

Abstract
We provide evidence that arsenic trioxide (As2O3) targets the BCR-ABL oncoprotein via a novel mechanism involving p62/SQSTM1-mediated localization of the oncoprotein to the autolysosomes and subsequent degradation mediated by the protease cathepsin B. Our studies demonstrate that inhibitors of autophagy or cathepsin B activity and/or molecular targeting of p62/SQSTM1, Atg7, or cathepsin B result in partial reversal of the suppressive effects of AS2O3 on BCR-ABL expressing leukemic progenitors, including primitive leukemic precursors from chronic myelogenous leukemia (CML) patients. Altogether, these findings indicate that autophagic degradation of BCR-ABL is critical for the induction of the antileukemic effects of As2O3 and raise the potential for future therapeutic approaches to target BCR-ABL expressing cells by modulating elements of the autophagic machinery to promote BCR-ABL degradation.
Introduction
Elements of the autophagic machinery have attracted recently considerable attention as a potential target for the development of novel approaches for the treatment of malignancies.1,2 Similar to apoptosis, autophagy is a programmed cell death mechanism, but it is distinguished by a self-catabolic process involving lysosomal proteolytic degradation of cellular components.3 This is initiated by the formation of a double-membrane enclosed structure, known as the autophagosome.4 On fusion with a lysosome, a cellular organelle characterized by low pH and hydrolytic enzymes,5,6 such structure eventually develops into the autophagolysosome where degradation of organelles occurs.
Under different circumstances, autophagy can either inhibit or promote malignant cell survival, but its precise role in tumorigenesis remains to be established.7,8 The role of inducible autophagy in BCR-ABL expressing leukemia cells is poorly understood. For example, there is previous evidence suggesting that autophagy may play regulatory roles in BCR-ABL leukemogenesis,9 whereas other studies have shown that pharmacologic inhibition of autophagy enhances the effects of imatinib mesylate and other targeted therapies in CML.10–12 There are also opposing lines of evidence, pointing toward tumor inhibitory effects of autophagy,13,14 although a recent study demonstrated that BCR-ABL exerts suppressive effects on autophagy via engagement of the PI3K/FoxO4/ATF5/mTOR pathway.15
Arsenic trioxide (AS2O3) exhibits potent antileukemic effects in vitro and in vivo and has major clinical activity in the treatment of patients with acute promyelocytic leukemia (APL).16–18 This agent was previously shown to target and eliminate leukemia initiating stem cells (LICs) in mouse models in vivo via PML targeting.19 Notably, there is evidence that AS2O3 degrades BCR-ABL,20 raising the possibility that this agent may provide an approach to target CML LICs. However, a major limiting factor for the incorporation of arsenic in non-APL malignancies has been the requirement of high concentrations for induction of cell death in non-APL cells and the incomplete understanding of the mechanisms by which it promotes antileukemic responses.
A major mechanism contributing to the antineoplastic effects of arsenic is induction of apoptosis,16–18 with upstream JNK activation being a prominent regulatory mechanism.21 In a recent study, we provided evidence that arsenic trioxide induces autophagy in AML leukemic progenitors and demonstrated that such autophagy is essential for generation of the inhibitory effects of arsenic on primitive leukemic precursors.22 However, the key downstream cellular events by which such arsenic-dependent induction of autophagy elicits antileukemic responses and its potential involvement in the generation of inhibitory responses in other types of leukemias have been unknown. As AS2O3 is known to induce BCR-ABL degradation,20,23 we examined the role of autophagy in the process and in the generation of antileukemic responses in BCR-ABL expressing cells. Our studies provide evidence for a novel mechanism, involving autophagic degradation of the transforming BCR-ABL oncoprotein. Inhibition of autophagy, pharmacologically or by siRNA-targeting of key elements of the autophagic machinery, reverses degradation of BCR-ABL and abrogates generation of arsenic-induced antileukemic effects. Our studies also establish that the function of the lysosome-localized protease cystein cathepsin-B exhibits regulatory effects on BCR-ABL degradation during the autophagic process, providing evidence for a novel mechanism for BCR-ABL targeting occurs in the autolysosomes.
Methods
Cells and reagents
The BCR-ABL expressing K562 human leukemia cell line was grown in RPMI 1640 supplemented with 10% fetal bovine serum and antibiotics. Pepstatin A, E-64d, and AS2O3 were purchased from Sigma-Aldrich, and CA-074 was obtained from Enzo Life Sciences. Antibodies against LC3B and Atg7 were from Cell Signaling Technologies. Antibodies against beclin 1 (H-300), p62/SQSTM1 (D-3), Hsp90 α/β (H-114), ABL (24-11), BCR, and cathepsin B (C-19), as well as normal mouse IgG, were obtained from Santa Cruz Biotechnology. An antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was obtained from Millipore. Control and cathepsin B shRNA lentiviral particles were obtained from Santa Cruz Biotechnology. Ba/F3-BCR-ABL and Ba/F3-T315I-BCR-ABL transfectants were provided by Dr Brian J. Druker (Howard Hughes Medical Institute and Oregon Health & Science University, Portland, OR).
Cell lysis and immunoblotting
Cells were incubated for the indicated times with the indicated concentrations of As2O3, and subsequently lysed in phosphorylation lysis buffer as in our previous studies.22,24–26 Immunoblotting using an enhanced chemiluminescence (ECL) methodology was performed as in our previous studies.24–27 In the experiments in which the pharmacologic inhibitors pepstatin A and E-64d were used, the cells were pretreated for 60 minutes with the indicated concentrations of the inhibitors before the addition of AS2O3 to the cultures. siRNA-mediated knockdown of Beclin 1 or Atg7 in human leukemic cells was performed using Nucleofector kits from Amaxa Biosystems. Cells were transfected with Beclin 1 or Atg7 specific siRNA pools from Dharmacon RNAi Technologies or single target RNAi from Ambion/Life Technologies.
Assays of primary hematopoietic progenitors from CML patients
Peripheral blood from or bone marrow samples from patients with CML were collected after obtaining informed consent approved by the Institutional Review Board of Northwestern University. This study was conducted in accordance with the Declaration of Helsinki. The effects of AS2O3 on leukemic progenitor colony formation were assessed by clonogenic assays in methylcellulose, as previously described.22,24,25,27
Acridine orange staining and flow cytometric analysis
Formation of acidic vesicular organelles (AVOs), a morphologic characteristic of autophagy was quantitated by acridine orange staining.28 Acridine orange (0.5 mg/mL; Invitrogen) was added 15 minutes before collection and after washing with phosphate buffered saline, cells were analyzed by flow cytometry. Once in the acidic compartments in acridine orange fluoresces red (FL3/PE-Cy5, Ex/Em 496/667 nm) and in the cytoplasm green (FL1/FITC, Ex/Em 494/520 nm) and dim red.29 Red fluorescence is proportional to the levels of acidity observed.
Immunofluorescence
Human leukemic cells were incubated in DQ-BSA (20 mg/mL; Invitrogen) for 16 hours. The cells were then collected and fixed with periodate/lysine/paraformaldehyde solution before staining.30 Fluorescence was detected using a Zeiss LSM 510 META, confocal microscope system and images were analyzed with ImageJ (National Institutes of Health).
Electron microscopy
K562 cells were treated as indicated and then fixed in BD cytofix/cytoperm fixation/permeabilization Solution (BD Bioscience) at 4°C for 30 minutes and stained with anti-p62/SQSTM1 (Sigma-Aldrich) and anti-ABL (Santa Cruz Biotechnology) antibodies, followed by 6 nm anti–mouse and 15 nm ant–rabbit EM-grade IgG gold conjugates (Aurion). Samples were then fixed in 0.1M sodium cacodylate buffer pH7.3 containing 2% paraformaldehyde and 2.5% glutaraldehyde and embedded in resin mixture of embed 812 and araldite. Samples were sectioned to 70 nm thin and poststained with 3% uranyl acetate and Reynolds lead citrate. Samples were examined under FEI Tecnai Spirit G2 TEM and digital images were captured on an FEI Eagle camera. Quantification of autophagic structures was performed using the principles of point counting as previously described.31
Results
Arsenic trioxide-dependent induction of autophagy in K562 leukemia cells
In initial studies we assessed the ability of As2O3 to induce autophagy in the BCR-ABL expressing K562 CML-blast crisis cell line. As expected based on previous reports,20 we found that As2O3-treatment of K562 cells results in significant reduction in the levels of BCR-ABL expression (Figure 1A), consistent with arsenic-dependent degradation of the protein.24 In addition, consistent with our previous work in different leukemia cell lines,22 we found that treatment of K562 cells with As2O3 results in induction of autophagy, as shown by immunoblotting experiments demonstrating up-regulation of LC3II (Figure 1B). When cells were incubated in the presence of the lysosomal specific protease inhibitors E64d and pepstatin A, there were increasing levels of LC3II (Figure 1C), consistent with autophagic flux. We also found that there was time-dependent decrease of p62/SQSMT1 levels that was seen after 72 hours of incubation (Figure 1D), whereas treatment the autophagy inhibitor bafilomycin level increased the levels of p62/SQSMT1 (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Arsenic trioxide-inducible autophagic structures could be seen by electron microscopy (Figure 1E), whereas flow cytometry studies established increased levels of acidic vesicular organelles (AVOs) after arsenic treatment (Figure 1F).
Figure 1.

As2O3–dependent degradation of BCR-ABL and induction of autophagy in the BCR-ABL expressing leukemia cells. (A) K562 cells were incubated with As2O3 for 24 hours, as indicated. Total cell lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (B) K562 cells were incubated with AS2O3 at the indicated final concentrations for 24 hours. Total cell lysates were resolved by SDS-PAGE and immunoblotted with anti-LC3 or anti-GAPDH antibodies, as indicated. (C) K562 cells were incubated with AS2O3 (2μM) in the presence or absence of E64d (10μM), and pepstatin A (10μM) for 24 hours, as indicated. Total cell lysates were resolved by SDS-PAGE and immunoblotted with anti-LC3 or anti-GAPDH antibodies, as indicated. (D) K562 cells were incubated for the indicated times with AS2O3 (2μM). Total cell lysates were resolved by SDS-PAGE and immunoblotted with anti-p62/SQSMT1 or anti-GAPDH antibodies, as indicated. (E) Electron microscopy analysis for autophagic compartments in untreated K562 cells or cells treated for 24 hours with As2O3 (2μM). Data from 5 independent measurements were quantitate and represent autophagic compartment (AC) accumulation in a 100μM2 cell surface area. Paired t test analysis showed P = .0207. (F) K562 cells were treated for 24 hours with As2O3 (2μM). The cells were then stained with acridine orange for quantitation of formation of acidic vesicular organelles (AVOs). An increase in AVOs formation is accompanied with an increase in FL3/PE-Cy5 fluorescence, reflecting induction of autophagy. Data from 3 independent experiments, including the one shown in the top panel, were quantitated are expressed as means ± SE. Paired t test analysis comparing AS2O3–treated cells versus control-untreated cells, demonstrated P = .016.
Degradation of BCR-ABL by arsenic trioxide can be reversed by inhibition of autophagy
As the mechanisms by which arsenic promotes degradation of the BCR-ABL oncoprotein are largely unknown, we sought to determine whether inhibition of the autophagic process exhibits regulatory effects. We targeted key components of the autophagic machinery using specific siRNAs and assessed their effects on AS2O3–dependent down-regulation of BCR-ABL protein. Selective knockdown of either Becn1 or Atg7 or p62SQSTM1 (Figure 2A) resulted in reversal of the suppressive effects of arsenic-treatment on BCR-ABL levels (Figure 2B, supplemental Figures 2-3). Treatment with pepstatin A and E64d resulted in reversal of BCR-ABL degradation (Figure 2C). In addition, siRNA-mediated targeting of elements of the autophagic machinery22 reversed the suppressive effects of As2O3 on K562-derived leukemic progenitor colony formation (CFU-L; Figure 2D-E).
Figure 2.
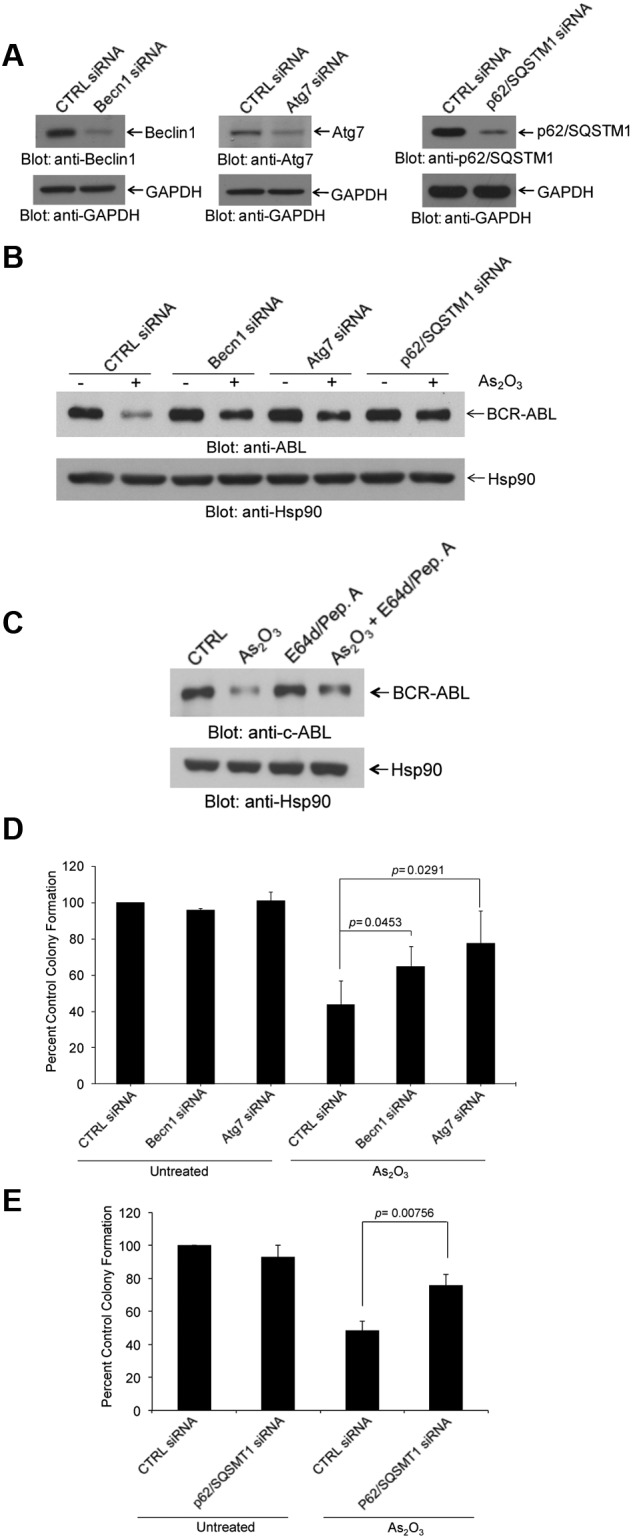
Arsenic trioxide-induced autophagic degradation of BCR-ABL. (A) K562 cells were transfected with control siRNA or siRNAs specifically targeting beclin 1 (Becn1) or Atg7 or p62/SQSTM1, as indicated. Cells were lysed, and total lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (B) K562 cells were transfected with control siRNA or siRNAs specifically targeting Becn1 or Atg7 or p62/SQSTM1, as indicated and were subsequently treated for 24 hours with As2O3 (2μM). Total lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (C) K562 cells were pretreated for 60 minutes with pepstatin A (10μM) and E64-d (10μM) as indicated, and were subsequently treated for 24 hours with As2O3 (2μM) in the continuous presence or absence of the inhibitors, as indicated. Total cell lysates were resolved by SDS-PAGE and immunoblotted with an anti-ABL or anti-Hsp90 antibodies, as indicated. (D-E) K562 cells were transfected with control-siRNA or Atg7 siRNA or Beclin1 siRNA (D) or p62/SQSMT1 siRNA (E) as indicated and the effects of AS2O3 (0.5μM) on leukemic progenitor (CFU-L) colony formation were assessed in clonogenic assays in methylcellulose. Data are expressed as percent control of CFU-L colony numbers for control siRNA-treated cells and represent means ± SE of 4 independent experiments. Paired t test analysis comparing AS2O3–treated control siRNA transfected cells versus AS2O3–treated Atg7 siRNA transfected cells or versus AS2O3–treated Beclin1 siRNA transfected cells or versus AS2O3–treated p62/SQSMT1 siRNA transfected cells and the corresponding paired P values are indicated.
Interaction of BCR-ABL with p62/SQSTM1
In subsequent studies, we sought to uncover potential mechanisms by which autophagy mediates arsenic-dependent BCR-ABL degradation. We examined whether BCR-ABL directly interacts with key elements of the autophagic machinery. Initially, we assessed whether BCR-ABL interacts with the ubiquitin-associated protein p62/SQSTM1. K562 cells were exposed to As2O3 for different times ranging from 8 to 48 hours and lysates were immunoprecipitated with an anti–anti-p62/SQSTM1 antibody and immunoblotted with an anti-ABL antibody to detect BCR-ABL (Figure 3A). There was arsenic-inducible time-dependent increase in the interaction between BCR-ABL and p62/SQSTM1. Such arsenic-inducible BCR-ABL–p62/SQSTM1 interaction was also seen in confocal imaging studies, including colocalization of this complex with lysosome targeting conjugates (Figure 3B, supplemental Figures 4-5). The interaction of p62/SQSTM1 with BCR-ABL was also detectable within autophagosome structures (Figure 3C) in immunogold electron microscopy studies. Collectively these findings physically linked BCR-ABL to p62/SQSTM1 in autophagosome structures and autolysosomes.
Figure 3.
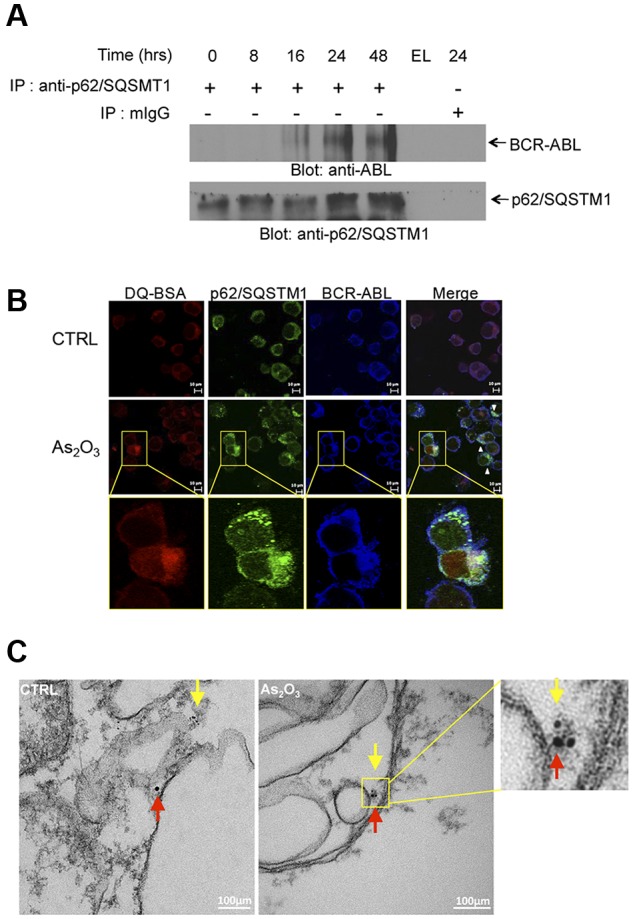
Lysosomal colocalization of BCR-ABL and p62/SQSTM1. (A) K562 cells were incubated in the presence of As2O3 for the indicated times. Cells were lysed and lysates were immunoprecipitated (IP) with an anti-p62/SQSTM1 antibody or control normal mouse IgG (mIgG), as indicated. EL indicates an empty lane. Immunoprecipitated proteins were resolved by SDS-PAGE and immunoblotted with anti-ABL or anti-p62/SQSTM1 antibodies, as indicate. (B) K562 cells were treated with As2O3 (2μM) for 24 hours. Before collection, cells were stained with quenched probe DQ-BSA (red), and after collection stained with either anti-ABL (blue) or anti-p62/SQSTM1 (green) and signals were detected by confocal microscopy. Merged panels indicate overlapping images of the 3 fluorescing signals, and arrows show the colocalization of p62/SQSTM1, BCR-ABL and lysosomal probe DQ-BSA. (C) As2O3–dependent colocalization of BCR-ABL with p62/SQSTM1, detected by electron microscopy. K562 cells were treated for 16 hours with AS2O3 (2μM) and were subsequently processed as described in “Electron microscopy.” Red arrows indicate BCR-ABL (15 nm gold-conjugate) and yellow arrows indicate p62 (6 nm gold-conjugate).
Requirement of Cathepsin B activity for autophagic degradation of BCR-ABL
Lysosomes are organelles that contain enzymes, and among the enzymes present in their structures are cathepsins.32 Cathepsin B has been implicated in autophagic flux-facilitation of the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm33 and in imatinib mesylate-induced CML cell death.34 This prompted us to determine whether cathepsin B activity plays a role in degradation BCR-ABL by arsenic. In initial studies, we found that cathepsin activity products colocalize with BCR-ABL and p62/SQSTM1 (Figure 4A). Interestingly in untreated samples cathepsin activity products appeared more locally concentrated in the cells, whereas after As2O3-treatment a more diffuse pattern of activity was seen throughout the cell, possibly reflecting autophagolysosome maturation (Figure 4A). Importantly, when cathepsin B activity was pharmacologically inhibited using a specific pharmacologic inhibitor (CA-074), we found that there was no longer colocalization of cathepsin products with BCR-ABL and the diffuse pattern of cathepsin activity products seen after arsenic treatment was reversed (Figure 4B).
Figure 4.

Cathepsin B activity is required for arsenic-induced autophagic degradation of BCR-ABL. (A) K562 cells were treated with AS2O3 (2μM) for 24 hours. The cells were then stained with a pan-cathepsin probe (Prosense 680; red), and aftercollection stained with either anti-ABL (blue) or anti-p62/SQSTM1 (green), and signals were detected by confocal microscopy. Merged panels indicate overlapping images of the 3 fluorescing signals, and magnified sections indicate colocalization of p62/SQSTM1, BCR-ABL, and cathepsins. (B) K562 cells were treated with or without As2O3 (2μM) or CA-074 (5μM) for 24 hours. The cells were then stained with quenched probe DQ-BSA (red), and after collection stained with either anti-ABL (blue), or anti-p62/SQSTM1 (red), and signals were detected by confocal microscopy. Merged panels indicate overlapping images of the 3 fluorescing signals, and arrows show the colocalization of p62/SQSTM1, BCR-ABL, and lysosomal probe DQ-BSA. (C left panel) Lysates from K562 cells stably expressing control (Ctrl)–shRNA or cathepsin B-shRNA where incubated with the biotinylated probe DCG-04 and, after resolution by SDS-PAGE, were immunoblotted with anti-streptavidin or anti-Hsp90 antibodies, as indicated. (Right panel) K562 cells stably expressing Ctrl-shRNA or Cathepsin B shRNA were treated with As2O3 (2μM) for 24 hours. Total lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (D left panel) K562 cells were incubated in the presence or absence of As2O3 (2μM) and/or CA-074 (5μM) for 24 hours. Cell lysates where incubated with the biotinylated probe DCG-04, resolved by SDS-PAGE and immunoblotted with anti-streptavidin or anti-Hsp90, as indicated. (Right panel) K562 cells were incubated in the presence or absence of As2O3 (2μM) and/or CA-074 (5μM) for 24 hours. Cell lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (E) K562 cells were plated in a methylcellulose assay system in the presence of either As2O3 (0.5μM) and/or CA-074 (5μM) as indicated. Data are expressed as percent control of CFU-L colony numbers for control untreated cells and represent means ± SE of 4 independent experiments. Paired t test analysis comparing the effects As2O3 in the absence or presence of CA-074 combination showed a paired P value = .0013. (F) K562 cells stably expressing control (CTRL)–shRNA or cathepsin B (CTSB)–shRNA were plated in a methylcellulose assay system in the presence of either AS2O3 (0.5μM) as indicated. Data are expressed as percent control of CFU-L colony numbers for control untreated cells and represent means ± SE of 4 independent experiments. Paired t test analysis comparing the effects AS2O3 showed a paired P value = .0249.
To determine the functional role of cathepsin B activity in arsenic-induced autophagic degradation of BCR-ABL, we stably knocked down cathepsin B in K562 cells using specific shRNA in a lentiviral vector (Figure 4C). Knockdown of cathepsin B stabilized BCR-ABL protein levels and reversed arsenic-mediated degradation of the oncoprotein (Figure 4C). These results were further verified by use of the specific and potent inhibitor of cathepsin B activity, CA-074. As shown in Figure 4D, concomitant treatment with CA-074 selectively blocked cathepsin B activity and reversed arsenic-induced autophagic degradation of BCR-ABL (Figure 4D). Inhibition of cathepsin B also partially reversed the frequency of autophagic cells (supplemental Figure 6). To assess the role of cathepsin B activity in the antileukemic properties of As2O3 we examined the effects of inhibition of cathepsin B activity on the suppressive effects of As2O3 on leukemic CFU-L progenitors. Concomitant treatment of cells with CA-074 partially reversed the effects of As2O3 on CFU-L growth (Figure 4E), indicating a key involvement of cathepsin B in the generation of antileukemic responses. Similar results were seen in studies in which the effects of stable knockdown of cathepsin B on K562-derived CFU-L leukemic colonies were assessed (Figure 4F).
Autophagy and the antileukemic effects of As2O3 on primitive leukemic progenitors
To further determine the significance of arsenic-induced autophagy and associated BCR-ABL degradation in a more pathophysiologically relevant system, studies were performed to define its role on primary leukemic precursors from patients with CML. Consistent with the cell line findings, using peripheral blood mononuclear cells isolated from a CML patient we observed colocalization of BCR-ABL with p62/SQSTM1 in an As2O3–dependent manner (Figure 5A). Treatment with pepstatin A and E64d (Figure 5B) or with the cathepsin B inhibitor CA-074 (Figure 5C) partially reversed the suppressive effects of arsenic on primary leukemic CFU-GM progenitors from CML patients, establishing a critical and essential role of autophagic degradation of BCR-ABL in the antileukemic properties of As2O3.
Figure 5.
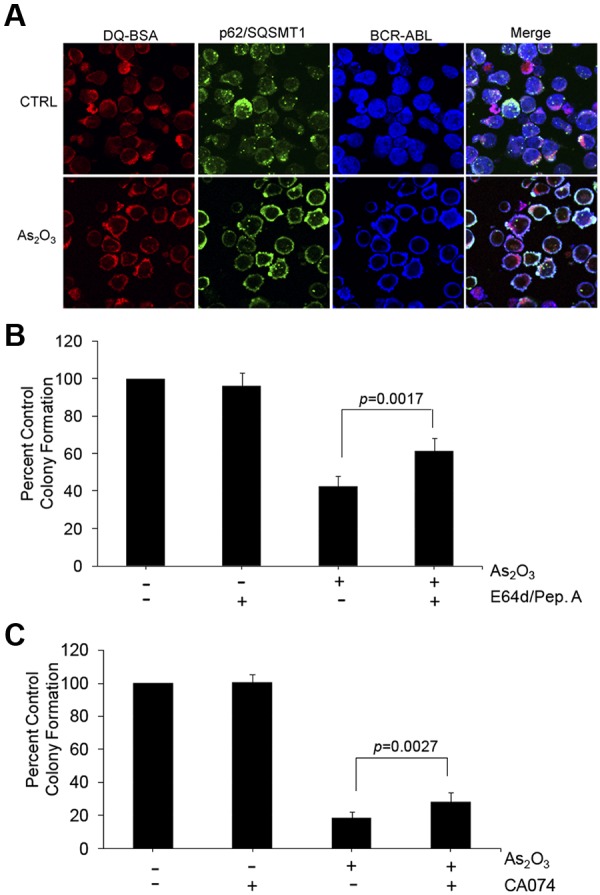
Autophagic degradation of BCR-ABL contributes to the generation of the antileukemic effects of As2O3 on primitive leukemic progenitors from CML patients. (A) Circulating leukemia cells from a CML patient were treated with As2O3 (2μM) for 24 hours. Before collection, cells were stained with quenched probe DQ-BSA (red), and after collection stained with either anti-ABL (blue) or anti-p62/SQSTM1 (green) as indicated, and signals were detected by confocal microscopy. Merged panels indicate overlapping images of the 3 fluorescing signals, and arrows show the colocalization of p62/SQSTM1, BCR-ABL, and lysosomal probe DQ-BSA. (B) Effects of As2O3 (0.5μM) or E64d/Pepstatin (10μM/10μM) or the indicated combinations on primitive leukemic progenitor (CFU-GM) colony formation from different CML patients were examined in clonogenic assays in methylcellulose. Data are expressed as percent control of CFU-GM colony numbers for control untreated cells and represent means ± SE of 6 independent experiments using samples from different patients. Paired t test analysis comparing the effects of As2O3 in the absence or presence of E64d/pepstatin demonstrated a paired value of P = .0017. (C) Effects of As2O3 (0.5μM) or CA-074 or the indicated combinations on primitive leukemic progenitor (CFU-GM) colony formation from different CML patients were examined in clonogenic assays in methylcellulose. Data are expressed as percent control of CFU-GM colony numbers for control untreated cells and represent means ± SE of 4 independent experiments using samples from different patients. Paired t test analysis comparing the effects of As2O3 in the absence or presence of E64d/pepstatin demonstrated a paired value of P = .0027.
Discussion
As2O3 is one of the most active agents in the treatment of APL16–18 and it is approved by the FDA for the treatment of patients suffering from this leukemia. There is also emerging evidence that this agent can target and eliminate LICs,19 making this metalloid derivative attractive for other hematologic malignancies and solid tumors. However, limitations in our overall understanding of its mechanisms of action have so far not allowed development of targeting approaches and the therapeutic combinations that would broaden its impact in non-APL malignancies. Extensive work has established that As2O3–dependent apoptosis occurs, in part, via induction of reactive oxygen species (ROS), which initiate downstream proapoptotic pathways.16,17,35–37 However, there is also evidence that ROS-independent apoptosis plays important roles38 and that arsenic can promote cell death in a caspase-independent manner.39 Cell-type specific targets of AS2O3 exist, including the PML-RAR fusion protein in APL cells40,41; the AML1/MDS1/EVI1 oncoprotein, which is present in the malignant cells of some patients with MDS, AML, or CML blast crisis42; and the BCR-ABL oncoprotein, which is the hallmark of CML.20 Targeting and degradation of such transforming oncoproteins by As2O3 is highly relevant and may be critical for the ability of arsenic to generate antileukemic responses, but the mechanisms by which such cellular events are initiated and regulated remain to be precisely defined.
We have recently shown that As2O3 induces autophagy of AML cells and demonstrated that such autophagy plays essential roles in the induction of its suppressive effects on primitive leukemic precursors from AML patients.22 Our previous studies have also raised the potential of modulating the autophagic machinery to promote arsenic-induced antileukemic effects, but the role of autophagy in specific arsenic-dependent targeting mechanisms in malignant cells remain to be defined. Notably, autophagy-modulating agents have become recently the focus of clinical-translational efforts,43 but the underlying mechanisms connecting autophagy and cancer remain unclear.
In this study, we demonstrate that degradation of the transforming oncoprotein BCR-ABL by arsenic trioxide occurs in autolysosomes in a cathepsin B-dependent manner. The localization of BCR-ABL to the autolysosome appears to occur via an As2O3–dependent interaction of BCR-ABL with p62/SQSTM1, ultimately resulting in proteolytic degradation of BCR-ABL. p62/SQSTM1, a ubiquitin binding protein, has been previously implicated in shuttling of ubiquitinated proteins and functions as a “cargo receptor” for autophagic degradation of targeted proteins.44–46 In fact, there is evidence that p62/SQSTM1 binds directly to LC3 to facilitate degradation of ubiquitinated protein targets,46 underscoring the importance of this protein in the process. Previous work has also shown that BCR-ABL can be ubiquitinated under certain circumstances47,48 suggesting a mechanism by which the p62/SQSTM1–BCR-ABL interaction occurs to target the oncoprotein to the autolysosomes. Our data firmly establish that the autophagic process is essential for selective targeting of BCR-ABL by arsenic, as demonstrated in studies in which selective knockdown of key elements of the autophagic machinery, such Atg7, Beclin1, or p62/SQSTM1 results in reversal of BCR-ABL degradation. Atg7 and Beclin1 play crucial roles in the early stages of autophagy and particularly during the formation of the autophagosome membrane structures. Importantly, our data demonstrate that inhibition of autophagic machinery also results in reversal of the suppressive properties of As2O3 on leukemic progenitors from patients with CML, indicating a critical and essential role for autophagy in the induction of arsenic responses in BCR-ABL–transformed leukemic precursors. Beyond reversal by targeting the autophagic machinery, such inhibitory effects on CML precursors are also reversible by pharmacologic inhibition of cathepsin B establishing that autolysosomal degradation the BCR-ABL oncoprotein is essential for this process. A proposed model for the sequence of events leading to autophagic degradation of the BCR-ABL oncoprotein in leukemia cells is shown in Figure 6.
Figure 6.
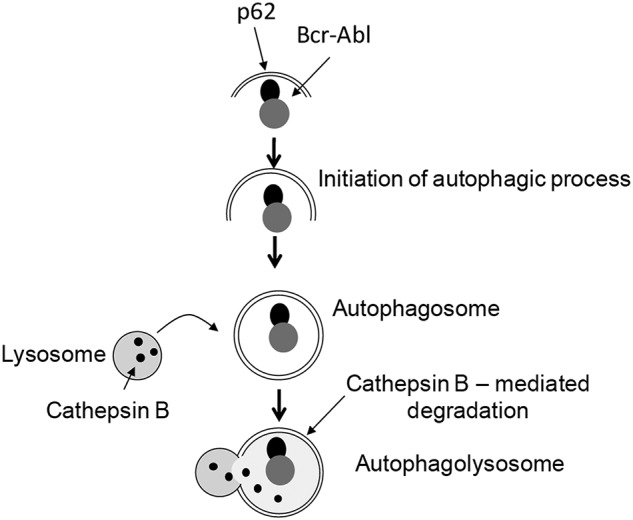
BCR-ABL degradation via the autophagic pathway. Proposed model for As2O3–dependent interaction of p62/SQSMT1 with BCR-ABL and subsequent autophagic degradation of BCR-ABL.
It remains to be determined whether cathepsin B-mediated autophagic degradation of other oncoproteins, such as PML-RARα,40,41 AML1/MDS1/EVI1,42 accounts for arsenic-induced antileukemic responses in different types of neoplastic cells. A recent study demonstrated that the autophagic process is involved in degradation of PML-RARα in response to all-trans retinoic acid (ATRA) or AS2O3 in the NB4 APL cell line and that induction of autophagy correlates with differentiation of APL cells.49 This raises the possibility that cathepsin B–mediated autolysosomal degradation of PML-RARα and potentially other targets may occur via a similar mechanism to the one we observed here in the case of BCR-ABL. Interestingly, there is also recent evidence that ATRA-induced myeloid differentiation is mediated by p62/SQSTM1–mediated degradation of PML-RARα,50 although the elements of the enzymatic process were not identified in that study.
The exact role of lysosomal enzymes in the autophagic process has been relatively unknown. Because cysteine cathepsins are broadly up-regulated in cancer, there has been an interest to pharmacologically target them for the treatment of malignancies.51 A notable target among these proteases is cathepsin B, a protease implicated in apoptosis via its cleavage-activating capacity aimed at Bcl-2 family members, resulting in disruption of the mitochondrial membrane and cytochrome c release.52 Before our study no equivalent downstream substrate for cathepsin B in the autophagy process had been identified. However, the Bcl-2 protein family is known to facilitate crosstalk between apoptosis and autophagy,53,54 raising the possibility for a role for cathepsins in cellular autophagy. In fact, cathepsin B is necessary for autophagic flux that promotes delivery of anthrax lethal factor into the cytoplasm33 and our data further implicate cathepsin B in autophagy-mediated destruction of targeted proteins.
Beyond providing a model by which As2O3 promotes elimination of BCR-ABL via autophagic degradation, the results of this work may ultimately prove to have important clinical-translational therapeutic implications. It remains to be determined whether rational targeting of other nonoverlapping cellular cascades may allow further potentiation of arsenic-induced autophagic degradation of BCR-ABL and other oncogenes that may be targeted by arsenic. For instance, it has been recently shown that pharmacologic inhibition of the PTP1B phosphatase promotes ubiquitination of BCR-ABL.48 Taken together with our studies, this raises the possibility that combinations of PTP1B inhibitors with AS2O3 may further promote formation of p62/SQSTM1–BCR-ABL complexes and their localization to autolysosomes. Other studies have shown that the flavonoid resveratrol up-regulates p62/SQSTM1 expression in a JNK-dependent manner and promotes autophagic cell death in BCR-ABL cells,55 suggesting that AS2O3-resveratrol combinations may result in enhancing effects and provide a novel therapeutic approach for the treatment of BCR-ABL malignancies. Such approaches may be particularly relevant and important in attempts to eliminate LICs, as there is already evidence that arsenic can target and eliminate leukemic stem cells in CML.19 On the other hand, the major BCR-ABL kinase inhibitor currently used in the treatment of CML, imatinib mesylate, does not eliminate LICs despite inhibition of BCR-ABL kinase activity.56 Thus, efforts to enhance arsenic-induced autophagic degradation of BCR-ABL in LICs may ultimately complement the use of kinase inhibitors in the treatment of CML and decrease the number of relapses seen after long-term responses. Similar translational approaches may be applicable in other leukemias with different molecular abnormalities and studies in that direction are warranted.
Supplementary Material
Acknowledgments
The authors thank Dr Brian Druker (Knight Cancer Institute, Oregon Health & Science University, Portland, OR) for providing the Ba/F3 transfectants.
This work was supported by National Institutes of Health R01 grants CA121192, CA77816, and CA155566; and by a Merit Review grant from the Department of Veterans affairs and by Leukemia and Lymphoma Society of America (LLS-6166-09).
Footnotes
There is an Inside Blood commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.J.G. designed and performed research, analyzed data, and wrote the paper; E.G. designed and performed research and analyzed data; E.J.W., E.V., B.S., and J.K.A. performed research and analyzed data; M.B. analyzed data and edited the paper; and L.C.P. conceived and designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Leonidas C. Platanias, Robert H. Lurie Comprehensive Cancer Center, 303 E Superior St, Lurie 3-107, Chicago, IL 60611; e-mail: l-platanias@northwestern.edu.
References
- 1.Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793(4):664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Kirisako T, Baba M, Ishihara N, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147(2):435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lucocq J, Walker D. Evidence for fusion between multilamellar endosomes and autophagosomes in HeLa cells. Eur J Cell Biol. 1997;72(4):307–313. [PubMed] [Google Scholar]
- 6.Dunn WA., Jr Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol. 1990;110(6):1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Klionsky DJ. The regulation of autophagy: unanswered questions. J Cell Sci. 2011;124(Pt 2):161–170. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 9.Altman BJ, Jacobs SR, Mason EF, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene. 2011;30(16):1855–1867. doi: 10.1038/onc.2010.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119(5):1109–1123. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamitsuji Y, Kuroda J, Kimura S, et al. The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and different modes of cell death execution in Bcr-Abl-positive leukemias. Cell Death Differ. 2008;15(11):1712–1722. doi: 10.1038/cdd.2008.107. [DOI] [PubMed] [Google Scholar]
- 12.Carew JS, Nawrocki ST, Kahue CN, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110(1):313–322. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6(6):505–510. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 15.Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118(10):2840–2848. doi: 10.1182/blood-2010-12-322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62(14):3893–3903. [PubMed] [Google Scholar]
- 17.Emadi A, Gore SD. Arsenic trioxide: an old drug rediscovered. Blood Rev. 2010;24(4-5):191–199. doi: 10.1016/j.blre.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platanias LC. Biological responses to arsenic compounds. J Biol Chem. 2009;284(28):18583–18587. doi: 10.1074/jbc.R900003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito K, Bernardi R, Morotti A, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453(7198):1072–1078. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang QY, Mao JH, Liu P, et al. A systems biology understanding of the synergistic effects of arsenic sulfide and Imatinib in BCR/ABL-associated leukemia. Proc Natl Acad Sci U S A. 2009;106(9):3378–3383. doi: 10.1073/pnas.0813142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davison K, Mann KK, Waxman S, Miller WH., Jr JNK activation is a mediator of arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells. Blood. 2004;103(9):3496–3502. doi: 10.1182/blood-2003-05-1412. [DOI] [PubMed] [Google Scholar]
- 22.Goussetis DJ, Altman JK, Glaser H, McNeer JL, Tallman MS, Platanias LC. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J Biol Chem. 2010;285(39):29989–29997. doi: 10.1074/jbc.M109.090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao JH, Sun XY, Liu JX, et al. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2010;107(50):21683–21688. doi: 10.1073/pnas.1016311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kannan-Thulasiraman P, Katsoulidis E, Tallman MS, Arthur JS, Platanias LC. Activation of the mitogen- and stress-activated kinase 1 by arsenic trioxide. J Biol Chem. 2006;281(32):22446–22452. doi: 10.1074/jbc.M603111200. [DOI] [PubMed] [Google Scholar]
- 25.Dolniak B, Katsoulidis E, Carayol N, et al. Regulation of arsenic trioxide-induced cellular responses by Mnk1 and Mnk2. J Biol Chem. 2008;283(18):12034–12042. doi: 10.1074/jbc.M708816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altman JK, Yoon P, Katsoulidis E, et al. Regulatory effects of mammalian target of rapamycin-mediated signals in the generation of arsenic trioxide responses. J Biol Chem. 2008;283(4):1992–2001. doi: 10.1074/jbc.M705227200. [DOI] [PubMed] [Google Scholar]
- 27.Kroczynska B, Kaur S, Katsoulidis E, et al. Interferon-dependent engagement of eukaryotic initiation factor 4B via S6 kinase (S6K)- and ribosomal protein S6K-mediated signals. Mol Cell Biol. 2009;29(10):2865–2875. doi: 10.1128/MCB.01537-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paglin S, Hollister T, Delohery T, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61(2):439–444. [PubMed] [Google Scholar]
- 29.Kanzawa T, Kondo Y, Ito H, Kondo S, et al. Induction of Autophagic Cell Death in Malignant Glioma Cells by Arsenic Trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 30.McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem. 1974;22(12):1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 31.Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009;452:143–164. doi: 10.1016/S0076-6879(08)03610-0. [DOI] [PubMed] [Google Scholar]
- 32.Tardy C, Codogno P, Autefage H, Levade T, Andrieu-Abadie N. Lysosomes and lysosomal proteins in cancer cell death (new players of an old struggle). Biochim Biophys Acta. 2006;1765(2):101–125. doi: 10.1016/j.bbcan.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Ha SD, Ham B, Mogridge J, Saftig P, Lin S, Kim SO. Cathepsin B-mediated autophagy flux facilitates the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm. J Biol Chem. 2010;285(3):2120–2129. doi: 10.1074/jbc.M109.065813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puissant A, Colosetti P, Robert G, Cassuto JP, Raynaud S, Auberger P. Cathepsin B release after imatinib-mediated lysosomal membrane permeabilization triggers BCR-ABL cleavage and elimination of chronic myelogenous leukemia cells. Leukemia. 2010;24(1):115–124. doi: 10.1038/leu.2009.233. [DOI] [PubMed] [Google Scholar]
- 35.Litzow MR. Arsenic trioxide. Expert Opin Pharmacother. 2008;9(10):1773–1785. doi: 10.1517/14656566.9.10.1773. [DOI] [PubMed] [Google Scholar]
- 36.Ralph SJ. Arsenic-based antineoplastic drugs and their mechanisms of action. Met Based Drugs. 2008;2008:260146. doi: 10.1155/2008/260146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumagai Y, Sumi D. Arsenic: signal transduction, transcription factor, and biotransformation involved in cellular response and toxicity. Annu Rev Pharmacol Toxicol. 2007;47:243–262. doi: 10.1146/annurev.pharmtox.47.120505.105144. [DOI] [PubMed] [Google Scholar]
- 38.Morales AA, Gutman D, Cejas PJ, Lee KP, Boise LH. Reactive oxygen species are not required for an arsenic trioxide-induced antioxidant response or apoptosis. J Biol Chem. 2009;284(19):12886–12895. doi: 10.1074/jbc.M806546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen GQ, Zhu J, Shi XG, et al. In vitro studies on cellular and molecular mechanisms of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia: As2O3 induces NB4 cell apoptosis with down-regulation of Bcl-2 expression and modulation of PML-RAR alpha/PML proteins. Blood. 1996;88(3):1052–1061. [PubMed] [Google Scholar]
- 40.McCafferty-Grad J, Bahlis NJ, Krett N, et al. Arsenic trioxide uses caspase-dependent and caspase-independent death pathways in myeloma cells. Mol Cancer Ther. 2003;2(11):1155–1164. [PubMed] [Google Scholar]
- 41.Shao W, Fanelli M, Ferrara FF, et al. Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer Inst. 1998;90(2):124–133. doi: 10.1093/jnci/90.2.124. [DOI] [PubMed] [Google Scholar]
- 42.Shackelford D, Kenific C, Blusztajn A, Waxman S, Ren R. Targeted degradation of the AML1/MDS1/EVI1 oncoprotein by arsenic trioxide. Cancer Res. 2006;66(23):11360–11369. doi: 10.1158/0008-5472.CAN-06-1774. [DOI] [PubMed] [Google Scholar]
- 43.Garber K. Inducing indigestion: companies embrace autophagy inhibitors. J Natl Cancer Inst. 2011;103(9):708–710. doi: 10.1093/jnci/djr168. [DOI] [PubMed] [Google Scholar]
- 44.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 45.Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8(13):1986–1990. doi: 10.4161/cc.8.13.8892. [DOI] [PubMed] [Google Scholar]
- 46.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 47.Sun H, Kapuria V, Peterson LF, et al. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011;117(11):3151–3162. doi: 10.1182/blood-2010-03-276477. [DOI] [PubMed] [Google Scholar]
- 48.Alvira D, Naughton R, Bhatt L, Tedesco S, Landry WD, Cotter TG. Inhibition of protein-tyrosine phosphatase 1B (PTP1B) mediates ubiquitination and degradation of Bcr-Abl protein. J Biol Chem. 2011;286(37):32313–32323. doi: 10.1074/jbc.M111.249060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116(13):2324–2331. doi: 10.1182/blood-2010-01-261040. [DOI] [PubMed] [Google Scholar]
- 50.Wang Z, Cao L, Kang R, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARalpha oncoprotein. Autophagy. 2011;7(4):401–411. doi: 10.4161/auto.7.4.14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci. 2008;29(1):22–28. doi: 10.1016/j.tips.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 52.Malla R, Gopinath S, Alapati K, et al. Downregulation of uPAR and cathepsin B induces apoptosis via regulation of Bcl-2 and Bax and inhibition of the PI3K/Akt pathway in gliomas. PLoS One. 2010;5(10):e13731. doi: 10.1371/journal.pone.0013731. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoka V, Turk B, Schendel SL, et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem. 2001;276(5):3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 55.Puissant A, Robert G, Fenouille N, et al. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70(3):1042–1052. doi: 10.1158/0008-5472.CAN-09-3537. [DOI] [PubMed] [Google Scholar]
- 56.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.