (IR) using infra-red labelling over several post-injury timepoints and compared our results with MRI detection. Our data indicate several changes over time in the areas ipsilateral to the lesion (Figure 3) and on the contralateral side (Figure 4). Quantification of AQP4-IR using infra-red labeling (Figure 3C, 4C) was confirmed at higher magnification with classical immunostaining (Figure 3B, 4B). The intensity of the AQP4-IR using infra-red labelling was not significantly changed within the ipsilateral lesion and perilesional cortex between jTBI and sham animals at 1d (Figure 3). However, AQP4-IR was significantly decreased in the contralateral striatum in jTBI compared to sham animals, but not in the contralateral cortex at 1d (Figure 4). When ADC values and T2 values normalized (Figure 1), AQP4-IR significantly increased within the lesion at 3 and 7d (Figure 3). No differences were observed in AQP4 staining at 3 and 7 d in the contralateral cortex or striatum (Figure 4). AQP4-IR normalized at 30 d in all regions (Figure 3C, 4C).
Figure 3.
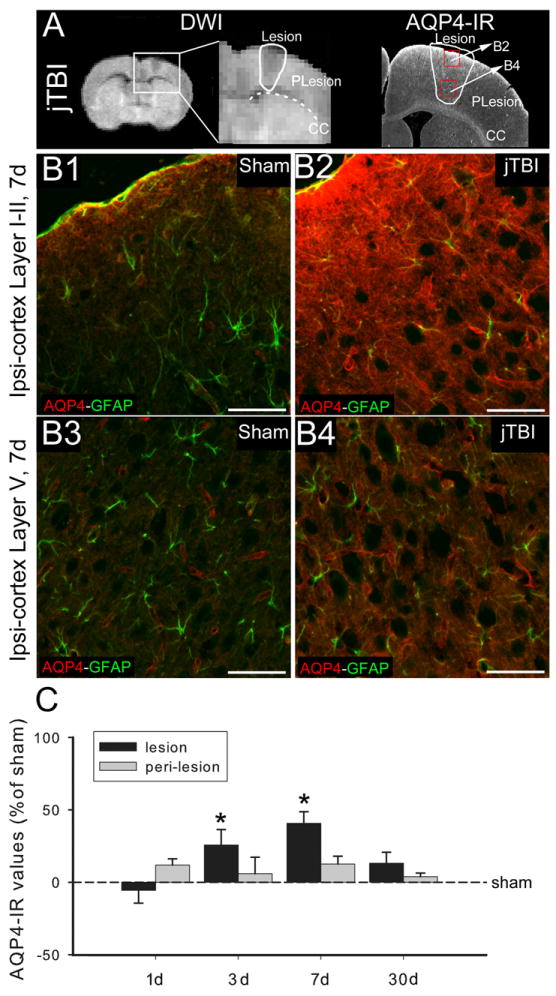
(A) DWI, (A left) and AQP4 immunoreactivity (A, right) at 7d after jTBI show the location of the lesion and perilesion ROIs used for the analysis and the location of the pictures, B2 and B4 in the red boxes.
(B1, B2, B3, B4) AQP4 (red) and GFAP (green) staining in the lesion area of the ipsilateral cortex near the surface in cortical layers I-II (B1, B2) and at the layer V (B3, B4) shows an increase of the AQP4 staining intensity at the glia limitans of jTBI (B2) compared to the shams (B1) and in the intracortical astrocytes in jTBI (B4) compared to sham (B3). (Scale bar = 100 μm.)
(C) AQP4 staining quantification was performed in the ROIs (lesion, perilesion, contralaral cortex and striatum, see figure 4). AQP4 immunoreactivity shows a significant increase of the in the lesion and perilesion at 3 and 7d (*p<0.05).
(DWI, diffusion weighted imaging; AQP4, aquaporin 4; d, days post injury; jTBI, juvenile traumatic brain injury; ROIs, regions of interest; GFAP, glial fibrillary acidic protein)
Figure 4.
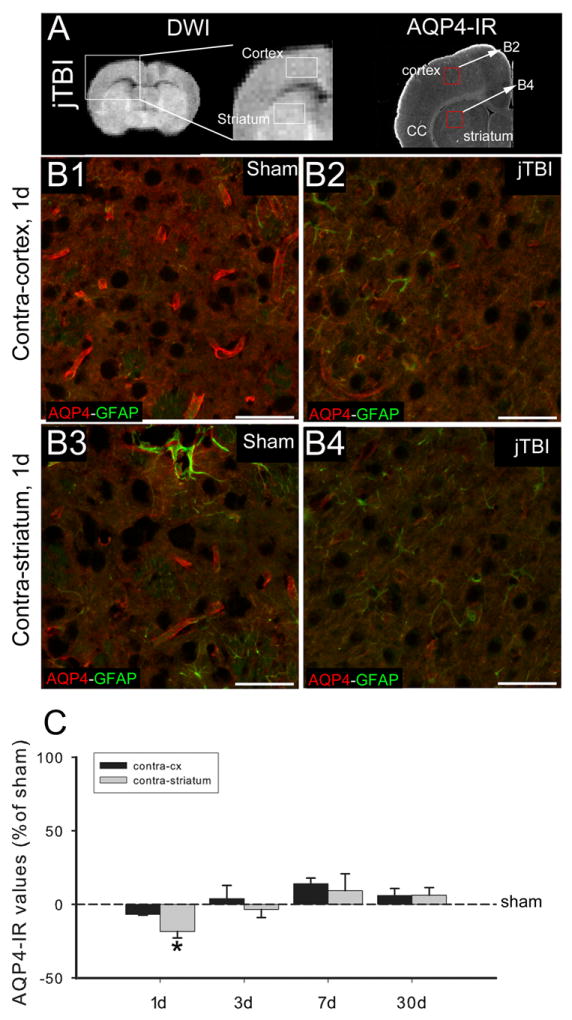
(A) DWI (left) and AQP4 immunoreactivity (right) at 1d after jTBI show the location of the contralateral cortex and striatum ROIs used for the analysis as well as the location of the pictures B2 and B4 in the red boxes.
(B1, B2, B3, B4) AQP4 (red) and GFAP (green) staining in the contra-cortex (B1, B2) and contra-striatum (B3, B4) show a decrease of the perivascular AQP4 staining in contra-cortex and contra-striatum of jTBI rats (B2, B4) compared to sham (B1, B3). Scale bar = 100 μm.
(C) AQP4 staining quantification shows a significant decrease of the AQP4 staining in the contra-striatum at 1d (*p<0.05).
(DWI, diffusion weighted imaging; AQP4, aquaporin 4; d, days post injury; jTBI, juvenile traumatic brain injury; ROIs, regions of interest; GFAP, glial fibrillary acidic protein)
More specifically, the higher magnification images show decreased AQP4 labeling on the astrocyte endfeet in contact with blood vessels in the contralateral striatum of jTBI compared to the sham rats at 1d (Figure 4B). At 3 and 7d, the increased AQP4 IR was not only on astrocyte endfeet but also on astrocyte processes and in the glia limitans (Figure 3B). The increase in immunoreactivity of AQP4-IR at 7 d was confirmed by western blot (Figure 5). We observed a significant increase in both isoforms of AQP4 (M23, M1) in jTBI compared to sham animals (Figure 5A). While the ratio of M23 to M1 did not differ between groups, the level of M23 expression was significantly higher than M1 expression in both sham and jTBI animals (Figure 5B).
At 60d, infra-red analysis showed no overall differences between groups; however, at higher magnification, AQP4 immunoreactivity revealed differences in the pattern of the staining (Figure 6). The density and distribution of AQP4 staining was categorized as Types 0-4 (see Methods and Figure 6C-F). Specifically, jTBI animals exhibited a scattered pattern of AQP4 immunostaining on the perivascular astrocyte endfeet in the dorsal parietal cortex region surrounding the lesion cavity at bregma -0.4, -1.4, -2.6 (p<0.05) (Figure 6G) at 60d. No changes were observed between sham and jTBI groups in the dorsal frontal cortex region at more anterior bregma levels (data not shown). Notably, the areas of reduced AQP4-staining correspond to the coronal slices with a visible lesion cavity at 60d. As expected, high AQP4-IR was observed in the glial scar immediately surrounding the lesional cavity in jTBI animals at 60d (Figure 6B).
Figure 6.
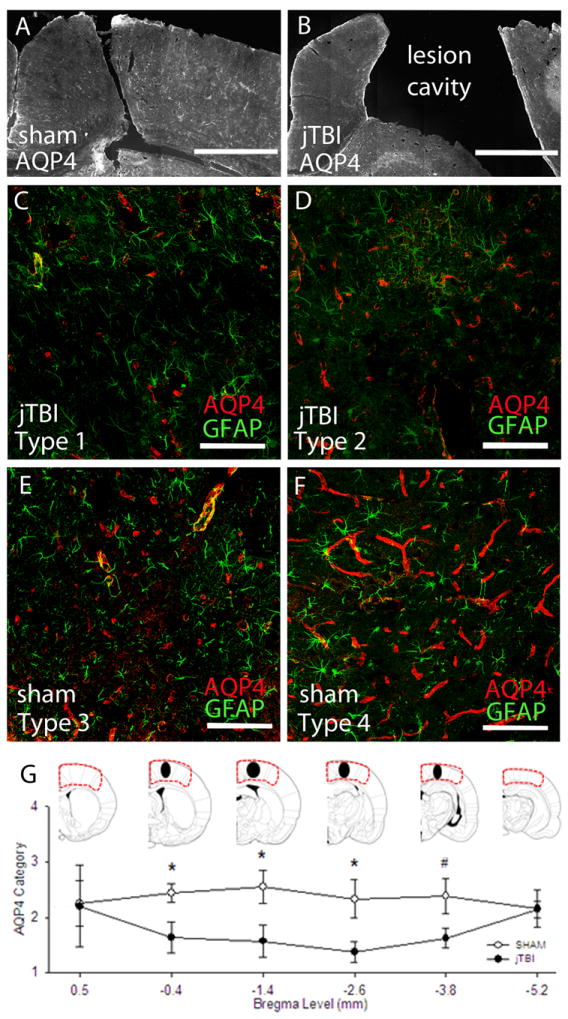
(A) A mosaic 10X image of representative Sham with AQP4 in perilesional cortex near Bregma level -2.6mm. (B) A mosaic 10X image of representative Sham with AQP4 in perilesional cortex near Bregma level -2.6mm. (bars in A,B= 500 μm) (C-F) Representative images from the parietal cortex showing four immunostaining patterns on astrocyte endfeet resting on micro- and macro-vessels. AQP4 was scored according to positive perivascular staining patterns as either Type 1, Type 2, Type 3, or Type 4. (bars in C-F = 100 μm) (G) Approximated drawing of the lesion size and location in jTBI animals at 60d (black ovals) located in the somatosensory cortex and spans a few coronal levels. Categorical AQP4 scores were assigned to the outlined area at each of the 6 coronal levels depicted. Different staining pattern of AQP4 protein expression was found in the ipsilateral dorsal cortex of jTBI nearest to the lesion cavity at Bregma -0.4mm, -1.4mm, -2.6mm, and a trend of smaller decrease more posteriorly at -3.8mm. (AQP4, aquaporin 4; jTBI, juvenile traumatic brain injury; d, days post injury; * p<0.05, #p<0.06)
Taken together, these data suggest that the delayed increase in AQP4 expression at 7 d is possibly related to the edema resolution (Figure 8) during the first week post-injury. However, by 60d when post-traumatic edema is no longer present, changes in AQP4 distribution may reflect other pathophysiological changes occurring near the lesion site and at distance from the impact (Ajao et al., in press).
Figure 8.
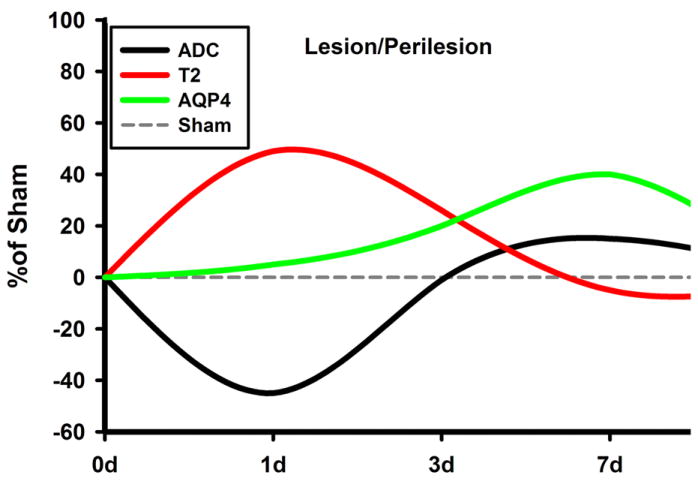
Summary Schematic of the Relative Changes in ADC, T2, and AQP4 at the region encompassing the site of impact: The values are normalized to the sham values, which is set at 0%. Notice the initial decrease of ADC and increase of T2, accompanied by a gradual increase in AQP4 values. As AQP4 increases, T2 and ADC start to normalize, signifying a possible resolution of edema.
AQP4, aquaporin 4; d, days post injury; ADC, apparent diffusion coefficient
Absence of astrocytic AQP9 changes after jTBI
Due to the involvement of several AQPs post-injury (Ribeiro Mde et al. 2006), we also evaluated the pattern of AQP9 in our model. AQP9 staining was observed in astrocytes of the corpus callosum (CC) and other white matter tracts such as the lateral olfactory tract (LOT) (Figure 7), in the astrocytes of the subfornical organs, tanycytes and ependymal cells as previously reported (Badaut et al., 2004). No significant differences in AQP9 staining were observed between the sham and jTBI animals at any time points.
Figure 7.
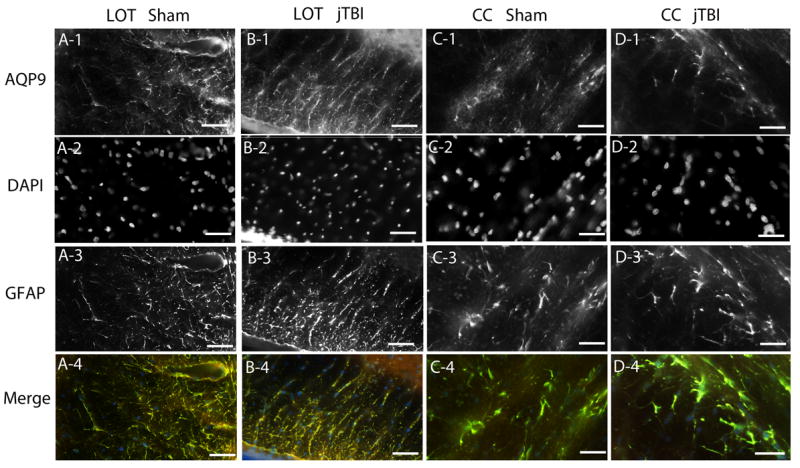
AQP9 showed no significant differences in intensity of staining or pattern of staining between sham and jTBI at any of the studied time points. (A1-A4) AQP9 staining of sham colocalized with DAPI and GFAP in the lateral olfactory tract. (B1-B4) AQP9 staining of jTBI colocalized with DAPI and GFAP in the lateral olfactory tract. (C1-C4) AQP9 staining of sham colocalized with DAPI and GFAP in the corpus callosum. (D1-D4) AQP9 staining of jTBI colocalized with DAPi and GFAP in the corpus callosum. (AQP9, aquaporin 9; GFAP, glial acidic fibrillary protein; jTBI, juvenile traumatic brain injury; LOT, lateral olfactory tract; CC, corpus callosum; scale bar = 100 μm)
DISCUSSION
Our data suggest that AQP4 plays a significant and more important role than AQP1 and AQP9 in regulating edema in the immature brain during the first week after jTBI. Similar to clinical observations, we observed a diffuse increase of brain water content and decrease of water mobility from regions close to, and far from, the lesion site in both hemispheres at 1 and 3d post-injury. By 7d, MRI values normalized in parallel with increases in AQP4 immunoreactivity, a pattern similar to stroke models (Ribeiro Mde et al., 2006, Badaut et al., 2007). At a distance from the lesion and primary sites of edema, we observed consistently higher AQP1 staining at 7, 30, and 60 d in neuronal filaments in the dorsolateral septum, thus highlighting the functional consequences of neuronal AQP1.
Clinically, diffusion weighted imaging (DWI) and T2 weighted imaging (T2WI) are useful modalities in the assessment of injury severity and outcome, particularly for edema formation and resolution (Obenaus and Ashwal, 2008, Chastain et al., 2009, Badaut et al., 2011b). At 1d, we observed bilateral increases in water content (higher T2) and decreases in water mobility, as shown by lower ADC values (Figure. 1). It is known that widespread edema at sites distant from the original impact are more common in juvenile than adult patients. These changes may be associated with differential susceptibility to blood-brain barrier (BBB) breakdown and cellular swelling following TBI (Pop and Badaut, 2011). Later, our ADC values returned to sham values by 3d but then increased by 30d, while T2 values normalized by 7d and remained stable. However, the pattern of acute ADC changes described in our study was not observed in a previously published paper using a rat jTBI model (Bertolizio et al., 2011). This discrepancy could be due to a difference in the magnet used for the MRI (11.7 T in our study vs 4.7T) and also to the definition of the ROI used for analysis (Bertolizio et al., 2011). In fact, few publications address both ADC and T2 changes in rodent jTBI. Comparable ADC and T2 changes to our animals were described in focal (Badaut et al., 2007) and global (Meng et al., 2004) rat pup models of hypoxia-ischemia. Overall, the patterns in our ADC and T2 data suggest that edema formation in the immature brain may have cytotoxic and vasogenic components at different post-injury timepoints.
AQPs have been proposed to account for MRI changes reflecting edema formation/resolution in several rodent models of brain injury (Meng et al., 2004, Tourdias et al., 2009, Badaut et al., 2011a, Badaut et al., 2011b, Tourdias et al., 2011). Here, we provided an extensive evaluation of several AQPs at multiple timepoints after jTBI, to address distinctive profiles during the post-traumatic period in the immature brain. At 1d, we observed stable levels of AQP4 near the lesion and decreased AQP4 in the contralateral striatum, in spite of MRI changes occurring bilaterally throughout the brain hemispheres. One explanation may be that stable AQP4 levels adjacent to the site of impact may contribute to water entry leading to cellular swelling (lower ADC) and increased edema (increased T2). At a distance, ADC changes may reflect cellular swelling secondary to reduced AQP4 (Figure 4), associated with transient water accumulation. Other models of normal or pathological brain show concomitant decreases in AQP4 expression and ADC (Meng et al., 2004, Badaut et al., 2011a), or increased AQP4 and ADC in hydrocephalus and inflammation models (Tourdias et al., 2009). Notably, a 30% decrease in AQP4 expression using small interference RNA led to a 50% decrease in ADC values in normal rats (Badaut et al., 2011a). However, in neonatal stroke models (Badaut et al., 2007) and neuroinflammation in adult rats (Tourdias et al., 2011), decreased ADCs did not correlate with AQP4 at 1d, similar to the discrepancies in our model. These global changes and heterogeneity of AQP4 expression underlie the complexity of molecular changes behind the fluctuations in water content and mobility during the edema process after jTBI.
Evidently, AQP4 may not be solely responsible for changes in water mobility associated with edema development after jTBI, suggesting the participation of other channel proteins. Two likely candidates are the gap junction protein Connexin-43 (Cx43) and inwardly rectifying potassium channel 4.1 (Kir4.1). Cx43 is down-regulated after silencing RNA against AQP4 in primary astrocyte cultures (Nicchia et al., 2005), while Kir4.1 co-localizes with AQP4 in astrocytic endfeet (Nagelhus et al., 2004). In addition, cellular potassium reuptake is impaired in an AQP4 knock-out epilepsy model (Binder et al., 2006) and potassium has been linked with water flux during astrocytic swelling (Dibaj et al., 2007). It is possible that AQP4, Cx43, and Kir 4.1 may be working in concert to address brain edema following jTBI.
Edema is a complex molecular process as shown in previous studies examining post-TBI AQP expression in adult TBI models. Reports of increased AQP4 (Sun et al., 2003, Guo et al., 2006, Ding et al., 2009, Higashida et al., 2011, Tomura et al., 2011) versus decreased AQP4 (Ke et al., 2001, Kiening et al., 2002, Zhao et al., 2005) are likely due to differences in injury type, rodent strains, and age at impact. We expect our juvenile cortical impact in P17 rats to differ from observations in adult animals, especially as early brain trauma may interfere with developmental phases and the evolution and mechanisms of injury. We observed increases in AQP4 at 3 and 7d near the lesion site in perivascular astrocyte endfeet, astrocyte processes, and the glia limitans. These changes may indicate that excess AQP4 could facilitate edematous fluid elimination through the subarachnoid space (Papadopoulos and Verkman, 2007, Tait et al., 2010, Tourdias et al., 2011). For example, increased AQP4 in the glia limitans may compensate for water accumulation at 1 and 3d (higher T2), with a gradual increase of AQP4 at 3d and normalization of both AQP and T2 values by 7d. This increase of AQP4 at 3 and 7d also parallels the restoration of the BBB with decreased IgG extravasation at 3d and no disruption by 7d (Pop and Badaut, 2011). Although AQP4 improves water removal, it can also regulate the initial formation of edema (based on increased T2 values at 1d) as previously proposed (Papadopoulos and Verkman, 2007, Tait et al., 2010). Thus, inhibiting AQP4 immediately after juvenile injury could be beneficial for edema reduction as shown in several other injury models (Manley et al., 2000, Papadopoulos and Verkman, 2005, Saadoun et al., 2008, Badaut et al., 2011b, Higashida et al., 2011, Igarashi et al., 2011).
Disproportionate levels of AQP4 isoforms and their ratios may interfere with the protein’s function, due to incorrect formation of the orthogonal array of particles, as described in stroke models (Badaut et al., 2011b). Moreover, although there is no general consensus and it remains controversial in vivo, some data show that the M1 isoform may have a higher water permeability than the M23 isoform (Fenton et al., 2010). This difference in water permeability may account for significant increases in the M1 isoform alone during stroke-related edema (Hirt et al., 2009). In our model, we detected higher protein levels of both isoforms on the impacted side compared to sham animals (Figure 5), higher levels of M23 than M1 in both groups, and no changes in isoform ratios. Together, these data suggest that the individual orthogonal array of particle arrangement may be similar between sham and jTBI by 7d, when the majority of edema and BBB disruption processes are nearly resolved.
We provide an evaluation at delayed post-injury timepoints, in contrast to reports in the adult literature focusing on the acute phase. Although AQP4-IR intensity was similar at 60d between groups, the pattern of AQP4 staining was different in jTBI animals. Notably, fewer scattered areas of perivascular AQP4 staining were observed in the parietal cortex near the lesion site in jTBI compared to sham by 60d. Long-term changes of AQP4 have been observed in other models such as spinal cord injury (Nesic et al., 2010), mouse models of Alzheimer disease (Wilcock et al., 2009, Yang et al., 2011), and Alzheimer disease patients (Wilcock et al., 2009, Moftakhar et al., 2010). These findings strengthen the concept that AQP4 may have additional roles that are not edema-related after certain injuries, such as cell migration (Saadoun et al., 2005, Auguste et al., 2007). In human and rat models of spinal cord injury, the ratio of GFAP to AQP4 in astrocytes around the lesion depend on both the time after injury as well as the severity of injury (Nesic et al., 2010). Thus, it is possible that the gliovascular unit as a whole is chronically affected. Further long-term studies of the gliovascular unit after jTBI should evaluate phenotypic changes in astrocytic networks in microvascular trees and functional correlations of those changes.
A few TBI studies describe increases in neuronal AQP1 (Tran et al., 2010, Oliva et al., 2011) and AQP9 (Ding et al., 2009, Oliva et al., 2011) after injury in adults. In our jTBI model, AQP1 expression in the choroid plexus did not differ between sham and jTBI at any time points. However, we found more intense AQP1 staining in neuronal filaments in the dorsolateral septum in jTBI animals at 7, 30, and 60 d. Neuronal septal tracts are involved in a number of cognitive pathways including learning, memory, sexual behavior, positive reinforcement, and pain (Gallagher et al., 1995, Singewald et al., 2011). Although consensus has not been established concerning the role of AQP1 in pain processing (Borsani, 2010), AQP1 knock-out mice had reduced pain responses versus wild-types (Oshio, 2006). TBI patients also report varying degrees of acute and chronic pain post-injury (Borsook, 2011; Nampiaparampil, 2008). Thus, septal increases in AQP1 and their influence on pain processing may be further elucidated in this jTBI model, by using appropriate nociceptive testing paradigms.
AQPs likely contribute to the evolution of several nervous system disorders, and can serve as potential therapeutic targets to improve clinical outcomes post-injury. Long-term changes in AQP4 and AQP1 after jTBI highlight differences in the edematous process occurring during a developmental period. Notably, edema can have different timelines for resolution in the young versus adult brain. Our data also confirm the importance of optimal timing for the administration of therapeutic agents targeting AQPs. While AQP therapies have been previously suggested for different pathologies such as stroke (Badaut et al., 2011b), optimal results can only be achieved when accounting for injury-specific parameters. It is important to consider age at injury (e.g. adult versus juvenile), type of injury model (e.g. cortical impact, fluid percussion, weight drop, craniotomy versus closed-head), and post-injury evaluation timepoints. Finally, no single model (e.g. TBI or stroke) can define the whole spectrum of a given pathology (e.g. edema) over time. Therefore, observational studies at multiple timepoints are valuable and necessary to adequately characterize post-injury sequelae, towards a comprehensive understanding of differential expression patterns of proteins involved in pathophysiological events. To determine the exact contribution of AQP4 on edema formation and resolution in parallel with our observations, it will be necessary to pursue a more functional study directed towards blocking or increasing AQP4 expression.
Highlights.
Diffuse edema formation in a rodent model of juvenile traumatic brain injury
Astrocytic AQP4 expression parallels the edema resolution after jTBI
Neuronal AQP1 expression is increased after jTBI
Long-term changes of AQP4 and AQP1 after jTBI
Acknowledgments
The authors thank Beatrice Ternon for her help for part of the histology and tissue analysis and Arash Adami and David Ajao for TBI induction. This work was supported in part by NINDS grant R01HD061946 (JB), the Loma Linda University Dept of Pediatrics Research Fund, the Swiss Science Foundation (FN 31003A-122166 and IZK0Z3-128973, JB), National Medical Test Bed (AO) and a NASA Cooperative Agreement NCC9-149 to the Radiobiology Program, Department of Radiation Medicine at Loma Linda University. A portion of this material was performed in the Loma Linda University School of Medicine Advanced Imaging and Microscopy Core that is supported by the National Science Foundation under Major Research Instrumentation, Division of Biological Infrastructure Grant No. 0923559 (Sean M Wilson) and the Loma Linda University School of Medicine.
LIST OF ABBREVIATIONS
- jTBI
juvenile traumatic brain injury
- TBI
traumatic brain injury
- AQP
aquaporin
- d
days post injury
- CCI
controlled cortical impact
- DWI
Diffusion Weighted Imaging
- T2WI
T2 weighted imaging
- ADC
Apparent Diffusion Coefficient
- MRI
Magnetic Resonance Imaging
- ROI
Region of Intertest
- GFAP
Glial Fibrillary Acidic Protein
- LOT
lateral olfactory tract
- CC
corpus callosum
Footnotes
Author’s contributions:
AF, AO, DS, JB and VP: generated the data, the analysis, and writing; experimental design and writing, AO, JB, SA
Conflict of Interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelson PD, Kochanek PM. Head injury in children. J Child Neurol. 1998;13:2–15. doi: 10.1177/088307389801300102. [DOI] [PubMed] [Google Scholar]
- Adelson PD, Nemoto E, Colak A, Painter M. The use of near infrared spectroscopy (NIRS) in children after traumatic brain injury: a preliminary report. Acta Neurochir Suppl. 1998;71:250–254. doi: 10.1007/978-3-7091-6475-4_72. [DOI] [PubMed] [Google Scholar]
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Badaut J. Aquaglyceroporin 9 in brain pathologies. Neuroscience. 2010;168:1047–1057. doi: 10.1016/j.neuroscience.2009.10.030. [DOI] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Adami A, Tone B, Recker R, Spagnoli D, Ternon B, Obenaus A. Brain water mobility decreases after astrocytic aquaporin-4 inhibition using RNA interference. J Cereb Blood Flow Metab. 2011a;31:819–831. doi: 10.1038/jcbfm.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Obenaus A. Aquaporins in cerebrovascular disease: a target for treatment of brain edema? Cerebrovasc Dis. 2011b;31:521–531. doi: 10.1159/000324328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Tone B, Regli L, Tian HR, Obenaus A. Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr Res. 2007;62:248–254. doi: 10.1203/PDR.0b013e3180db291b. [DOI] [PubMed] [Google Scholar]
- Badaut J, Petit JM, Brunet JF, Magistretti PJ, Charriaut-Marlangue C, Regli L. Distribution of Aquaporin 9 in the adult rat brain: preferential expression in catecholaminergic neurons and in glial cells. Neuroscience. 2004;128:27–38. doi: 10.1016/j.neuroscience.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Bauer R, Fritz H. Pathophysiology of traumatic injury in the developing brain: an introduction and short update. Exp Toxicol Pathol. 2004;56:65–73. doi: 10.1016/j.etp.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Bertolizio G, Bissonnette B, Mason L, Ashwal S, Hartman R, Marcantonio S, Obenaus A. Effects of hemodilution after traumatic brain injury in juvenile rats. Paediatr Anaesth. 2011;21:1198–1208. doi: 10.1111/j.1460-9592.2011.03695.x. [DOI] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Chastain CA, Oyoyo UE, Zipperman M, Joo E, Ashwal S, Shutter LA, Tong KA. Predicting outcomes of traumatic brain injury by imaging modality and injury distribution. J Neurotrauma. 2009;26:1183–1196. doi: 10.1089/neu.2008.0650. [DOI] [PubMed] [Google Scholar]
- Dibaj P, Kaiser M, Hirrlinger J, Kirchhoff F, Neusch C. Kir4.1 channels regulate swelling of astroglial processes in experimental spinal cord edema. J Neurochem. 2007;103:2620–2628. doi: 10.1111/j.1471-4159.2007.04979.x. [DOI] [PubMed] [Google Scholar]
- Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. 2009;453:68–72. doi: 10.1016/j.neulet.2009.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Vulnerability of developing brain not explained by cell number/cell size hypothesis. Early Hum Dev. 1981;5:227–231. doi: 10.1016/0378-3782(81)90030-x. [DOI] [PubMed] [Google Scholar]
- Faul M, X L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations and deaths 2002-2006. Atlanta (GA): Centers for Disease Control and Prevention, National center for Injury Prevention and Control; 2010. [Google Scholar]
- Fenton RA, Moeller HB, Zelenina M, Snaebjornsson MT, Holen T, MacAulay N. Differential water permeability and regulation of three aquaporin 4 isoforms. Cell Mol Life Sci. 2010;67:829–840. doi: 10.1007/s00018-009-0218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JP, Zheng F, Hasuo H, Shinnick-Gallagher P. Activities of neurons within the rat dorsolateral septal nucleus (DLSN) Prog Neurobiol. 1995;45:373–395. doi: 10.1016/0301-0082(95)98600-a. [DOI] [PubMed] [Google Scholar]
- Gomori E, Pal J, Abraham H, Vajda Z, Sulyok E, Seress L, Doczi T. Fetal development of membrane water channel proteins aquaporin-1 and aquaporin-4 in the human brain. Int J Dev Neurosci. 2006;24:295–305. doi: 10.1016/j.ijdevneu.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sayeed I, Baronne LM, Hoffman SW, Guennoun R, Stein DG. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp Neurol. 2006;198:469–478. doi: 10.1016/j.expneurol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, Ding JY, Dornbos D, 3rd, Li X, Guthikonda M, Rossi NF, Ding Y. The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- Hirt L, Ternon B, Price M, Mastour N, Brunet JF, Badaut J. Protective role of early aquaporin 4 induction against postischemic edema formation. J Cereb Blood Flow Metab. 2009;29:423–433. doi: 10.1038/jcbfm.2008.133. [DOI] [PubMed] [Google Scholar]
- Hsu MS, Seldin M, Lee DJ, Seifert G, Steinhauser C, Binder DK. Laminar-specific and developmental expression of aquaporin-4 in the mouse hippocampus. Neuroscience. 2011;178:21–32. doi: 10.1016/j.neuroscience.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi H, Huber VJ, Tsujita M, Nakada T. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci. 2011;32:113–116. doi: 10.1007/s10072-010-0431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke C, Poon WS, Ng HK, Pang JC, Chan Y. Heterogeneous responses of aquaporin-4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett. 2001;301:21–24. doi: 10.1016/s0304-3940(01)01589-0. [DOI] [PubMed] [Google Scholar]
- Kiening KL, van Landeghem FK, Schreiber S, Thomale UW, von Deimling A, Unterberg AW, Stover JF. Decreased hemispheric Aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci Lett. 2002;324:105–108. doi: 10.1016/s0304-3940(02)00180-5. [DOI] [PubMed] [Google Scholar]
- Kochanek PM. Pediatric traumatic brain injury: quo vadis? Dev Neurosci. 2006;28:244–255. doi: 10.1159/000094151. [DOI] [PubMed] [Google Scholar]
- Lang DA, Teasdale GM, Macpherson P, Lawrence A. Diffuse brain swelling after head injury: more often malignant in adults than children? J Neurosurg. 1994;80:675–680. doi: 10.3171/jns.1994.80.4.0675. [DOI] [PubMed] [Google Scholar]
- Lee DJ, Amini M, Hamamura MJ, Hsu MS, Seldin MM, Nalcioglu O, Binder DK. Aquaporin-4-dependent edema clearance following status epilepticus. Epilepsy Res. 2011 doi: 10.1016/j.eplepsyres.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Meng S, Qiao M, Lin L, Del Bigio MR, Tomanek B, Tuor UI. Correspondence of AQP4 expression and hypoxic-ischaemic brain oedema monitored by magnetic resonance imaging in the immature and juvenile rat. Eur J Neurosci. 2004;19:2261–2269. doi: 10.1111/j.0953-816X.2004.03315.x. [DOI] [PubMed] [Google Scholar]
- Moftakhar P, Lynch MD, Pomakian JL, Vinters HV. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2010;69:1201–1209. doi: 10.1097/NEN.0b013e3181fd252c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM, Ottersen OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. 2004;129:905–913. doi: 10.1016/j.neuroscience.2004.08.053. [DOI] [PubMed] [Google Scholar]
- Nesic O, Guest JD, Zivadinovic D, Narayana PA, Herrera JJ, Grill RJ, Mokkapati VU, Gelman BB, Lee J. Aquaporins in spinal cord injury: the janus face of aquaporin 4. Neuroscience. 2010;168:1019–1035. doi: 10.1016/j.neuroscience.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesic O, Lee J, Unabia GC, Johnson K, Ye Z, Vergara L, Hulsebosch CE, Perez-Polo JR. Aquaporin 1 - a novel player in spinal cord injury. J Neurochem. 2008;105:628–640. doi: 10.1111/j.1471-4159.2007.05177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicchia GP, Srinivas M, Li W, Brosnan CF, Frigeri A, Spray DC. New possible roles for aquaporin-4 in astrocytes: cell cytoskeleton and functional relationship with connexin43. Faseb J. 2005;19:1674–1676. doi: 10.1096/fj.04-3281fje. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Ashwal S. Magnetic resonance imaging in cerebral ischemia: focus on neonates. Neuropharmacology. 2008;55:271–280. doi: 10.1016/j.neuropharm.2008.06.010. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Kang Y, Truettner JS, Sanchez-Molano J, Furones C, Yool AJ, Atkins CM. Fluid-percussion brain injury induces changes in aquaporin channel expression. Neuroscience. 2011;180:272–279. doi: 10.1016/j.neuroscience.2011.02.020. [DOI] [PubMed] [Google Scholar]
- Oshio K, Watanabe H, Yan D, Verkman AS, Manley GT. Impaired pain sensation in mice lacking Aquaporin-1 water channels. Biochem Biophys Res Commun. 2006;341:1022–1028. doi: 10.1016/j.bbrc.2006.01.062. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Binder DK, Manley GT, Krishna S, Verkman AS. Molecular mechanisms of brain tumor edema. Neuroscience. 2004;129:1011–1020. doi: 10.1016/j.neuroscience.2004.05.044. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem. 2005;280:13906–13912. doi: 10.1074/jbc.M413627200. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 and brain edema. Pediatr Nephrol. 2007;22:778–784. doi: 10.1007/s00467-006-0411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Badaut J. A Neurovascular Perspective for Long-Term Changes After Brain Trauma. Transl Stroke Res. 2011;2:533–545. doi: 10.1007/s12975-011-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro Mde C, Hirt L, Bogousslavsky J, Regli L, Badaut J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res. 2006;83:1231–1240. doi: 10.1002/jnr.20819. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Bell BA, Verkman AS, Papadopoulos MC. Greatly improved neurological outcome after spinal cord compression injury in AQP4-deficient mice. Brain. 2008;131:1087–1098. doi: 10.1093/brain/awn014. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Schneier AJ, Shields BJ, Hostetler SG, Xiang H, Smith GA. Incidence of pediatric traumatic brain injury and associated hospital resource utilization in the United States. Pediatrics. 2006;118:483–492. doi: 10.1542/peds.2005-2588. [DOI] [PubMed] [Google Scholar]
- Singewald GM, Rjabokon A, Singewald N, Ebner K. The modulatory role of the lateral septum on neuroendocrine and behavioral stress responses. Neuropsychopharmacology. 2011;36:793–804. doi: 10.1038/npp.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skucas VA, Mathews IB, Yang J, Cheng Q, Treister A, Duffy AM, Verkman AS, Hempstead BL, Wood MA, Binder DK, Scharfman HE. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J Neurosci. 2011;31:6392–6397. doi: 10.1523/JNEUROSCI.6249-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MC, Honey CR, Berk C, Wong NL, Tsui JK. Regulation of aquaporin-4 in a traumatic brain injury model in rats. J Neurosurg. 2003;98:565–569. doi: 10.3171/jns.2003.98.3.0565. [DOI] [PubMed] [Google Scholar]
- Tait MJ, Saadoun S, Bell BA, Verkman AS, Papadopoulos MC. Increased brain edema in aqp4-null mice in an experimental model of subarachnoid hemorrhage. Neuroscience. 2010;167:60–67. doi: 10.1016/j.neuroscience.2010.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura S, Nawashiro H, Otani N, Uozumi Y, Toyooka T, Ohsumi A, Shima K. Effect of decompressive craniectomy on aquaporin-4 expression after lateral fluid percussion injury in rats. J Neurotrauma. 2011;28:237–243. doi: 10.1089/neu.2010.1443. [DOI] [PubMed] [Google Scholar]
- Tourdias T, Dragonu I, Fushimi Y, Deloire MS, Boiziau C, Brochet B, Moonen C, Petry KG, Dousset V. Aquaporin 4 correlates with apparent diffusion coefficient and hydrocephalus severity in the rat brain: a combined MRI-histological study. Neuroimage. 2009;47:659–666. doi: 10.1016/j.neuroimage.2009.04.070. [DOI] [PubMed] [Google Scholar]
- Tourdias T, Mori N, Dragonu I, Cassagno N, Boiziau C, Aussudre J, Brochet B, Moonen C, Petry KG, Dousset V. Differential aquaporin 4 expression during edema build-up and resolution phases of brain inflammation. J Neuroinflammation. 2011;8:143. doi: 10.1186/1742-2094-8-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran ND, Kim S, Vincent HK, Rodriguez A, Hinton DR, Bullock MR, Young HF. Aquaporin-1-mediated cerebral edema following traumatic brain injury: effects of acidosis and corticosteroid administration. J Neurosurg. 2010;112:1095–1104. doi: 10.3171/2009.8.JNS081704. [DOI] [PubMed] [Google Scholar]
- Wen H, Nagelhus EA, Amiry-Moghaddam M, Agre P, Ottersen OP, Nielsen S. Ontogeny of water transport in rat brain: postnatal expression of the aquaporin-4 water channel. Eur J Neurosci. 1999;11:935–945. doi: 10.1046/j.1460-9568.1999.00502.x. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Vitek MP, Colton CA. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience. 2009;159:1055–1069. doi: 10.1016/j.neuroscience.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LM, Nilsson LN, Lannfelt L, Xu Y, Amiry-Moghaddam M, Ottersen OP, Torp R. Loss of Astrocyte Polarization in the Tg-ArcSwe Mouse Model of Alzheimer’s Disease. J Alzheimers Dis. 2011 doi: 10.3233/JAD-2011-110725. [DOI] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Clifton GL, Dash PK. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J Neurosci Res. 2005;82:499–506. doi: 10.1002/jnr.20649. [DOI] [PubMed] [Google Scholar]
- Zheng GQ, Li Y, Gu Y, Chen XM, Zhou Y, Zhao SZ, Shen J. Beyond water channel: aquaporin-4 in adult neurogenesis. Neurochem Int. 2010;56:651–654. doi: 10.1016/j.neuint.2010.01.014. [DOI] [PubMed] [Google Scholar]