

Abstract
Two synthetic approaches to psymberin have been accomplished. A highly convergent first generation synthesis led to the complete stereochemical assignment and demonstrated that psymberin and irciniastatin A are identical compounds. This synthesis featured a diastereoselective aldol coupling between the aryl fragment and a central tetrahydropyran core, and a novel one-pot procedure to convert an amide, via intermediacy of a sensitive methyl imidate, to the N-acyl aminal reminiscent of psymberin. The highlights of the second generation synthesis include an efficient iridium-catalyzed enantioselective bis-allylation of neopentyl glycol, and a stepwise Sonogashira coupling/cycloisomerization/reduction sequence to construct the dihydroisocoumarin unit. The two synthetic avenues were achieved in 17–18 steps (longest linear sequence, ~14–15 isolations) from 3 fragments prepared in 7–8 steps (1st generation) and 3–8 steps (2nd generation) each. This convergent approach allowed for the preparation of sufficient amounts of psymberin (~ 0.5 g) for follow-up biological studies. Meanwhile, our highly flexible strategy enabled the design and synthesis of multiple analogs, including a psymberin-pederin hybrid termed psympederin that proved crucial to a comprehensive understanding of the chemical biology of psymberin and related compounds that will be described in a subsequent manuscript.
INTRODUCTION
In 2004, Crews and Pettit independently reported the isolation of structurally novel, constitutionally identical cytotoxins. Psymberin (1) was obtained from the marine sponge Psammocinia sp.,1 whereas irciniastatin A (2) was isolated from Irciniaramosa sp.2 Multidimensional NMR studies substantiated the assigned relative configuration for psymberin as shown in 1, save for the undefined configuration at C4. The absolute configuration was assured through observation of a well-defined positive Cotton effect at the n → Π* transition (280 nm) of the dihydroisocoumarin unit. The relative stereochemistry of irciniastatin A (2) was only resolved for the C8–C13 aminal fragment. Given the differing relative configuration (C8–C9) and producing organisms, the structures formulated for irciniastatin and psymberin thus define two different natural products. The overall structural features of these natural products most closely resemble those of the pederin family of natural products including pederin (3),3 and mycalamide A (4)4 (Fig. 1).5 However, psymberin is uniquely extended with a dihydroisocoumarin unit not found in any of the other >36 members of the pederin family isolated to date, and lacks this family’s signature acetal-containing pederate side chain.5c
Figure 1.

Psymberin and other representative natural products of the pederin family.
Pederin, mycalamide and other members of the family are potent eukaryotic protein synthesis inhibitors and cytotoxic agents, which exhibit strong blistering activity upon contact with the skin.5 An indication that psymberin might be endowed with an alternative mode of action came from the observation that psymberin, unlike pederin and mycalamide, displayed a highly differential cytotoxicity profile with >10,000-fold potency differences in the NCI 60 cell human tumor cell line panel.1 In contrast, the material isolated by Pettit and coworkers did not exhibit this differential activity and uniformly inhibited the growth of a different selection of human cancer cell lines with single digit nanomolar potency. Irciniastatin also potently arrested the growth of human umbilical vein endothelial cells (HUVEC) with a GI50 of 0.5 nM with no evidence of tube formation, an indication it could be useful as an antivascular agent.2
Due to uncertainties regarding the structural relation between psymberin and irciniastatin, significant structural divergence from the pederin family of natural products, low natural abundance, and impressive biological activities, psymberin/irciniastatin has become an attractive target for synthetic pursuit. Through the total synthesis of several diastereoisomers consistent with partially assigned structures of 1 and 2, our group concluded that psymberin and irciniastatin are actually identical compounds.6 Several other total syntheses,7 formal syntheses,8 fragment syntheses,9 as well as analog syntheses, 10 have appeared during recent years. A combined supply of natural psymberin and material prepared by our group enabled further in vivo evaluation by the NCI Developmental Therapeutics Program, which indicated encouraging therapeutic efficacy.11 Additionally, our synthetic psymberin was explored as an antibody drug conjugate in collaboration with Seattle Genetics.12 In a quest to discover the mode of action, and an interest in a more comprehensive preclinical evaluation of psymberin, we have continued our study of this fascinating natural product. In this paper, we describe a full account of the total synthesis, an improved second generation total synthesis, and the synthesis of strategically designed analogs of psymberin. In a subsequent article, we detail our biological investigations that led to the target identification of psymberin, and attributes that distinguish it from the pederin/mycalamide family of natural products.13
RESULTS AND DISCUSSION
First Generation Synthesis
Psymberin (1) is a complex polyketide comprising nine stereocenters, a geminal dimethyl, and a dihydroisocoumarin fragment. Its tetrahydropyranyl core is appended with a 2-hydroxy-3-methoxy-5-methyl-hex-5-enoic acid (psymberic acid) through an N-acylaminal linkage. As outlined in scheme 1, we anticipated to intercept an intermediate N-acyl-methoxyimidate 5 with a reducing agent to provide the C8-S and C8-R N-acyl aminals corresponding to the assigned structures of psymberin and irciniastatin, respectively. Given the unknown configuration at C4, intermediate 5 would result from acylation of imidate 7, to be prepared from the corresponding primary amide, with diastereomeric acid chlorides 6a or 6b. Both epimeric acid chlorides in turn will be accessible from a common intermediate to be derived from D-mannitol via a stereocontrolled methallylation. Our approach to 7 hinged on combing arylaldehyde 9, to be derived from commercially available 2,4-dimethoxy-1-methylbenzene (10) and ethyl ketone 8 via a substrate-controlled aldol coupling to set the correct stereochemistry of the C15-C17 stereotriad. Ethyl ketone 8 was envisioned to be derived from aldehyde 11, in turn accessible through an oxidative cleavage of C2-symmetric bis-homoallyl alcohol 12.
Scheme 1.

Synthetic Plan.
Given the unknown configuration at C4, we needed to prepare both diastereomers of the psymberic acid side chain (cf. 6a/6b, Scheme 1). We initially approached this problem starting from known triol 13, prepared in two steps from D-tartaric acid (Scheme 2, panel A).14 Oxidative diol-cleavage of this material, 15 followed by treatment of the crude aldehyde 14 delivered an inseparable mixture of homoallylic alcohols. Selective protection of the primary alcohol and methylation of the secondary alcohol provided compounds 15a and 15b, which could be separated at this stage (ratio = ~1:1.4). Independent processing via desilylation and a two-step oxidation then provided protected psymberic acids 16a and 16b. In panel B, we delineate a stereoselective gram-scale synthesis of benzoyl-protected psymberic acids 20a and 20b. Starting from protected glyceraldehyde 17, available via a one-step oxidation from D-mannitol,16 asymmetric methallylation with a borane reagent derived from isobutenyllithium and (−)-Ipc2BOMe,17 followed by methylation delivered methyl ether 18 in 65% yield (2 steps) and 97:3 dr, a significant improvement over the 4:3 ratio obtained by Williams using the corresponding Grignard reagent.9a Acetonide hydrolysis, silylation of the primary and benzoylation of the secondary alcohol, followed by an acidic aqueous work-up provided 6.6 g of alcohol 19 over a three steps sequence from 18. Finally, a two-step oxidation procedure yielded gram quantities of psymberic acid 20a (7 steps from aldehyde 17; 47% yield). The relative stereochemistry set during the methallylation step was ascertained through 1H-NMR analysis of acetal 21, obtained via ozonolysis of the terminal olefin 19, followed by acetal formation (→ 21). Diastereomeric acid 20b was synthesized as outlined for acid 20a, except that methallylation of aldehyde 17 exploited the antipodal borane reagent.
Scheme 2. Synthesis of Psymberic Acidsa.

a Reagents and conditions: Panel A: (a) NaIO4, CH2Cl2, aq. NaHCO3; (b) CH2CCH3CH2MgBr, THF, −78 °C; (c) TBDPSCl, imidazole, DMF; (d) KH, MeI, THF, 77% (4 steps); (e) TBAF, THF, 85%; (f) DMP, CH2Cl2, 95%; (g) NaH2PO4, NaClO2, 2-methyl-2-butene, t-BuOH/H2O, 99%. Panel B: (a) (−)-(Ipc)2BOMe ((+)-(Ipc)2BOMe for 20b), CH2CMeCH2Li, Et2O, −78 °C, 69%; (b) NaH, MeI, THF, 95%; (c) PPTS, MeOH/H2O, 50 °C, 93%; (d) TBSCl, imidazole, CH2Cl2, 95%; (e) BzCl, py; then aq. 3 N HCl, 95%; (f) DMP, CH2Cl2; (g) NaH2PO4, NaClO2, 2-methyl-2-butene, tBuOH/H2O, 87% over two steps; (h) O3, CH2Cl2; Me2S; (i) TsOH, CH(OMe)3, MeOH, 80% over two steps. Abbreviations: DMP, Dess-Martin Periodinane; Ipc, isopinocampheyl.
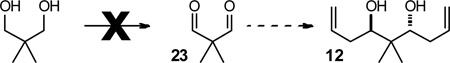
As we will detail later, our approach towards the central tetrahydropyranyl core of psymberin was inspired by the possibility to engage C2-symmetrical bis-homoallyl alcohol 12 in an oxidative desymmetrization reaction. On paper, this compound should be available from an enantioselective bis-allylation of dialdehyde 23 (Eq. 1).18 A practical problem arises when one considers that malondialdehydes are unstable, sensitive to hydrate formation, self-condensation and oligomerization. In fact, all attempts to generate pure dialdehyde 23 from neopentyl glycol met with failure. In light of this, we originally settled for a slightly longer approach starting from isobutyraldehyde (24, Scheme 3). According to a known literature procedure,19 this material was converted in three steps to 3,3-dimethoxy-2,2-dimethylpropanal 25, a mono-protected version of the corresponding malondialdehyde 23. An enantioselective allylation of aldehyde 25 using Leighton’s silane reagent 27 provided homoallyl alcohol 26 in 94% ee 20 Noteworthy, the dimethyl acetal was unmasked during the work-up conditions, enabling for a subsequent allylation under the same reaction conditions. The corresponding bishomoallyl alcohol 12 was thus obtained in 77% yield and >17:1 dr. Since our original route to this compound,6 Krische and coworkers developed a creative two-directional carbonyl allylation from the alcohol oxidation level to circumvent the use of difficult to handle malondialdehydes.21 As reported by Krische, treatment of diol 29 with allyl acetate employing a cyclometallated catalyst formed in situ from [Ir(cod)Cl]2, (R )-Cl-MeO-BIPHEP 28, 4-chloro-3-nitrobenzoic acid and Cs2CO3 in degassed dioxane, furnished diol 12 in 42% yield (99% ee , 20:1 dr ) on a 4 gram scale. A mono-allylated intermediate was also isolated (not shown) in 24–30% yield. This material could be allylated under the same reaction conditions to obtain additional bis-allylated product 12 in 30% yield (51% combined yield from 29).
Scheme 3.

Synthesis of Bis-Homoallyl Alcohol 12.
 |
(1) |
At this point, we were hopeful that we could differentiate the termini of diene 12 via an oxidative cleavage of the terminal olefins, after which one of the resulting aldehydes would be trapped as a lactol. As shown in Scheme 4, this concept was best put to practice after mono-protection of diol 12 (silyl or benzyl) followed by ozonolytic cleavage of both double bonds to yield lactols 30a (92%) and 30b (76%), respectively. At this point, we needed to homologate the aldehyde to an ethyl ketone, and activate the lactol for introduction of the cyano group as a masked primary amide. In the event, acylation of the benzylated lactol 30b provided acetate 11b in 60% yield. Under the reaction conditions, dioxabicyclononane byproduct 31 was formed in 30% yield. Lewis acid-catalyzed acetate displacement with trimethylsilyl cyanide22 yielded cyano acetal 32 wherein the aldehyde was trapped as a cyanohydrin (60% yield).23 Hydrolysis of the cyanohydrin was more difficult than anticipated and provided aldehyde 33 in modest yields (31–36%). Treatment of this material with diethylzinc in the presence of a diaminocyclohexane-ligated titanium(IV) catalyst24 was followed by Dess-Martin oxidation to 25 yield the corresponding ethyl ketone 8b in 80% yield for this two-step procedure. In order to avoid cyanohydrin formation, we changed the order of events and first employed the diethylzinc addition to aldehyde 11b yielding carbinol 34 in 87% yield. Unfortunately, cyanide displacement of acetate 34 was now accompanied by the formation of dioxabicyclononane byproduct 36, resulting from intramolecular acetate displacement with the secondary alcohol. Preventing alcohol participation via oxidation of 34 to ethyl ketone 37, obtained in 88% yield, now led to dehydrative dioxabicyclononane formation upon treatment with trimethylsilyl cyanide22 and boron trifluoride etherate to yield bicycle 38 as the major product isolated in 47% yield, together with 14% of the desired ethyl ketone 8b.
Scheme 4. Synthesis of Ethyl Ketone 8 Through Desymmetrization of Bis-homoallylic Alcohol 12.a.

a Reagents and Conditions: (a) TBSOTf, 2,6-lutidine, CH2Cl2; (b) BnBr, NaH, THF; (c) O3, CH2Cl2; PPh3; (d) Ac2O, Et3N, DMAP, CH2Cl2; (e) TMSCN, BF3·Et2O, MeCN; (f) aq. NaOH, Et2O; (g) Ti(OiPr)4, N,N’-(1R,2R-cyclohexane-1,2-diyl)bis(trifluoromethanesulfonamide), Et2Zn, PhMe; (h) DMP, CH2Cl2; (i) TMSCN, RT; then ZnI2, MeCN; then aq. 1 N HCl.
So far, we learned from the above synthetic exercises that activation of the anomeric acetate with boron trifluoride etherate not only enables cyanide introduction, but also leads to a facile formation of dioxabicyclononane and cyanohydrin byproducts in the presence of an aldehyde or ketone. Ultimately, we formulated a solution that started with the silylated lactol 30a (Scheme 4, bottom). Acylation of this material was not accompanied by dioxabicyclononane ring formation (cfr. 31), presumably because of the larger steric hindrance of the TBS protecting group. Ethyl ketone formation with diethylzinc using the Kobayashi24 conditions also proceeded with higher yield, providing ethyl carbinol 39 in 68% yield (2 steps). To avoid formation of potential bicyclic ring products, we modified the conditions for introduction of the cyano group. In a one-pot procedure, trimethylsilyl cyanide22 was added to alcohol 39 (neat) in the absence of Lewis acid to allow protection of the secondary alcohol as a silyl ether, followed by addition of a solution of zinc iodide in acetonitrile to initiate oxonium formation (40), followed by axial cyanide attack. After acidic aqueous workup, compound 41 was obtained in 91% yield and further oxidized to target ethyl ketone 8a in 95% yield. Crystallographic analysis of crystals obtained from 8a fully confirmed the assigned structure and relative stereochemistry. Using this optimized sequence, ethyl ketone 8a was obtained in seven steps from bis-homoallylic alcohol 12 in 54% overall yield, versus ~7% overall yield (6–7 steps) for the synthesis of the corresponding benzylated ethyl ketone 8b.
The synthesis of the aryl fragment 9 and coupling with ethyl ketone 8a is shown in Scheme 5. Starting from the commercially available 2,4-dimethoxy-1-methylbenzene 10, formylation, 26 oxidation to the corresponding carboxylic acid, and amidation afforded diethylamide 42 in 66% yield for the three-step sequence. Ortho-directed allylation27 of this material was followed by deprotection of the phenolic methyl ethers with boron tribromide. Formation of the methyl ester 43 was best executed according to a protocol developed by Keck and coworkers,28 i.e. treatment of the amide with trimethyloxonium tetrafluoroborate followed by aqueous hydrolysis of the incipient methylimidate. The overall yield for this three-step sequence was 45%. Subsequent protection of the phenols and a two-step oxidative cleavage of the double bond (dihydroxylation; diol-cleavage) delivered coupling partner aldehyde 9 in an eight-step sequence and 24% overall yield from commercially available starting material 10.
Scheme 5. Synthesis of Dihydroisocoumarin Fragments.a.

a Reagents and Conditions: (a) DMF, POCl3, 80%; (b) NaH2PO4, NaClO2, 2-methyl-2-butene, t-BuOH/H2O, 85%; (c) SOCl2, benzene; Et2NH, 97%; (d) sec-BuLi, CuBrSMe2, allylBr, THF, −78 °C, 76%; (e) BBr3, CH2Cl2, −78 to 25 °C, 81%; (f) Me3OBF4, CH2Cl2; Na2CO3, MeOH, 73%; (g) PMBCl, Bu4NI, K2CO3, DMF, 80 °C, 92%; (h) cat. OsO4, NMO, THF/H2O; NaIO4, aq MeOH, 81%; (i) PhBCl2, iPr2NEt, CH2Cl2, −78 °C, 88%, 12:1 dr ; (j) catecholborane, THF, 0 °C; (k) (MeO)MeCCH2, PPTS, 89% over two steps; (l) catecholborane, THF, 0 °C; aq 2 N NaOH, 99%; (m) TBAF, THF, 99%; (n) TBSOTf, 2,6-lutidine, CH2Cl2, 92%; (o) cat. [PtH(PMe2OH)(PMe2O)2H] (50), EtOH/H2O, 80 °C, 99%; (p) DDQ, CH2Cl2/H2O, 99%, (q) H2, Pd/C, EtOH, 95%, (r) BzCl, iPr2NEt, PhMe, 91%.
Having accessed the psymberin fragments in high yield and enantiomerically pure form enabled us to explore their union via a convergent coupling strategy. First, we needed to accomplish a stereoselective aldol coupling between ethyl ketone 8a and arylaldehyde 9. Although initial explorations with titanium enolates (TiCl4, iPr2NEt)29 only delivered mixtures of dia-stereoisomers (~1:1 ratio), we found that the Z-chlorophenylboryl enolate30 44 added to aldehyde 9 with high facial selectivity producing the syn-aldol product 45 in 88% yield and 12:1 dr.31 The stereochemical outcome was predicted based on the inherent facial bias of enolate 44 when combined with aldehyde 9 (Si-face) through a chair-like transition state.30 Stereoselective reduction of β-hydroxyketone 45 with catecholborane32 provided lactone 47 after basic workup (99%). Quenching the reducing mixture with aqueous Na,K-tartrate permitted isolation of the 1,3-syn-diol for derivatization as acetonide 46.33 1H an 13C NMR analysis of this derivative confirmed the 1,3-syn configuration.34 Silylation of the secondary alcohol (→ 49, 92%) set the stage for a mild nitrile hydrolysis exploiting a platinum(II)-catalyst (50) developed by Ghaffar and Parkins to deliver compound 51 in 99% yield.35 Treatment of this compound with excess DDQ only removed the ortho-phenolic p-methoxybenzyl protecting group (99% yield). Fortunately, the second p-methoxybenzyl group could be removed via hydrogenolysis to deliver diol 53 (95%), which was transformed to mono-benzoate 54 in 91% yield.
The stage was now set to execute a reductive fragment coupling of dihydroisocoumarin 54 with the psymberic acid side chain. Our plan was to acylate imidate 55 with acid chloride 56 (from acid 16a) and intercept the incipient acylimidate with a reducing agent (Scheme 6), a tactic employed for the synthesis of the structural relative pederin (3).36 However, we were unable to prepare and handle imidate 55 using Me3OBF4 as reported.36 Treatment of 54 with Me3OBF4 resulted in decomposition, methyl ester formation, and N-methylation. Reasoning that the acidity of Meerwein’s salt and water are potential culprits, we screened for additives including Et3N, NaHCO3, proton sponge, molecular sieves, and pyridine to no avail. Extensive experimentation identified a uniquely beneficial effect of adding poly(4-vinylpyridine) (25% cross-linked) during the imidate formation with Me3OBF4 (CH2Cl2). After stirring at room temperature, the crude imidate reaction mixture was filtered and concentrated, followed by dissolving the crude imidate 55 in toluene, addition of Hunig’s base and acid chloride 56. The mixture was heated to 40 °C for 2 h, cooled to 0 °C and treated with an ethanolic sodium borohydride solution. After workup, the crude compounds were saponified to afford a separable mixture of 57a and 57b (~1:2 ratio)37 in 57% yield from 54. This saponification step was necessary because some acylation of the free phenol occurred during the acylation step with acid chloride 56. Since bis-phenols 57a and 57b were unstable towards the oxidative conditions to remove the p-methoxybenzyl protecting group present in the psymberate side chain, and hydrogenolytic conditions resulted in saturation of the terminal 1,1-disubstituted olefin, these compounds were benzoylated to afford benzoates 58a and 58b respectively (70%). Oxidative removal of the p-methoxybenzyl protecting group now smoothly yielded alcohols 59a and 59b.
Scheme 6. Initial Attempts Toward Psymberin.a.

a Reagents and conditions: (a) Me3OBF4, CH2Cl2, PVP, rt; filter, concentrate; (b) PhMe, iPr2NEt, 56, 40 °C, 2 h, cool to 0 °C; then add NaBH4in EtOH; (c) LiOH, MeOH, rt; (d) BzCl, i Pr2NEt, PhMe, 70%; (e) DDQ, CH2Cl2/H2O, 70%; (f) TBAF, THF. Abbreviations: PVP, poly(4-vinylpyridine) (25% cross-linked).
We anticipated an uneventful deprotection of both silyl ethers. Instead, treatment of 59a and 59b with tetrabutylammonium fluoride first removed the phenolic benzoate, followed by a slow but partial desilylation of one of the silyl ethers (crude NMR). Only a small amount of fully deprotected material was observed, even when a large excess of fluoride was added. We were unable to convert, nor purify, the crude mixture of compounds 60–62a or 60–62b to a globally deprotected material. However, thorough analysis of the crude 1H-NMR spectra hinted that the mixture containing 62a provided a spectral fingerprint congruent with that reported for psymberin.1
Although we had defined an endgame strategy toward psymberin, it was clear that protecting group issues were plaguing an efficient execution. From the above described initial forays toward psymberin, it became obvious that the p-methoxybenzyl protecting groups needed to be removed before installing the olefin-containing psymberic acid side chain, and that we needed more easily removable replacements for the silylether protecting groups. Toward this end, we processed diol 48, obtained from silylether 47 via fluoride-mediated deprotection (TBAF, 99%, Scheme 5). As shown in Scheme 7, nitrile 48 was hydrolyzed with the Ghaffar-Parkins catalyst 50,35,38 followed by hydrogenolytic removal of the p-methoxybenzyl protecting groups and peracylation with acetic anhydride. The corresponding crystalline amide 63, obtained in 88% yield (3 steps), permitted an unambiguous confirmation of the full stereochemistry as shown via crystallography. As before (Scheme 6), stirring a solution of amide 63 (CH2Cl2) with Me3OBF4 in the presence of poly(4-vinylpyridine) yielded a crude imidate mixture (→ 64) that was filtered, concentrated, and resuspended in toluene. After addition of Hunig’s base, acid chloride 6a or 6b was added, followed by heating to 40 °C for 2 h. The reaction mixture was then cooled to 0 °C and treated with an ethanolic sodium borohydride solution, providing after workup and saponification of the acetate protecting groups with a methanolic LiOH solution a separable mixture of 1 and 65 (71:29 ratio) in 56% yield from 6a and an inseparable mixture of 66 and 67 (75:25 ratio) in 50% yield from 6b.37 As noted in the introduction, a constitutional identical natural product termed irciniastatin was isolated by the Pettit group.2 Although the relative stereochemistry of irciniastatin was only partly resolved, the assignment dictated it to be a different compound than psymberin (structure 2 in Fig 1). Because the NMR spectra of psymberin and irciniastatin were recorded in different solvents, we recorded the spectra of psymberin/irciniastatin diastereoisomers 1, 65–67 in both solvents reported for psymberin and irciniastatin respectively. Careful analysis of all the spectral data obtained for the four synthetic diastereoisomers indicated that 1 represents the true structure of natural psymberin as well as irciniastatin; i.e psymberin and irciniastatin are identical compounds.
Scheme 7. Synthesis of Psymberin and Diastereomers. a.

a Reagents and Conditions: (a) cat. [PtH(PMe2OH)(PMe2O)2)H] (50), EtOH/H2O, 80 °C, 97%; (b) 10% Pd/C, H2, EtOH, 99%; (c) Ac2O, pyridine, 92%; (d) 63, Me3OBF4, CH2Cl2, PVP, rt; filter, concentrate; (e) crude 64, PhMe, i Pr2NEt, 6a or 6b, 40 °C, 2 h, cool to 0 °C; then add NaBH4 in EtOH; (f) LiOH, MeOH.
Second Generation Synthesis
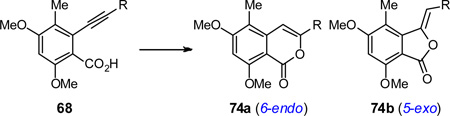
Although our first generation synthesis served us well in producing ~0.5 g of psymberin (1), introducing the dihydroisocoumarin fragment required an eight-step synthesis of aromatic aldehyde 9, followed by a stereoselective aldol coupling and reductive lactonization. To facilitate future SAR studies around the aromatic fragment, we designed an alternative more flexible approach that would rely on a late-stage introduction of an aromatic electrophile such as triflate 69 via Sonogashira cross-coupling with an alkynyl partner such as 70 (see Scheme 8). The resulting alkyne-substituted benzoic acid derivative 68 would enable dihydroisocoumarin formation (63) via cycloisomerization followed by hydrogenation of the resulting isocoumarin. This approach has the advantage that many functionalized aromatic halides and triflates (from the phenol) are commercially available. For the synthesis of psymberin, aromatic triflate 69 is a known compound available in 3 steps from commercial trimethoxytoluene (71).39 Alkynyl fragment 70 in turn would be accessible from lactol 30a via a Marshall propargylation.40
Scheme 8.

Alternative Synthetic Strategy.
As illustrated in Scheme 9, acylation of lactol 30a delivered aldehyde 11a, which was converted to anti-propargylic alcohol 72 via treatment with an in situ prepared allenylindium species derived from mesylate 71 according to Marshall and coworkers in good yield (70%) and excellent diastereoselectivity (dr >10:1, separable).40, 41 The corresponding reaction with the allenylzinc species derived from 71 (Pd(OAc)2, PPh3, Et2Zn in THF) led to decomposition.40b Acetate displacement with TMSCN22 under conditions described for the corresponding reaction leading to 41 (Scheme 4), gave cyanotetrahydropyran 73 in 81% yield. Initial efforts to invert the stereochemistry of anti-alcohol 73 using contemporary Mitsunobu conditions (diethyl azodicarboxylate or diisopropyl azodicarboxylate, PPh3, in THF) faied,42 and led to elimination (enyne) or recovered starting material. However, exploiting condition developed by Tsunoda et al. (N,N,N',N'-tetramethylazodicarboxamide, PBu3, benzene) efficiently provided inverted p-nitrobenzoate 70 in 90% yield.43 Initial model studies to construct the isocoumarin via a one-pot Pdcatalyzed heteroannulation between alkyne 70 and o-iodobenzoic acid indicated low conversion and significant amounts of phthalide formation.44 Therefore, a stepwise construction of the isocoumarin was investigated.
Scheme 9. Alternative Synthesis of Dihydroisocoumarin 63.a.

a Reagents and Conditions: (a) Ac2O, Et3N, DMAP, CH2Cl2, 0 °C, 81%; (b) 55, PdCl2(dppf)2, InI, HMPA, THF, rt, 70%, dr > 10:1; (c) TMSCN (neat); ZnI2, MeCN; aq. 1N HCl, 81%; (d) TMAD, Bu3P, p-NO4BzOH, benzene, 90%; (e) 51, PdCl2(PPh3)2, CuI, Et3N, DMF, 40 °C, 83%; (f) aq. 1N LiOH, MeOH, 0 °C, 99%; (g) AuNTf2(Xphos) (5 mol%), CH2Cl2, rt; (h) H2 (1 atm), cat. [Ir(cod)(PCy3)(py)]PF6 (2 mol%), CH2Cl2, rt, 79% over two steps; (i) BBr3, CH2Cl2, −78 to 25 °C, 77%; (j) HF/Pyridine, THF, 95%; (k) cat. [PtH(PMe2OH)(PMe2O)2H] (50), EtOH/H2O, 80 °C, 97%; (l) Ac2O, pyridine, 92%. Abbreviations: TMAD, N,N,N',N'-tetramethylazodicarboxamide.
Sonogashira coupling of alkyne 70 with penta-substituted aryl triflate 69 followed by saponification yielded benzoic acid-substituted alkyne 68 in 83% yield.45
As shown in Table 1, extensive experimentation was required to obtain the desired isocoumarin 74a. Besides solving the issue of regioselectivity (5-exo to 74b vs 6-endo to 74a), the presence of the homopropargylic alcohol also could create issues with competing hydroalkoxylation and elimination to the enyne. Furthermore, the isocoumarin 74a and isomeric alkylideneisobenzofuran-1(3H)-one 74b were unstable to chromatography. Various cycloisomerization conditions46 including Bronsted acid47 (entries 1, 2), InBr348 (entry 3), and AuCl3(entry 4) gave complex mixtures from which no characterizable compounds could be observed by crude NMR. Silver(I)-mediated cycloisomerization proceeded more smoothly, but afforded an equimolar mixture of 5-exo and 6-endo products (entry 5, ~70% mass balance) similar to result reported in the literature.49 Inspired by the cycloisomerization methodology developed by our group, we explored Zeise’s dimer ([Pt(CH2CH2)Cl2]2).50 Although 5 mol% of this catalyst now promoted the cycloisomerization at room temperature, the undesired 5-exo product dominated (3:7 ratio of 74a:74b, ~50% mass balance, entry 6). Switching to JohnPhos-ligated AuCl51 provided a slight improvement, but still favoring the undesired 5-exo product 74b (entry 7).52 In the end, we could overturn the regioselectivity favoring the desired 6-endo product when cationic Au(I) (PPh3PAuCl, AgSbF6) was engaged as the catalyst (2:1 ratio of 74a:74b, ~60% mass balance, entry 8). The use of (Ph3P)AuNTf251 further improved the selectivity for the isocoumarin product 74a (4:1 ratio, 75% mass balance, entry 9). Finally, dihydroisocoumarin 75 was obtained in 79% isolated yield and >95:5 regioselectivity by stirring a room temperature solution of alkyne 68 with 5 mol% of Xphosligated AuNTf253 (entry 10, ~80% mass balance), followed by hydrogenation of 74a with Crabtree’s catalyst (>99%). Continuing with the psymberin synthesis, a two-step methyl (BBr3) and silyl ether (HF/py) deprotection provided dihydroisocoumarin 76 in 73% yield for the two step sequence. Ghaffar-Parkins35 nitrile hydrolysis followed by peracetylation provided a material (compound 63) that was identical to that obtained via the route outlined in Scheme 5. Single crystal X-ray analysis of compound 63 obtained via this route further confirmed the stereochemistry as assigned. Overall, this synthetic approach towards psymberin entails an 18 steps longest linear sequence from three fragments each prepared in 3, 7, and 8 steps respectively. Although the overall yields using the two alternative synthetic approaches are similar, this 2nd generation approach offers an attractive late stage introduction of aromatic fragments for SAR around the dihydroisocoumarin structure.
Table 1.
Cycloisomerization of Alkynyl Benzoic Acid 68.a
 | |||||
|---|---|---|---|---|---|
| Entry | Reagent | Solvent | Time(h) | Temp | 74a:b |
| 1 | pTsOHb | EtOH | 0.1 | 100 °C | - |
| 2 | TFAb | THF | 3 | Δ | - |
| 3 | InBr3b | THF | 1 | Δ | - |
| 4 | AuCl3c | aq. ACN | 2 | 50°C | - |
| 5 | AgSbF6c | DMF | 4 | 60°C | 1:1 |
| 6 | Pt(C2H4)Cl2]2c | DMF | 0.5 | rt | 3:7 |
| 7 | ClAuL1c | CH2Cl2 | 1 | rt | 2:5 |
| 8 | Ph3PAuCl/AgSbF6c | CH2Cl2 | 1 | rt | 2:1 |
| 9 | Ph3PAuNTf2c | CH2Cl2 | 1 | rt | 4:1 |
| 10 | Tf2NAuL2c | CH2Cl2 | 1 | rt | >95:5 |
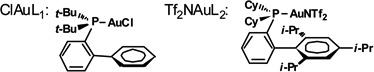 |
|||||
All reactions were performed with 50 µmol 68.
20 mol%.
5 mol%.
Ratios were determined by 1H-NMR.
Analog Synthesis
As noted in the introduction, psymberin is structurally distinct from other pederin family members in two important aspects: (1) it lacks the typical cyclic pederate side chain present in all other family members; and (2) psymberin is the only member containing a dihydroisocoumarin side chain. Here, we describe the synthesis of psymberin analogs that were useful for the structure-function and mode-of-action studies that will be detailed in a subsequent article.13 The terminal olefin of 1 is distinct from the exocyclic double bond present in other pederin family members. In the subsequent article, we demonstrate that the homoallylic ether in pederin is responsible for the irritant/blistering activity associated with this class of compounds.5 Although psymberin does not possess blistering activity,13 hydrogenation of the terminal olefin of psymberin would allow for the preparation of a radio-labeled variant for biological studies. Thus, hydrogenation of psymberin (1) over platinum provides 95% of the corresponding dihydropsymberin 79 (Scheme 10). Alternatively, C phenolic methyl ether 78 or acetate 77 was prepared via methylation or acetylation of psymberin 1 (90–95% yield).
Scheme 10.

Synthesis of Methyl-, Acetyl-, and Dihydro-Psymberin.
As mentioned above, to fully assess the importance of psymberin’s unique dihydroisocoumarin moiety, we designed a truncated psymberin analog 85 (Scheme 11) that lacks this fragment, which is in essence also an analogue of pederin with the acyclic “psymberate” (C1–C6) side chain substituting for the cyclic “pederate” fragment reminiscent of the pederin/mycalamide natural products. The synthesis of this analog, which we term psympederin, commenced from the C2-symmetrical bis-homoallylic alcohol 12, which was mono-acetylated via cyclic orthoacetate formation and hydrolysis under acidic conditions, followed by ozonolytic double bond cleavage to yield the desymmetrized lactol 30c in 84% yield for the two-step process. Acylation of lactol 30c then permitted olefination of the aldehyde, after which anomeric acetatenide22 in the presence of ZnI2 to yield a single axial nitrile 81 in 94% yield. The dihydroxylation of terminal olefin 81 required extensive experimentation to yield an acceptable diastereoselectivity. Dihydroxylation using the UpJohn process (cat. OsO4, N-methylmorpholine) revealed an intrinsic facial bias slightly favoring the undesired diastereomer 82b(82a:82b = 1:1.4), whereas Sharpless asymmetric dihydroxylation using the (DHQ)2PYR or (DHQ)2PHAL ligand was nonselective (1:1 ratio).54 After some experimentation, we found that hydroxyquinine 9-phenanthryl ether (HQP ether) was the optimal ligand for the asymmetric dihydroxylation of 81,55 providing a ~3:1 mixture favoring the desired C15-S configuration (82a). Kocienski and coworkers had previously screened various ligands for the asymmetric dihydroxylation of a closely related substrate (TBS-protected version of 81) and found HQP ether also to be optimal, although selectivity for the desired diastereomer was lower (1.5:1) with their substrate.56 This inseparable mixture of epimers was treated with Meerwein’s salt and proton sponge to afford a separable mixture of methyl ethers 83a and 83b. The stereochemistry was determined by chemical correlation of acetate 83a to the corresponding known TBS-ether.56 Nitrile hydrolysis of the major methyl ether 83a with use of the Ghaffar-Parkins catalyst35 80 (mixture of anomers) was treated with trimethylsilyl cya-provided acetylpedamide 84 in 95% yield.57 The final introduction of the C1–C6 “psymberate” side chain was accomplished via the protocol outlined for the synthesis of psymberin (Scheme 7). Thus, acylation of the imidate derived from 84 with the acid chloride 6a derived from carboxylic acid 20a followed by reduction and saponification yielded a separable 1:4 mixture of psympederin 85 and epi-psympederin in 60% yield from acetylpedamide 84.58,59 This result is in sharp contrast with the corresponding psymberin result where the natural methoxyaminal epimer dominated (3:1), and indicates that diastereoselectivity associated with the N-acylimidate reduction is highly dependent on the presence or absence of the dihydroisocoumarin fragment.
Scheme 11. Synthesis of a Psymberin-Pederin Hybrid, Psympederina.

a Reagents and Conditions: (a) MeC(OMe)3, p-TsOH, MeCN; AcOH/H2O, 88%; (b) O3, CH2Cl2; PPh3, 95%; (c) Ac2O, Et3N, DMAP, CH2Cl2; (d) Ph3PCH2, CH2Cl2, 60% (2 steps); (e) TMSCN, ZnI2, MeCN, 94%; (f) hydroxyquinine 9-phenanthryl ether, K3Fe(CN)6, K2CO3, t-BuOH/H2O, 0 °C; OsO4, 92%; (g) Me3OBF4, CH2Cl2, 2,6-lutidine, 93%; (h) cat. [PtH(PMe2OH)(PMe2O)2H] (50), EtOH/H2O, 80 °C, 95%; (i) Me3OBF4, CH2Cl2, PVP; filter, concentrate; (j) PhMe, iPr2NEt, 6a, 40 °C, 2 h, cool to 0 °C; then add NaBH4 in EtOH; (k) LiOH, MeOH, 60% (3 steps).
CONCLUSION
With its dihydroisocoumarin and acyclic N-acyl side chains, psymberin represents a structural and perhaps functional outlier of the pederin/mycalamide family of natural products. In this manuscript, we have described a detailed synthetic study of psymberin, including two alternative total syntheses and the design of analogs and probe reagents for biological studies. We also assigned the full stereostructure of psymberin and demonstrated that psymberin and irciniastatin are actually identical compounds. Our total syntheses are based on a convergent, flexible strategy that delivered ~ 0.5 g of psymberin in 17–18 steps (longest liner sequence) from three fragments prepared in 7–8 steps (1st generation) or 3–8 steps (2nd generation) each. The synthesis of the central tetrahydropyranyl fragment via a double dehydrogenative allylation developed by Krische, followed by an oxidative desymmetrization was crucial to the successful implementation of an efficient total synthesis. Other highlights include: (1) a cyanide displacement avoiding dioxabicycloalkane ring formation, (2) a stereoselective aldol coupling/one-pot reduction-dihydroisocoumarin formation, (3) exploitation of the Ghaffar-Parkins Pt(II) catalyst for nitrile hydrolysis in complex molecular settings, (4) an optimized procedure for the reductive N-acyl aminal fragment coupling showing a uniquely beneficial effect of polyvinylpyridine during methylimidate formation with Me3OBF4, and (5) a regioselective gold(I)-catalyzed isocoumarin formation from ortho-alkynyl benzoic acids. Founded on this synthetic footing, we were able to study the biology and identify the molecular target of psymberin, which will be detailed in a subsequent manuscript.13
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by the National Institutes of Health (grant CA90349) and the Robert A. Welch Foundation (grant I-1422). We thank Muhammed Yousufuddin (UT Arlington) for crystallographic analysis.
Footnotes
Supporting Information. Experimental procedures, characterization data, copies of NMR spectra, and X-Ray crystal structure data (PDF, CIF). This material is available free of charge via the Internet at http://pubs.acs.org
REFERENCES
- 1.Cichewicz RH, Valeriote FA, Crews P. Org. Lett. 2004;6:1951. doi: 10.1021/ol049503q. [DOI] [PubMed] [Google Scholar]
- 2.Pettit GR, Xu JP, Chapuis JC, Pettit RK, Tackett LP, Doubek DL, Hooper JNA, Schmidt JM. J. Med. Chem. 2004;47:1149. doi: 10.1021/jm030207d. [DOI] [PubMed] [Google Scholar]
- 3.Pavan M, Bo G. Physiol. Comput. Oecol. 1953;3:307. [Google Scholar]
- 4.(a) Perry NB, Blunt JW, Munro MHG. J. Am. Chem. Soc. 1988;110:4850. [Google Scholar]; (b) West LM, Northcote PT, Hood KA, Miller JH, Page MJ. J. Nat. Prod. 2000;63:707. doi: 10.1021/np9904511. [DOI] [PubMed] [Google Scholar]
- 5.For selected reviews, see: Narquizian R, Kocienski PJ. The Role of Natural Products in Drug Discovery. In: Mulzer J, Bohlmann R, editors. Ernst Schering Research Foundation Workshop 32. New York: Springer; 2000. pp. 25–56. Piel J, Butzke D, Fusetani N, Hui D, Platzer M, Wen G, Matsunaga S. J. Nat. Prod. 2005;68:472. doi: 10.1021/np049612d. (c) For a complete listing of pederin related natural products, see the Supporting Information of ref. 1.
- 6.Jiang X, Garcia-Fortanet J, De Brabander JK. J. Am. Chem. Soc. 2005;127:11254. doi: 10.1021/ja0537068. [DOI] [PubMed] [Google Scholar]
- 7.(a) Huang X, Shao N, Palani A, Aslanian R, Buevich A. Org. Lett. 2007;9:2597. doi: 10.1021/ol071068n. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, III, Jurica JA, Walsh SP. Org. Lett. 2008;10:5625. doi: 10.1021/ol802466t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Crimmins MT, Stevens JM, Schaaf GM. Org. Lett. 2009;11:3990. doi: 10.1021/ol901655e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Watanabe T, Imaizumi T, Chinen T, Nagumo Y, Shibuya M, Usui T, Kanoh N, Iwabuchi Y. Org. Lett. 2010;12:1040. doi: 10.1021/ol1000389. [DOI] [PubMed] [Google Scholar]; (e) Wan S, Wu F, Rech JC, Green ME, Balachandran R, Horne WS, Day BW, Floreancig PE. J. Am. Chem. Soc. 2011;133:16668. doi: 10.1021/ja207331m. [DOI] [PubMed] [Google Scholar]; (f) Byeon SR, Park H, Kim H, Hong J. Org. Lett. 2011;13:5816. doi: 10.1021/ol2024289. [DOI] [PubMed] [Google Scholar]
- 8.Shangguan N, Kiren S, Williams LJ. Org. Lett. 2007;9:1093. doi: 10.1021/ol063143k. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kiren S, Williams LJ. Org. Lett. 2005;7:2905. doi: 10.1021/ol0508375. [DOI] [PubMed] [Google Scholar]; (b) Green ME, Rech JC, Floreancig PE. Org. Lett. 2005;7:4117. doi: 10.1021/ol051396s. [DOI] [PubMed] [Google Scholar]; (c) Rech JC, Floreancig PE. Org. Lett. 2005;7:5175. doi: 10.1021/ol0520267. [DOI] [PubMed] [Google Scholar]; (d) Huang X, Shao N, Palani A, Aslanian R, Buevich A, Seidel-Dugan C, Huryk R. Tetrahedron Lett. 2008;49:3592. [Google Scholar]; (e) Lachance H, Marion O, Hall DG. Tetrahedron Lett. 2008;49:6061. [Google Scholar]; (f) Brown LE, Landaverry YR, Davies JR, Milinkevich KA, Ast S, Carlson JS, Oliver AG, Konopelski JP. J. Org. Chem. 2009;74:5405. doi: 10.1021/jo9009003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Pietruszka J, Simon RC. Eur. J. Org. Chem. 2009:3628. [Google Scholar]; (h) Kiren S, Shangguan N, Williams L. Tetrahedron Lett. 2007;48:7456. [Google Scholar]; (i) Henssen B, Kasparyan E, Marten G, Pietruszka J. Heterocycles. 2007;74:245. [Google Scholar]; (j) Pietruszka J, Simon RC. Eur. J. Org. Chem. 2009;21:3628. [Google Scholar]; (k) Buffham WJ, Swain NA, Kostiuk SL, Goncalves TP, Harrowven DC. Eur. J. Org. Chem. 2012:1217. [Google Scholar]
- 10.(a) Jiang X, Williams N, De Brabander JK. Org. Lett. 2007;9:227. doi: 10.1021/ol062656o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang X, Shao N, Huryk R, Palani A, Aslanian R, Seidel-Dugan C. Org. Lett. 2009;11:867. doi: 10.1021/ol802772s. [DOI] [PubMed] [Google Scholar]; (c) Shao N, Huang X, Palani A, Aslanian R, Buevich A, Piwinski J, Huryk R, Seidel-Dugan C. Synthesis. 2009:2855. [Google Scholar]
- 11.Robinson S, Tenney K, Yee DF, Martinez L, Media JE, Valeriote FA, van Soest RWM, Crews P. J. Nat. Prod. 2007;70:1002. doi: 10.1021/np070171i. [DOI] [PubMed] [Google Scholar]
- 12.Jeffrey SC, De Brabander JK, Miyamoto J, Senter PD. ACS Med. Chem. Lett. 2010;1:277. doi: 10.1021/ml100039h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu CY, Feng Y, Cardenas ER, Williams N, Floreancig PE, De Brabander JK, Roth MG. doi: 10.1021/ja3057002. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steuer B, Wehner V, Lieberknecht A, Jäger V. Org. Synth. 1997;74:1. [Google Scholar]
- 15.Sugisaki CH, Ruland Y, Baltas M. Eur. J. Org. Chem. 2003:672. [Google Scholar]
- 16.Schmid CR, Bryant JD. Org. Synth. 1995;72:6. [Google Scholar]
- 17.Jadhav PK, Bhat KS, Perumal PT, Brown HC. J. Org. Chem. 1986;51:432. [Google Scholar]
- 18.Linclau B, Cini E, Oakes CS, Josse S, Light M, Ironmonger V. Angew. Chem. Int. Ed. 2012;51:1232. doi: 10.1002/anie.201107370. [DOI] [PubMed] [Google Scholar]
- 19.Johnson PR, White JD. J. Org. Chem. 1984;49:4424. [Google Scholar]
- 20.Kubota K, Leighton JL. Angew. Chem. Int. Ed. 2003;42:946. doi: 10.1002/anie.200390252. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew. Chem. Int. Ed. 2009;48:5018. doi: 10.1002/anie.200901648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CAUTION! TMSCN is very toxic. All reactions using this reagent should be performed in a well-ventilated hood.
- 23.Nakata T, Nagao S, Mori N, Oishi T. Tetrahedron Lett. 1985;26:6461. [Google Scholar]
- 24.Takahashi H, Kawakita T, Ohno M, Yoshioka M, Kobayashi S. Tetrahedron. 1992;48:5691. [Google Scholar]
- 25.Dess DB, Martin JC. J. Am. Chem. Soc. 1991;113:7277. [Google Scholar]
- 26.Lambooy LP. J. Am. Chem. Soc. 1956;78:771. [Google Scholar]
- 27.(a) Kamila S, Mukherjee C, Mondal SS, De A. Tetrahedron. 2003;59:1339. [Google Scholar]; (b) Casas R, Cave´ C, d’Angelo J. Tetrahedron Lett. 1995;36:1039. [Google Scholar]
- 28.Keck GE, McLaws MD, Wager TT. Tetrahedron. 2000;56:9875. [Google Scholar]
- 29.Evans DA, Rieger DL, Bilodeau MT, Urpi F. J. Am. Chem. Soc. 1991;113:1047. [Google Scholar]
- 30.Evans DA, Calter M. Tetrahedron Lett. 1993;34:6871. [Google Scholar]
- 31.For a comprehensive study of aldol reactions between a range of protected arylaldehyde fragments and ethyl ketone fragments see the Supporting Information.
- 32.Evans DA, Hoveyda AH. J. Org. Chem. 1990;55:5190. [Google Scholar]
- 33.Reduction with Zn(BH4)2 provided a 5:1 ratio of syn:anti diols in 81% yield.
- 34.(a) Evans DA, Rieger DL, Gage JR. Tetrahedron Lett. 1990;31:7099. [Google Scholar]; (b) Rychnovsky SD, Rogers B, Yang G. J. Org. Chem. 1993;58:3511. [Google Scholar]
- 35.(a) Ghaffar T, Parkins AW. Tetrahedron Lett. 1995;36:8657. [Google Scholar]; (b) Ghaffar T, Parkins AW. J. Mol. Catal. A. 2000;160:249. [Google Scholar]
- 36.Takemura T, Nishii Y, Takahashi S, Kobayashi J, Nakata T. Tetrahedron. 2002;58:6359. [Google Scholar]
- 37.We have not been able to determine if this represents a kinetic or thermodynamic ratio as all attempts to equilibrate the individual N-acylaminal diastereomers led to decomposition. We favor the interpretation of kinetic product formation under the reductive basic reaction conditions because N-acylaminals are known to equilibrate only under acidic conditions. For a review see: Speckamp WN, Hiemstra H. Tetrahedron. 1985;41:4367.
- 38.Amide hydrolysis using phase transfer conditions reported by Magnus and coworkers (aq. H2O2, aq. NaOH, Bu4NHSO4, CH2Cl2) gave incomplete conversion, providing the amide in 50% isolated yield. For a reference, see: Venit JJ, DiPierro M, Magnus P. J. Org. Chem. 1989;54:4298.
- 39.Solladie G, Gehrold N, Maignan J. Tetrahedron: Asymmetry. 1999;10:2739. and ref. 7a. [Google Scholar]
- 40.(a) Johns BA, Grant CM, Marshall JA. Org. Synth. Coll. 2004;10:170. ;or Org. Synth. 2002, 79, 59. [Google Scholar]; (b) Marshall JA, Eidam P, Eidam HS. J. Org. Chem. 2006;71:4840. doi: 10.1021/jo060542k. [DOI] [PubMed] [Google Scholar]
- 41.Absolute stereochemistry confirmed by Mosher ester analysis, see Supporting Info.
- 42.Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 43.(a) Tsunoda T, Otsuka J, Yamamiya Y, Ito S. Chem. Lett. 1994:539. [Google Scholar]; (b) Tsunoda T, Yamamiya Y, Kawamura Y, Ito S. Tetrahedron Lett. 1995;36:2529. [Google Scholar]
- 44.(a) Barry RD. Chem. Rev. 1964;64:229. [Google Scholar]; (b) Hauser FM, Baghdanov VM. J. Org. Chem. 1988;53:4676. [Google Scholar]; (c) Ogawa Y, Maruno M, Wakamatsu T. Heterocycles. 1995;41:2587. [Google Scholar]; (d) Kundu NG, Pal M, Nandi B. J. Chem. Soc. Perkin Trans. 1. 1998:561. [Google Scholar]; (e) Izumi T, Nishimoto Y, Kohei K, Kasahara A. J. Heterocycl. Chem. 1990;27:1419. [Google Scholar]; (f) Liao H-Y, Cheng C-H. J. Org. Chem. 1995;60:3711. [Google Scholar]; (g) Subramanian V, Batchu VR, Barange D, Pal M. J. Org. Chem. 2005;70:4778. doi: 10.1021/jo050440e. [DOI] [PubMed] [Google Scholar]
- 45.Toyota M, Komori C, Ihara M. J. Org. Chem. 2000;65:7110. doi: 10.1021/jo000816i. [DOI] [PubMed] [Google Scholar]
- 46.Cyclization of acetylenic acids, see: Diederich F, Stang PJ, Tykwinski RR, editors. Acetylene Chemistry: Chemistry, Biology, and Material Science. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA; 2005. pp. 59–62. Alonso F, Beletskaya IP, Yus M. Chem. Rev. 2004;104:3079. doi: 10.1021/cr0201068.
- 47.(a) Bras GL, Hamze A, Messaoudi S, Provot O, Calvez P-BL, Brion JD, Alami M. Synthesis. 2008:1607. [Google Scholar]; (b) Hellal M, Bourguignon JJ, Bihel FJJ. Tetrahedron Lett. 2008;49:62. [Google Scholar]; (c) Uchiyama M, Ozawa H, Takuma K, Matsumot Y, Yonehara M, Hiroya K, Sakamoto T. Org. Lett. 2006;8:5517. doi: 10.1021/ol062190+. [DOI] [PubMed] [Google Scholar]
- 48.Sakai N, Annaka K, Fujita A, Sato A, Konakahara T. J. Org. Chem. 2008;73:4160. doi: 10.1021/jo800464u. [DOI] [PubMed] [Google Scholar]
- 49.(a) Yoshikawa T, Mori S, Shindo M. Org. Lett. 2009;11:5378. doi: 10.1021/ol902086t. [DOI] [PubMed] [Google Scholar]; (b) Marchal E, Uriac P, Legouin B, Toupet L, van de Weghe P. Tetrahedron. 2007;63:9979. [Google Scholar]
- 50.Liu B, De Brabander JK. Org. Lett. 2006;8:4907. doi: 10.1021/ol0619819. [DOI] [PubMed] [Google Scholar]
- 51.Mézailles N, Ricard L, Gagosz F. Org. Lett. 2005;7:4133. doi: 10.1021/ol0515917. [DOI] [PubMed] [Google Scholar]
- 52.For selected reviews on gold-catalysis, see: Jiménez-Núñez E, Echavarren AM. Chem. Commun. 2007:333. doi: 10.1039/b612008c. Fürstner A, Davies PW. Angew. Chem. Int. Ed. 2007;46:3410. doi: 10.1002/anie.200604335. Gorin DJ, Toste FD. Nature. 2007;446:395. doi: 10.1038/nature05592. Hashmi ASK. Chem. Rev. 2007;107:3180. doi: 10.1021/cr000436x. Shen HC. Tetrahedron. 2008;64:3885.
- 53.Buzas AK, Istrate FM, Gagosz F. Org. Lett. 2007;9:985. doi: 10.1021/ol063031t. [DOI] [PubMed] [Google Scholar]
- 54.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483. [Google Scholar]
- 55.Sharpless KB, Amberg W, Beller M, Chen H, Hartung J, Kawanami Y, Lübben D, Manoury E, Ogino Y, Shibata T, Ukita T. J. Org. Chem. 1991;56:4585. [Google Scholar]
- 56.Kocienski PJ, Narquizian R, Raubo P, Smith C, Farrugia LJ, Muir K, Boyle FT. J. Chem. Soc. Perkin Trans. 1. 2000:2357. [Google Scholar]
- 57.Our synthesis of acetylpedamide, accomplished in 8 steps from neopentylglycol 29, compares favorable to Nakata’s36 and Kocienski’s 55 15-step syntheses of benzoylpedamide (from S-malic acid) and tert-butyldimethylsilylpedamide (from ethyl isobutyrate), respectively.
- 58.A similar coupling between benzoylpedamide and the pederin “pederate” fragment in Nakata’s36 pederin total synthesis also yielded a 1:3 mixture of pederin and epi-pederin (38% yield).
- 59.The natural C8-Sconfiguration of 85 was assigned based upon a detailed analysis of 1H-1H coupling constants and 1D- and 2D-NOE correlations, see ref. 10a for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.