

Abstract
Optimization of the imidazo[4,5-b]pyridine-based series of Aurora kinase inhibitors led to the identification of 6-chloro-7-(4-(4-chlorobenzyl)piperazin-1-yl)-2-(1,3-dimethyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine (27e), a potent inhibitor of Aurora kinases (Aurora-A Kd = 7.5 nM, Aurora-B Kd = 48 nM), FLT3 kinase (Kd = 6.2 nM), and FLT3 mutants including FLT3-ITD (Kd = 38 nM) and FLT3(D835Y) (Kd = 14 nM). FLT3-ITD causes constitutive FLT3 kinase activation and is detected in 20–35% of adults and 15% of children with acute myeloid leukemia (AML), conferring a poor prognosis in both age groups. In an in vivo setting, 27e strongly inhibited the growth of a FLT3-ITD-positive AML human tumor xenograft (MV4–11) following oral administration, with in vivo biomarker modulation and plasma free drug exposures consistent with dual FLT3 and Aurora kinase inhibition. Compound 27e, an orally bioavailable dual FLT3 and Aurora kinase inhibitor, was selected as a preclinical development candidate for the treatment of human malignancies, in particular AML, in adults and children.
Introduction
Aurora kinases, a family of three serine-threonine kinases designated A, B, and C, play key and distinct roles in different stages of mitosis.1−3 At the early stages of mitosis, Aurora-A forms a complex with TPX2 (targeting protein for Xklp2) that regulates centrosome maturation and mitotic spindle assembly.4,5 Aurora-B forms complexes with the inner centromere protein (INCENP), survivin and borealin, thereby regulating chromosome condensation, chromosome alignment, mitotic checkpoint, and cytokinesis.6−9 Overexpression of Aurora-A and -B has been reported in a wide range of human malignancies, including breast, colorectal, ovarian, glioma, thyroid carcinoma, and seminoma.10−16 The function of Aurora-C during mitosis is less well understood. However, high expression of Aurora-C has been reported in the testis.17,18
In recent years, small-molecule targeting of the Aurora kinases has become a common strategy for the discovery of new molecularly targeted cancer therapeutics for the treatment of solid tumors and hematological malignancies, including AML.19,20 Some inhibitors of Aurora kinases were reported to inhibit FLT3,18 and high expression of Aurora kinases has been demonstrated in leukemia cell lines and patient cohorts.21−24 A number of structurally diverse inhibitors of Aurora kinase activity have been reported,18,25,26 including 1 [VX-680 (MK-0457)],272 (AZD1152),283 (PHA-739358),29,30 and 4 (AMG 900)31 (Figure 1).
Figure 1.

Inhibitors of Aurora kinases.
We have previously reported the discovery of 6 (Figure 2), a novel, potent, and orally bioavailable inhibitor of Aurora kinases that inhibits growth of the SW620 human colon carcinoma xenograft with concomitant biomarker modulation consistent with target engagement.32 However, the preclinical development of this compound was limited by a narrow safety margin against hERG (IC50 = 3.0 μM)32 and low human liver microsomal stability (86% metabolized after a 30 min incubation, unpublished data). Herein, we report the evolution of the medicinal chemistry program to identify orally bioavailable dual FLT3/Aurora kinase inhibitors with higher metabolic stability and wider therapeutic index against hERG suitable for preclinical evaluation.
Figure 2.

Imidazo[4,5-b]pyridine-based Inhibitors of Aurora kinases.
Chemistry
Compounds 10a–e shown in Table 1 were prepared from 7 or 8(32) by treatment with the appropriate benzaldehyde 9a–e in the presence of Na2S2O4 (Scheme 1).32,33
Table 1. N-Methylpiperazine Modificationsd,e.

Results are mean values for samples run in triplicate.
From ref (32).
MLM/HLM: Percentage of parent compound metabolized after a 30 min incubation.
For both Aurora-A IC50 and SW620 human colon cancer cell GI50 determinations, the results are mean values of two independent determinations or mean (±SD) for n > 2 unless specified otherwise.
n.d. = not determined.
Scheme 1.

Reagents and conditions: (a) EtOH, 1 M aq Na2S2O4, heating.
2-Amino-4,5-dichloro-3-nitropyridine (12) and 2-amino-5-bromo-4-chloro-3-nitropyridine (13), precursors for 7 and 8, were synthesized as previously described32 or by halogenation of 2-amino-4-chloro-3-nitropyridine (11) as shown in Scheme 2.
Scheme 2.

Reagents and conditions: (a) NCS, CH3CN, 80 °C, 1 h; (b) NBS, CH3CN, 80 °C, 1 h.
The benzaldehyde derivatives 9b–e were prepared as shown in Scheme 3. Acetylation of 4-(piperazin-1-yl)benzaldehyde afforded 9b, whereas 9c and 9d were obtained from 15 via a Pd-catalyzed amination reaction34 followed by acid-mediated acetal hydrolysis. Nucleophilic displacement of the F of aldehyde 18 with (S)-1,2-dimethylpiperazine afforded the corresponding 1,2-dimethylpiperazine derivative 9e.
Scheme 3.

Reagents and conditions: (a) TFA, CH2Cl2, rt, 1.5 h; (b) CH2Cl2, acetyl chloride, iPr2NEt, 0 °C to rt; (c) (i) toluene, Pd2(dba)3, BINAP, NaOtBu, 100 °C, (ii) 1 M aq HCl; (d) MeOH/THF, 33% HCHO in water, Na(OAc)3BH, rt, 18 h; (e) (i) TFA/CH2Cl2, (ii) iPr2NEt, DMSO, 120 °C, 2 h.
Access to imidazo[4,5-b]pyridine derivatives 20a–f (Table 2) was also gained by reacting 7 (Scheme 1) with the appropriately substituted benzaldehyde in the presence of Na2S2O4.32,33 3-Fluoro-4-(4-methylpiperazin-1-yl)benzaldehyde (19a) and 2-fluoro-4-(4-methylpiperazin-1-yl)benzaldehyde (19b) were prepared by reacting 1-methylpiperazine with 3,4-difluorobenzaldehyde and 2,4-difluorobenzaldehyde, respectively. 3-(4-Methylpiperazin-1-yl)benzaldehyde (19f) was obtained from 1-bromo-3-(diethoxymethyl)benzene via a Pd-catalyzed amination reaction followed by acid-mediated acetal hydrolysis34 in a manner similar to that described for synthesis of 9c and 9d (Scheme 3). Compounds 21a–i and 22a–e respectively presented in Tables 3 and 4 were readily accessed by reaction of 7 or 8 with the requisite commercially available aldehyde in the presence of Na2S2O4.32,33
Table 2. Basic Nitrogen Positional Effect and pKa Modulationc.

Results are mean values for samples run in triplicate.
MLM/HLM: Percentage of parent compound metabolized after a 30 min incubation.
For both Aurora-A IC50 and SW620 human colon cancer cell GI50 determinations, the results are mean values of two independent determinations or mean (±SD) for n > 2 unless specified otherwise.
Table 3. R2 Modificationsb.

Results are mean values for samples run in triplicate.
Results are mean values of two independent determinations or mean (±SD) for n > 2 unless specified otherwise.
Table 4. R2-Pyrazole Modificationsd.
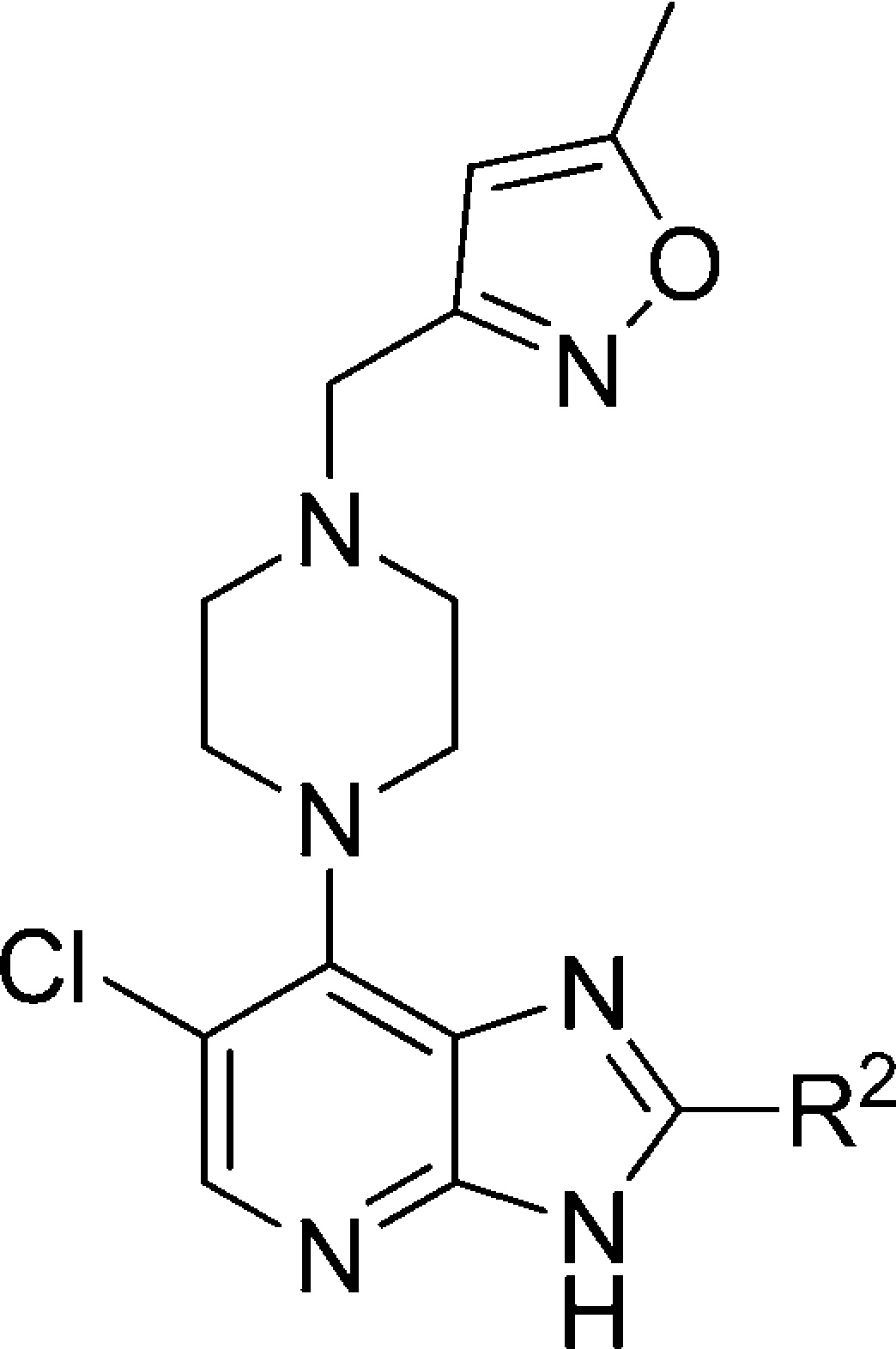

Results are mean values for samples run in triplicate.
MLM/HLM: Percentage of parent compound metabolized after a 30 min incubation.
Calculated log P.(46)
For both Aurora-A IC50 and SW620 human colon cancer cell GI50 determinations, the results are mean values of two independent determinations or mean (±SD) for n > 2 unless specified otherwise.
The synthesis of 1,3-dimethylpyrazole derivatives 27a–g (Table 5) is shown in Scheme 4. The key 2-amino-3-nitropyridine intermediates 26a–g, were obtained by nucleophilic displacement of the C-4 chloride of 12 with the appropriate piperazine derivative. Boc-piperazines 24f and 24g were prepared by a reductive alkylation of 1-Boc-piperazine (23) with 5-formylpyrimidine and pyrazine-2-carbaldehyde, respectively. The Boc-piperazine derivatives 24b and 24d were obtained by a substitution reaction of 1-Boc-piperazine (23) with 3-(chloromethyl)-1,2,4-oxadiazole and 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole, respectively (Scheme 4).
Table 5. R3-Isoxazole Replacementsd.
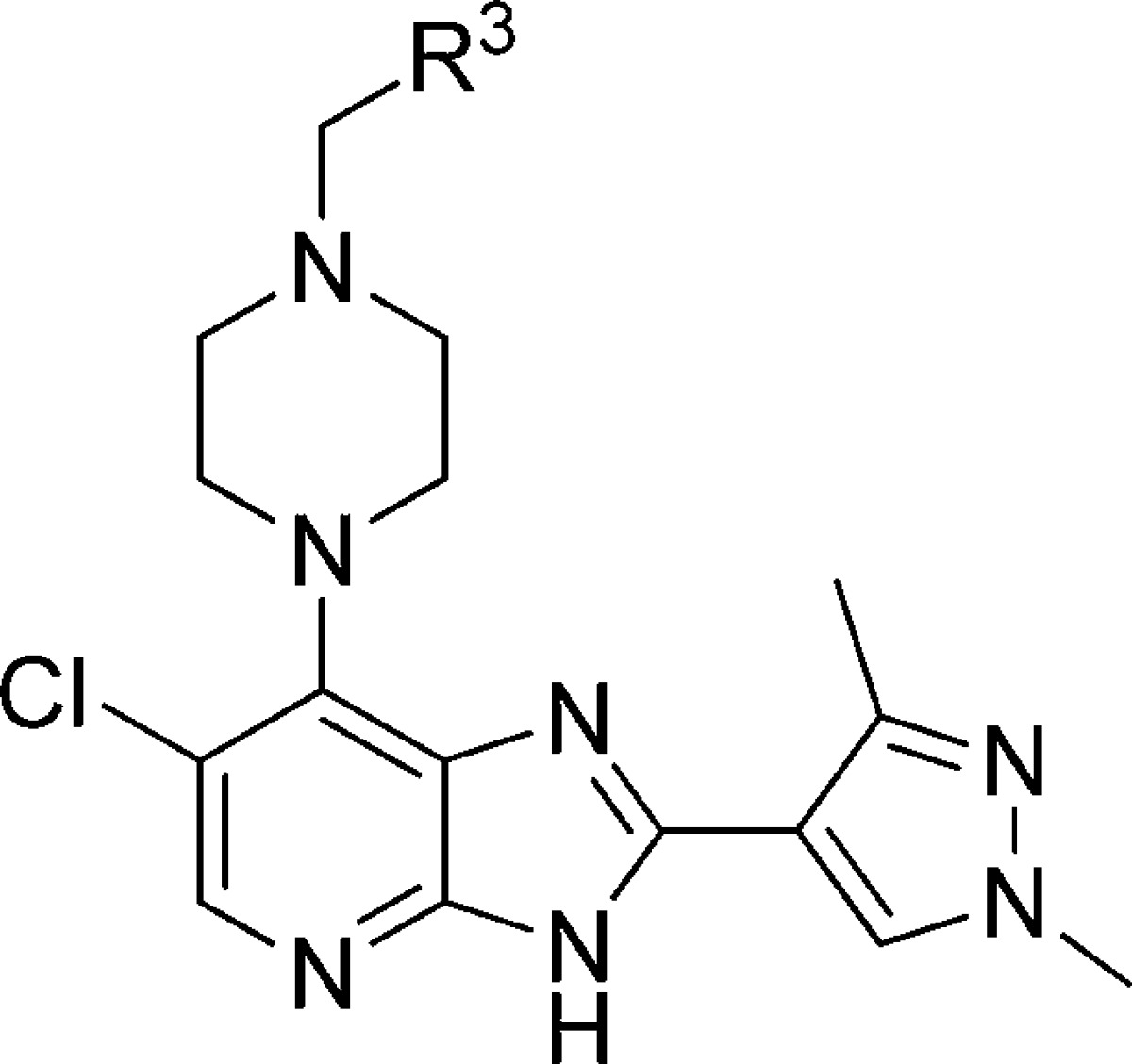

Results are mean values for samples run in triplicate.
MLM/HLM: percentage of parent compound metabolized after a 30 min incubation.
Calculated log P(46)
For both Aurora-A IC50 and SW620 human colon cancer cell GI50 determinations, the results are mean values of two independent determinations or mean (±SD) for n > 2 unless specified otherwise.
Scheme 4.

Reagents and conditions: (a) for 24b,d, CH2Cl2, 3-(chloromethyl)-1,2,4-oxadiazole or 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole, Et3N, 50 °C; for 24f,g, CH2Cl2, 5-formylpyrimidine or pyrazine-2-carbaldehyde, NaBH(OAc)3, AcOH; (b) TFA, CH2Cl2, rt; (c) iPrOH, heating; (d) 1,3-dimethyl-1H-pyrazole-4-carbaldehyde, EtOH, 1 M aq Na2S2O4, 80 °C.
Results and Discussion
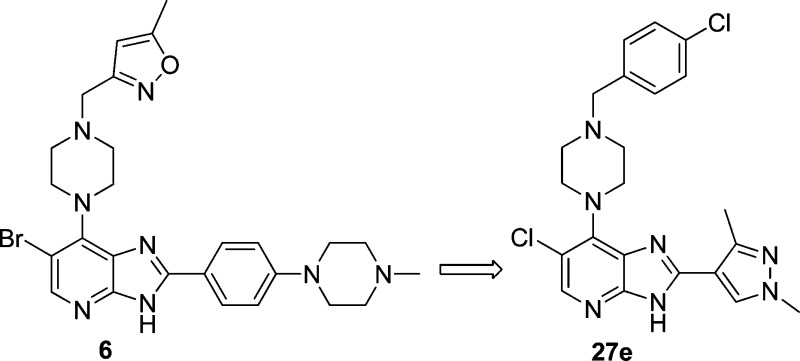
Compound 6 and its 6-Cl counterpart 5 (Figure 2) served as starting points for our investigation aimed at identifying orally bioavailable inhibitors of Aurora kinases suitable for preclinical evaluation. The crystal structure of 6 bound to Aurora-A32 and the existing structure–activity relationship in this series32,35 provided us with a clear understanding of the interactions of this class of compounds with Aurora kinases. The N4 pyridine hydrogen bond acceptor and the N3 imidazole hydrogen bond donor of the imidazo[4,5-b]pyridine scaffold form hydrogen bonds to Ala213 in the hinge region of the kinase.32 The R2 (4-methylpiperazin-1-yl)phenyl substituent points into the solvent accessible area with the lipophilic phenyl ring residing in close proximity to Gly216. The R3 5-methylisoxazol-3-yl group makes contact with the Gly-rich loop, and in vitro compound profiling and subsequent Free–Wilson analysis36 identified p-chlorophenyl, and 5-methylisoxazol-3-yl as the R3 substituents most frequently observed in highly active compounds. The incorporation of a Br or Cl substituent at the C6 position results in a significant increase in enzyme inhibitory activity with 6-Br/Cl derivatives >8-fold more potent relative to their unsubstituted counterparts.32,35 Selected physicochemical properties for this class of compounds were also studied. The measured log D7.4 for compound 6 is 2.58,37 and for the analogue of 6 where R2 = p-methoxyphenyl (i.e., 6-bromo-2-(4-methoxyphenyl)-7-[4-(5-methyl-isoxazol-3-ylmethyl)-piperazin-1-yl]-3H-imidazo[4,5-b]pyridine),32 the pKa of the piperazine nitrogen bearing the 5-methylisoxazol-3-yl substituent was measured as 5.39 and the acidic pKa of the imidazopyridine N3 proton was 9.51, consistent with a strong hydrogen bond donor.37 The pKa of the N-Me piperazine nitrogen in 6 was calculated as 8.50.38,39
In pharmacophore models for hERG blockade, it is reported that a basic nitrogen in lipophilic molecules can increase hERG inhibitory potency via π-cation stacking.40−42 As a consequence, one of the most common strategies for designing out hERG affinity involves the reduction of the pKa and/or the introduction of steric bulk or shielding around the basic nitrogen.41 The concomitant reduction of both pKa and clogP often leads to a reduction in hERG activity.41 It has been suggested that a 1 log unit reduction in clogP leads to 0.8 log unit reduction in hERG activity.41 In a recent study by Waring and Johnstone, the relationship between hERG and log P for acids, bases, neutrals, and zwitterions was investigated.43 In that study, it was reported that a higher log P is associated with an increasing potential for hERG affinity, with basic compounds being more likely to have hERG liabilities than neutrals.43 With this in mind, our approaches to lower hERG affinity involved modulation of the pKa of the N-Me piperazine nitrogen, introduction of steric bulk around this nitrogen, and replacement of the R2 (4-methylpiperazin-1-yl)phenyl substituent with a weakly basic or neutral five-membered heteroaromatic moiety. The latter approach could potentially lead to a neutral molecule of lower molecular mass and clogP with the potential to reduce both hERG affinity and susceptibility to oxidative metabolism.44,45
Replacement of the N-methylpiperazine moiety in 6 with a morpholine ring (compound 10a) was beneficial in reducing affinity for hERG, consistent with our hypothesis that hERG affinity is driven by the presence of a piperazine basic center. However, this change was detrimental to human liver microsomal stability (99% of parent compound was metabolized following 30 min incubation, Table 1). Tactics to introduce steric bulk and lower the pKa of the piperazine nitrogen by acetylation (nonbasic, compound 10b) or introduction of a pendant methoxyethyl side chain (compound 10d, calculated pKa = 8.0638,39) did not lower hERG affinity. The introduction of steric bulk around the N-methylpiperazine nitrogen (compounds 10c and 10e, Table 1) resulted in retention of the Aurora-A, and tumor cell growth inhibitory activity but failed to reduce hERG inhibitory potency. Analogues 10c,e displayed affinities for hERG similar to those observed with the parent compounds 5 and 6 (Table 1). In addition, the high susceptibility to human liver metabolism remained a concern, with HLM stability values for compounds 10b–e similar to those for the parent compounds 5 and 6 (Table 1).
In a further attempt to lower the pKa of the N-Me piperazine nitrogen in 5, a fluoro substituent was introduced on the phenyl ring ortho or meta to the piperazine (Table 2, compounds 20a, 20b, respectively). The o-F substituted analogue 20a (piperazine N-Me nitrogen calculated pKa = 7.9838,39) was less potent in inhibiting hERG compared to 5; however, its human liver microsomal stability remained low (68% metabolized after a 30 min incubation). The m-F substituted derivative 20b (piperazine N-Me nitrogen calculated pKa = 8.5038,39) displayed a similar affinity for hERG compared to the parent compound 5. The m-F substituted analogue 20b was a less potent inhibitor of Aurora-A compared to its o-regioisomer 20a, a trend consistent with previously reported SAR in this series.35
The positional effect of the basic nitrogen on human microsomal stability and hERG affinity was next investigated (Table 2, compounds 20c–f). The m-CH2NMe2 and m-OCH2CH2NMe2 analogues (compounds 20d, 20c, respectively) displayed similar hERG inhibitory activities to 5 (Tables 1 and 2). The moderately basic morpholino derivative 20e exhibited low affinity for hERG, but was rapidly metabolized in mouse and human liver microsomes. However, the introduction of the N-methyl piperazine moiety at the m-position (compound 20f, Table 2) resulted in a desirable outcome in relation to both hERG inhibitory activity and MLM/HLM metabolic stability. Compound 20f inhibited hERG with an IC50 value of 9.50 μM (Table 2; see also general experimental) and displayed high human liver microsomal stability (24% metabolized after a 30 min incubation versus 89% for compound 5 and 86% for compound 6).
Subsequently, our chemical efforts were focused on the introduction of neutral or weakly basic R2 heteroaryl substituents. To this end, the Aurora-A and tumor cell growth inhibitory effect of replacing the R2 (4-methylpiperazin-1-yl)phenyl moiety in 5 and 6 with a range of five-membered heteroaromatic rings was studied (Table 3). With the exception of 1-methyl-1H-imidazol-5-yl and 1-methyl-1H-pyrazol-4-yl derivatives 21a and 21i, all analogues presented in Table 3 were considerably less potent inhibitors of Aurora-A compared to 5 or 6 (Table 1). In particular, the incorporation of a 1-methyl-1H-imidazol-2-yl, 2,5-dimethyloxazol-4-yl, 5-methylisoxazol-3-yl, and 1,2,3-thiadiazol-4-yl as an R2 substituent (compounds 21b, 21d, 21e, 21f, respectively) resulted in a significant drop in Aurora-A inhibitory potency. To unequivocally establish the binding mode of these 2-heteroaryl-based analogues to Aurora-A, compound 21a was cocrystallized with the catalytic domain of Aurora-A (residues 122–403). The crystal structure of 21a bound to Aurora-A was determined to a resolution of 2.5 Å (Figure 3, Table S1, Supporting Information), and shows that 21a occupies the ATP-binding site in a mode similar to that described for 6.32 As was observed for 6, the isoxazole group of 21a interacts with the Gly-rich loop of Aurora-A between residues 141 and 146; in addition, the side chain of Lys143 makes an additional contact, although the significance of this is unclear. As expected, the R2 imidazole ring of 21a sits close to Gly216, in an equivalent position to the phenyl ring of 6, with the methyl substituent pointing away from the hinge to avoid steric clashes with the main chain Ala213 and Pro214. The Aurora-A affinity observed for compounds 21a (IC50 = 0.015 μM), 21c (IC50 = 0.068 μM), and 21i (IC50 = 0.045 μM) is consistent with a preferred, sterically restricted, lipophilic interaction with Ala213, which is satisfied by the C4–H of the imidazole ring of 21a (Figure 3) but is precluded in 21d (IC50 = 2.2 μM) owing to the presence of either a polar nitrogen atom or more sterically demanding methyl moiety in the same vector.
Figure 3.
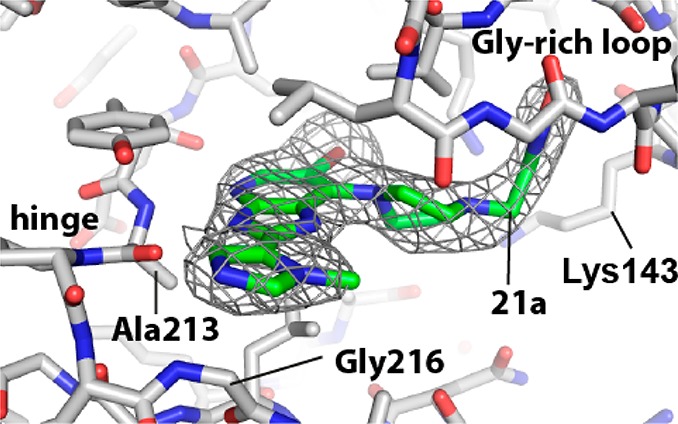
2.5 Å resolution crystal structure of compound 21a bound to Aurora-A (PDB code 4B0G). The color scheme used is nitrogen, blue; oxygen, red; bromine, brown; carbon in Aurora-A, white; carbon in 21a, green. The final 2mFo-DFc electron density map surrounding 21a is shown as a gray wire-mesh, contoured at 1.0 σ.
The two most promising compounds (i.e., 21a and 21i, Table 3) were subject to additional in vitro profiling which revealed the 1-methyl-1H-imidazol-5-yl derivative 21a to be an inhibitor of hERG (94% inhibition at 10 μM). The 1-methyl-1H-pyrazol-4-yl analogue 21i was also an inhibitor of hERG (67% at 10 μM), but exhibited high stability in both MLM (20% metabolized after a 30 min incubation) and HLM (10% metabolized after a 30 min incubation). The tPSA and the clogP for 21i were calculated as 80.42 and 2.37, respectively.46 On the basis of these findings, 21i was selected as a scaffold for an optimization study aimed at increasing the Aurora-A/tumor cell growth inhibitory potencies and reducing hERG affinity.
Replacement of the N1-methyl in the R2 pyrazole subsituent of 21i with an ethyl, isopropyl, or difluoroethyl substituent (compounds 22a, 22b, 22c, respectively, Table 4) gave no Aurora-A or tumor cell growth inhibitory benefit, although high stability to human liver microsomal metabolism was maintained (Table 4). On the other hand, the introduction of an additional methyl group at the 3-position of the pyrazole ring in 21i (compound 22d, Table 4) resulted in an enhancement to Aurora-A inhibition and SW620 cell growth inhibitory potency. In addition, the inhibitory activity of 22d against hERG was determined as 48% at 10 μM (IC50 = 6.3 μM, Table 4). Subsequent introduction of a C3-cyclopropyl group, a bulkier and more lipophilic substituent, led to a marginal increase in Aurora-A inhibitory potency (22e vs 22d, Table 4). However, compound 22e inhibited hERG with an IC50 value of 2.5 μM (Table 4). On balance, the data presented in Table 4 point to 1,3-dimethyl-1H-pyrazol-4-yl as an optimal R2 substituent.
Having identified 1,3-dimethyl-1H-pyrazol-4-yl as a preferred R2 substituent, our efforts focused on exploring the R3 substituent. Our aim was to further improve the in vitro profile, in particular to reduce hERG affinity, and to subsequently progress the most promising analogues to in vivo PK characterization and evaluation in a human tumor xenograft model. The R3 5-methylisoxazol-3-yl group in 22d (Table 4) was replaced with a range of five- and six-membered aromatic heterocycles and the p-chlorophenyl previously identified by Free–Wilson analysis as a preferred substituent (Table 5). The 4-methyl-1,2,5-oxadiazole derivative 27a was a more potent inhibitor of Aurora-A and SW620 tumor cell growth relative to 22d but highly susceptible to metabolism, in particular in mouse liver microsomes. On the other hand, the 1,2,4-oxadiazole analogues 27b and 27c exhibited lower Aurora-A and SW620 tumor cell inhibitory potency relative to 22d. Both 27b and 27c displayed similar inhibitory activities against hERG (IC50 values of 11.0 and 9.5 μM, respectively; Table 5). The 1,2,4-triazole analogue 27d displayed considerably lower Aurora-A and SW620 tumor cell inhibitory potencies relative to 22d, though its affinity for hERG was significantly reduced (14% inhibition at 10 μM). The introduction of the R3p-chlorophenyl substituent was, however, beneficial versus inhibition of SW620 tumor cell growth (compound 27e (CCT241736), GI50 = 0.283 μM). In addition, 27e inhibited Aurora-A with an IC50 value 0.038 μM, was highly stable in mouse and human microsomes (34% and 10% metabolized after a 30 min incubation respectively), and displayed low affinity for hERG (IC50 > 25 μM). Finally, two more polar six-membered heteroaromatic substituents were explored, i.e., pyrimid-5-yl and pyrazin-2-yl (27f and 27g, respectively); both derivatives exhibited low affinity for hERG (32% and 23% inhibition at 10 μM respectively) but also low stability in both mouse and human liver microsomes. Although a trend to lower hERG inhibition was observed with less lipophilic compounds (compare compounds 22b,c in Table 4 with compounds 27d,f,g in Table 5), structural modifications that increase log P could also interestingly reduce affinity for hERG, as illustrated for compound 27e (clogP = 4.81,47 hERG IC50 > 25 μM) compared to 22d (clogP = 2.34, hERG IC50 = 6.3 μM). Considering the impact of these R3 modifications (Table 5) on MLM stability, all four R3 five-membered heteroaromatic-substituted compounds, 27a (clogP = 2.34), 27b (clogP = 1.45), 27c (clogP = 1.72), and 27d (clogP = 1.21), were more susceptible to mouse liver metabolism compared with 22d despite displaying lower or equal clogP (22d, clogP = 2.34). A similar trend was observed with the R3 six-membered heteroaromatic compounds 27f (clogP = 1.64) and 27g (clogP = 1.64), both being significantly more susceptible to mouse liver metabolism compared with p-chlorophenyl derivative 27e. On the basis of a desirable set of in vitro properties, compound 27e was selected for in depth in vitro and in vivo characterization. Kinase selectivity was assessed by profiling 27e in a 442-kinase panel (containing 386 nonmutant kinases) at a concentration of 1 μM using the KINOMEScan technology (see Table S2, Supporting Information).48−50 The S(10) selectivity score, which is calculated by dividing the number of nonmutant kinases for which >90% competition of control ligand is observed (this is measured as <10% of control) by the total number of nonmutant kinases tested, was determined as 0.057, i.e., 22 hits from the 386 nonmutant kinases tested. As expected, Aurora-A, -B, and -C were potently inhibited with percent control values of 3.4, 1, and 16, respectively, at 1 μM. Consistent with previous data on this imidazo[4,5-b]pyridine series,51 screening also revealed greater than 94% competition for wild-type FLT3 kinase and clinically relevant FLT3-resistant mutants including FLT3-ITD and FLT3(D835Y) which was confirmed by Kd determination (Table 6). Taken together, these data indicate that 27e is a potent dual inhibitor of FLT3 and Aurora kinases with few off-target kinase activities across the kinome (Figure 4).
Table 6. Kd Values for Compound 27e.
| kinase | Kd (nM) |
|---|---|
| Aurora-A | 7.5 |
| Aurora-B | 48 |
| FLT3 | 6.2 |
| FLT3(D835H) | 11 |
| FLT3(D835Y) | 14 |
| FLT3-ITD | 38 |
| FLT3(K663Q) | 5.1 |
| FLT3(N841I) | 16 |
| FLT3(R834Q) | 110 |
Figure 4.
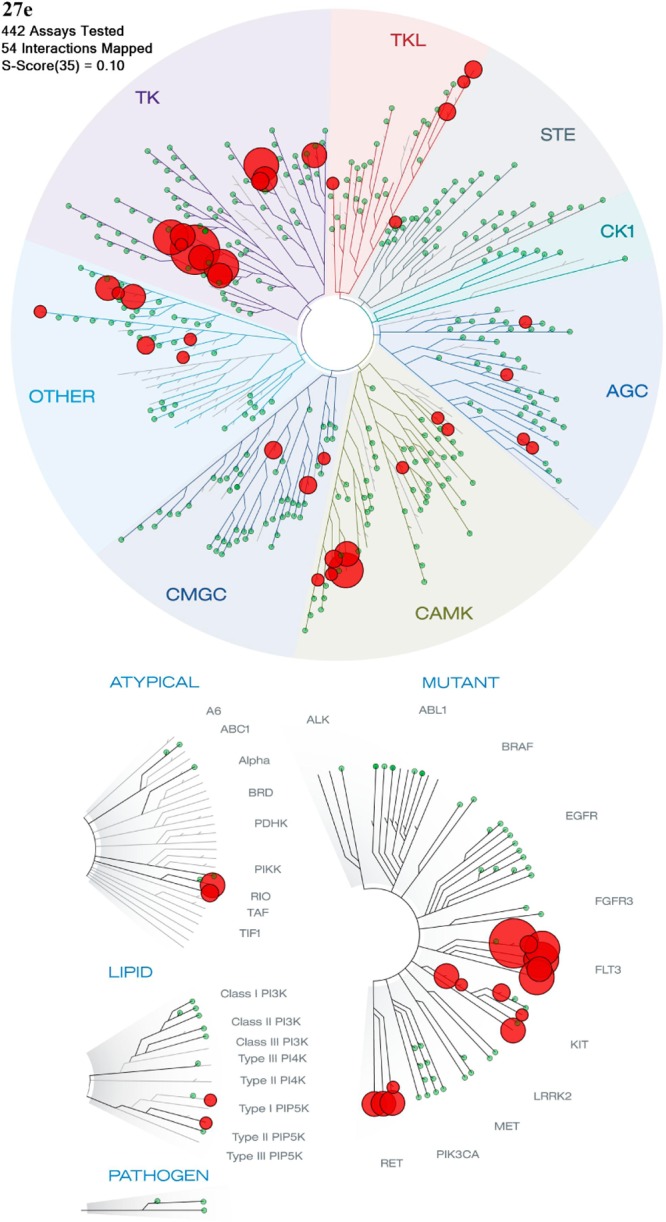
TREEspot interaction map48 depicting the selectivity profile for 27e. Significant off-target inhibition was observed for FLT1, JAK2, RET, and PDGFRB with percent control values of 0.3, 1.3, 1.8, and 4 at 1 μM respectively (Supporting Information, Table S2).
Selected examples from the imidazo[4,5-b]pyridine series including compound 6, which we recently reported as a dual FLT3/Aurora inhibitor,51 were also tested against FLT3 and FLT3-ITD. All compounds tested displayed potent affinity for both forms of FLT3 kinase (Table 7), indicating that FLT3 binding is a common feature for this series of imidazo[4,5-b]pyridines. FLT3 is a trans-membrane kinase of the class III receptor tyrosine kinase (RTK) family. Binding of FLT3-ligand (FL) to its receptor leads to dimerization, autophosphorylation, and subsequent activation of downstream signaling pathways, playing a role in the survival and proliferation of leukemic cells.52 High levels of FLT3 expression have been found in AML blasts, and two major classes of activating mutations, i.e. internal-tandem duplications (ITDs) and tyrosine kinase domain (TKD) point mutations, have been identified in AML patients.52,53 ITDs are detected in 20–25% of AML patients, and TKD point mutations in 5–10% of AML patients.52,53 Internal tandem duplication of the FLT3 gene (FLT3-ITD) results in constitutive FLT3 kinase activation.54 Meshinchi et al. reported that FLT3-ITD is detected in 20–35% of adults and 15% of children with AML, conferring a poor prognosis in both age groups.55 Over the past decade FLT3 inhibition has emerged as a therapeutic strategy of interest for the treatment of AML, and a number of small-molecule inhibitors have been evaluated in clinical trials.53,56
Table 7. FLT3 and FLT3-ITD Kd Values (nM).
| compd | FLT3 | FLT3-ITD |
|---|---|---|
| 6 | 1.2 | 4.9 |
| 21a | 14 | 62 |
| 21i | 12 | 66 |
| 20f | 5.4 | 26 |
| 22d | 2.4 | 10 |
| 27b | 4.4 | 14 |
| 27c | 5.6 | 26 |
| 27f | 5.9 | 15 |
Consistent with dual FLT3/Aurora inhibitory activity, compound 27e displayed antiproliferative activity in a range of human tumor cell lines, including HCT116 human colon carcinoma (GI50 = 0.300 μM) and the human FLT3-ITD positive AML cell lines MOLM-13 (GI50 = 0.104 μM) and MV4–11 (GI50 = 0.291 μM). In Hela cervical cancer cells, 27e inhibited both the autophosphorylation of Aurora-A at T288 (a biomarker for Aurora-A inhibition: IC50 = 0.030 μM) and histone H3 phosphorylation at S10 (a biomarker for Aurora-B inhibition: IC50 = 0.148 μM),57,58 consistent with potent cellular activity versus both Aurora-A and -B. Compound 27e also inhibited Aurora-A in MOLM-13 cells with concomitant inhibition of FLT3 signaling (FLT3 phosphorylation, data not shown).
The inhibition of cytochrome P450 isoforms by 27e was also investigated. Compound 27e did not significantly inhibit the major cytochrome P450 isoforms (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP3A4); all IC50 values were greater than 10 μM. Compound 27e performed well in the Caco-2 assay, giving a permeability of 18.6 × 10–6 cm/s with no efflux. The desirable set of in vitro properties led to the in vivo evaluation of 27e. The mouse plasma protein binding for 27e was determined as 97.3%, and the PK profile of 27e in mouse revealed a highly orally bioavailable compound (F = 100%) with moderate clearance (0.058 L/h, 48 mL/min/kg) and Vd (0.066 L, 3.3 L/kg) (Table 8). Pharmacokinetic evaluation in rats also showed high oral bioavailability (79%), low clearance (0.057 L/h, 4.57 mL/min/kg), and moderate volume of distribution (0.369 L, 1.79 L/kg) (Table 8).
Table 8. PK Parameters in Mouse and Rat for Compound 27e (iv dosing, 5 mg/kg; oral dosing, 5 mg/kg).
| T1/2 (iv) (h) | Cl (iv) (L/h) | AUCinf (iv) (h nmol/L) | Vd (L) | F (po) (%) | |
|---|---|---|---|---|---|
| mouse | 0.84 | 0.058 | 3753 | 0.066 | 100 |
| rat | 4.62 | 0.057 | 39853 | 0.369 | 79 |
The encouraging in vivo pharmacokinetic profile coupled with dual FLT3/Aurora inhibitory activity prompted us to evaluate 27e in a human AML subcutaneous xenograft model. As shown in Figure 5, 27e strongly inhibited the growth of MV4–11 human tumor xenografts in a dose-dependent manner with no observed toxicity as defined by body weight loss and general condition. When therapy was discontinued after 11 days, tumors were undetectable in mice treated with an oral dose of 100 mg/kg po b.i.d. 27e and had decreased to 42% of the initial volume in those treated with 50 mg/kg b.i.d. Control mice were culled on day 18 from the start of therapy when the mean tumor volume had increased by over 500%. In contrast, individual mice were culled when tumors progressed to this stage as follows: days 28 and 31 at 50 mg/kg and days 46 and 56 at 100 mg/kg. Three out of five mice in each treatment group (60%) did not develop progressively growing tumors at the time the study was terminated on day 60, indicative of long-term growth control. As a result of this potent in vivo antitumor effect, tumors from treated animals were too small to provide material for a pharmacokinetic/pharmacodynamic analysis.
Figure 5.

Oral efficacy of 27e against MV4–11 human FLT3-ITD positive AML tumor xenografts in athymic mice (dosing interval day 0–11; twice daily for 7 days, and once daily for a further 4 days): (A) relative tumor volumes ± SEM and (B) mouse body weights. N = 5 per group.
A 4-day PK/PD study (27e po at 50 and 100 mg/kg b.i.d.) showed clear inhibition of both histone H-3 phosphorylation and Stat5 phosphorylation (a direct downstream target of FLT3 kinase) at 2 h after the final dose, consistent with dual inhibition of Aurora and FLT3 kinases in the tumor (Figure 6).51,59,60 In addition, average free drug concentrations in plasma samples obtained 2 h after the final dose (222 and 488 nM for the 50 and 100 mg/kg dosing schedules, respectively; Figure 6) significantly exceed Kd values of 27e against the relevant kinases, i.e., Aurora-A (Kd = 7.5 nM), Aurora-B (Kd = 48 nM), FLT3 (Kd = 6.2 nM), FLT3-ITD (Kd = 38 nM). These findings demonstrate that 27e significantly inhibits the growth of a clinically relevant FLT3-ITD-positive AML human tumor xenograft model in vivo, with biomarker modulation and free drug exposure consistent with dual FLT3 and Aurora kinase target inhibition.
Figure 6.

PK/PD study in mice bearing MV4–11 human FLT3-ITD-positive AML tumor xenografts treated with 27e (50 and 100 mg/kg po, b.i.d., for 4 days). (A) Total plasma and tumor concentrations and free plasma concentrations; all samples were taken 2 h after the final dose. (B) Inhibition of histone H-3 phosphorylation at S10 and inhibition of Stat5 phosphorylation at Y694. Tumor samples were obtained 2 h after the final dose. Total histone H3, total Stat5, and GAPDH were used as loading controls.
Conclusion
Compound 6 and its 6-Cl counterpart 5 were used as starting points for medicinal chemistry optimization aimed at the identification of an orally bioavailable preclinical development candidate. In the course of this work we paid particular attention to minimization of hERG inhibition and human liver microsomal instability. We found that the switch of the N-methylpiperazine moiety in compound 5 to the m-position was beneficial in reducing affinity for hERG and significantly increasing the human liver microsomal stability. Replacement of the R2 (4-methylpiperazin-1-yl)phenyl substituent with five-membered heteroaromatics led to the identification of 1,3-dimethyl-1H-pyrazol-4-yl as an optimal R2 substituent. Subsequent property refinement by R3 substituent modification led to 27e, an orally bioavailable, dual FLT3/Aurora kinase inhibitor with high selectivity within the tested kinome. Compound 27e also potently inhibits mutant FLT3 kinases, including FLT3-ITD, which causes constitutive FLT3 kinase activation and is detected in 20–35% of adults and 15% of children with AML, conferring a poor prognosis in both age groups. In an in vivo setting, 27e strongly inhibited the growth of a FLT3-ITD-positive AML human tumor xenograft with biomarker modulation and plasma free drug exposures consistent with dual FLT3 and Aurora kinase target inhibition. Compound 27e, an orally bioavailable dual FLT3/Aurora kinase inhibitor, has been selected as a preclinical development candidate for the treatment of human malignancies, with particular relevance for the treatment of AML in adults and children who become resistant to existing therapies.61
Experimental Section
Aurora Kinase Assays
Aurora kinase IC50 values were determined as previously described.32,62
Kinase Selectivity Profiling
Kinase profiling was performed using the KINOMEScan technology, and Kd determinations were performed by KINOMEscan, a Division of DiscoveRx Corp., San Diego, CA (www.kinomescan.com).
Cell Viability Assay
GI50 values (50% cell growth inhibitory concentration) were determined as previously described.32,62
Determination of Cellular IC50 Values of 27e for Aurora-A and -B Inhibition
Myc-tagged Aurora-A was transfected in Hela cells using Lipofectamine LTX in 24 well plates. Twenty-four hours after transfection, cells were treated with different concentrations of 27e for 2 h. Cells were then lysed in 2× LDS sample buffer. Proteins from different samples were resolved by 4–12% Bis-Tris NuPage (Invitrogen) gels and analyzed by Western blotting using P-histone H3 (S10) and P-Aurora-A (T288) antibodies. The bands for P-histone H3 and P-Aurora-A were quantified using Image J software, and IC50 values were calculated using Graphpad Prism.
Cocrystallization of Aurora-A with Ligand
Wild-type Aurora-A catalytic domain (residues 122–403) was expressed and purified as previously described.5 Cocrystals with 21a were produced using 0.1 M sodium acetate pH 4.5, 0.2 M (NH4)2SO4, 25% PEG 4000/2000 MME as crystallization buffer. Structures were solved by molecular replacement using Aurora-A (PDB code 1MQ4) as a model. Ligand fitting and model rebuilding was carried out using Coot63 and refinement was carried out using Phenix.64 Coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4B0G.
Mouse Liver Microsomal Stability
Compounds (10 μM) were incubated with male CD1 mouse liver microsomes (1 mg mL–1) protein in the presence of NADPH (1 mM), UDPGA (2.5 mM), and MgCl2 (3 mM) in phosphate-buffered saline (10 mM) at 37 °C. Incubations were conducted for 0 and 30 min. Control incubations were generated by the omission of NADPH and UDPGA from the incubation reaction. The percentage compound remaining was determined after analysis by LC–MS.
Human Liver Microsomal Stability
Compounds (10 μM) were incubated with mixed gender pooled human liver microsomes (1 mg mL–1) protein in the presence of NADPH (1 mM), UDPGA (2.5 mM), and MgCl2 (3 mM) in phosphate-buffered saline (10 mM) at 37 °C. Incubations were conducted for 0 and 30 min. Control incubations were generated by the omission of NADPH and UDPGA from the incubation reaction. The percentage compound remaining was determined after analysis by LC–MS.
Inhibition of Cytochrome P450 Isoforms
Inhibition of human liver CYP isozymes was assessed in human liver microsomes (pool of 50 individuals) as previously described65 with the following modifications: microsomal protein concentration 0.5 mg/mL, incubation time 10 min, mephenytoin as the CYP2C19 substrate, and metabolite detection by LC–MSMS ESI+ on an Agilent 1290 Infinity Series LC system with 6410 triple quadrupole mass spectrometer (4-hydroxytolbutamide, hydroxymephenytoin) or Waters Acquity UPLC connected to a QTRAP 4000 (Applied Biosystems).
hERG Inhibition
All hERG percentage inhibitions at 10 μM compound concentration were determined by Millipore in a high-throughput cell-based electrophysiology assay for inhibition of hERG tail current,66 and values are reported as a mean of multiple determinations. 0.3% DMSO aqueous vehicle negative control gave 7–16% inhibition. Cisapride (1 μM) positive control gave 96–104% inhibition. hERG IC50 values were determined by Millipore,66 and the hERG IC50 for compound 27e was also determined by Cyprotex plc.67 The hERG IC50 value for compound 20f (Table 2) was determined by Cyprotex plc measuring hERG tail currents by whole-cell voltage clamping,67 and in this assay, the hERG IC50 value for compound 5 was determined as 4.3 μM.
Physicochemical Properties
log D and pKa measurements were performed by Pharmorphix Solid State Services, Member of the Sigma-Aldrich Group, Cambridge, UK.
In Vivo Mouse PK (Compound 27e)
Mice (female Balb/C) were dosed po or iv with 27e (5 mg kg–1) in 10% DMSO, 5% Tween 20 in saline. After administration, mice were sacrificed at 5, 15, and 30 min and 1, 2, 4, 6, and 24 h. Blood was removed by cardiac puncture and centrifuged to obtain plasma samples. Plasma samples (100 μL) were added to the analytical internal standard (Olomoucine; IS), followed by protein precipitation with 300 μL of methanol. Following centrifugation (1200g, 30 min, 4 °C), the resulting supernatants were analyzed for 27e levels by LC–MS using a reverse-phase Acquity UPLC C18 (Waters, 50 × 2.1 mm) analytical column and positive ion mode ESI MRM on an Agilent 1200 liquid chromatography system coupled to a 6410 triple quadrupole mass spectrometer (Agilent Ltd.).
In Vivo Rat PK (Compound 27e)
Rats (female Sprague–Dawley) were dosed po or iv with 27e (5 mg kg–1) in 10% DMSO, 5% Tween 80, 20% PEG400 in water (po) or 10% DMSO, 5% Tween 20, 85% saline, 0.3% 1 M HCl (iv). Blood was removed by serial tail vein bleed at 15 and 30 min and 2, 4, 6, and 24 h and centrifuged to obtain plasma samples. Plasma samples (50 μL) were added to the analytical internal standard (Olomoucine; IS), followed by protein precipitation with 150 μL of methanol. Following centrifugation (1200g, 30 min, 4 °C), the resulting supernatants were analyzed for 27e levels by LC–MS using a reverse-phase Acquity UPLC C18 BEH (Waters, 50 × 2.1 mm) analytical column and positive ion mode ESI MRM on an Acquity H-Class UPLC coupled to Xevo TQ-S triple quadrupole mass spectrometer (Waters Ltd.).
Human Tumor Xenograft Efficacy Study
Procedures involving animals were carried out within guidelines set out by The Institute of Cancer Research’s Animal Ethics Committee and in compliance with national guidelines.68 Female adult CrTacNCr-Fox1(nu) athymic mice were implanted subcutaneously with 107FLT3-ITD-positive MV4–11 human leukemia cells. When the tumor xenografts were well-established (10 days after implantation, mean tumor volumes of at least 100 mm3), animals were treated with either vehicle (10% DMSO, 20% PEG 400, 5% Tween 80 and 65% water) or 27e administered orally at two doses, 50 and 100 mg/kg (n = 5 per group). Dosing was twice daily for 7 days, and once daily for a further 4 days.
PK/PD study
A 4-day PK/PD study was performed by oral administration of vehicle as above or 50 and 100 mg/kg of 27e twice daily in athymic mice bearing well-established MV4–11 AML tumor xenografts (17 days after implantation). Plasma and tumor samples were collected 2 and 6 h after the final doses.
Chemistry
Commercially available starting materials, reagents, and dry solvents were used as supplied. Flash column chromatography was performed using Merck silica gel 60 (0.025–0.04 mm). Column chromatography was also performed on a FlashMaster personal unit using ISOLUTE flash silica columns or a Biotage SP1 purification system using Biotage flash silica cartridges. Preparative TLC was performed on Analtech or Merck plates. Ion exchange chromatography was performed using acidic ISOLUTE Flash SCX-II cartridges. 1H NMR spectra were recorded on a Bruker Avance-500. Samples were prepared as solutions in a deuterated solvent and referenced to the appropriate internal nondeuterated solvent peak or tetramethylsilane. Chemical shifts were recorded in ppm (δ) downfield of tetramethylsilane. LC–MS analysis was performed on a Waters LCT with a Waters Alliance 2795 separations module and Waters 2487 dual-wavelength absorbance detector coupled to a Waters/Micromass LCT time-of-flight mass spectrometer with ESI source. Analytical separation was carried out at 30 °C either on a Merck Chromolith SpeedROD column (RP-18e, 50 × 4.6 mm) using a flow rate of 2 mL/min in a 3.5 min gradient elution with detection at 254 nm or on a Merck Purospher STAR column (RP-18e, 30 × 4 mm) using a flow rate of 1.5 mL/min in a 3.5 min gradient elution with detection at 254 nm. The mobile phase was a mixture of methanol (solvent A) and water (solvent B) both containing formic acid at 0.1%. Gradient elution was as follows: 1:9 (A/B) to 9:1 (A/B) over 2.25 min, 9:1 (A/B) for 0.75 min, and then reversion back to 1:9 (A/B) over 0.3 min, and finally 1:9 (A/B) for 0.2 min.
LC–HRMS analysis was performed on an Agilent 1200 series HPLC and diode array detector coupled to a 6520 quadrupole-time of flight mass spectrometer with dual multimode APCI/ESI source. Analytical separation was carried out at 30 °C on a Merck Purospher STAR column (RP-18e, 30 × 4 mm) using a flow rate of 1.5 mL/min in a 4 min gradient elution with detection at 254 nm. The mobile phase was a mixture of methanol (solvent A) and water (solvent B) both containing formic acid at 0.1%. Gradient elution was as follows: 1:9 (A/B) to 9:1 (A/B) over 2.5 min, 9:1 (A/B) for 1 min, and then reversion back to 1:9 (A/B) over 0.3 min, finally 1:9 (A/B) for 0.2 min. The following references masses were used for HRMS analysis: caffeine, [M + H]+ 195.087 652; hexakis(1H,1H,3H-tetrafluoropentoxy)phosphazene, [M + H]+ 922.009 798); and hexakis(2,2-difluoroethoxy)phosphazene, [M + H]+ 622.028 96, or reserpine, [M + H]+ 609.280 657.
Analytical HPLC analysis was performed on a Thermo-Finnigan Surveyor HPLC system or an Agilent Technologies 1200 series HPLC system at 30 °C using a Phenomenex Gemini C18 column (5 μm, 50 × 4.6 mm) and 10 min gradient of 10→90% MeOH/0.1% formic acid, visualizing at 254, 309, or 350 nm. The purity of final compounds was determined by analytical HPLC as described above and is ≥95% unless specified otherwise.
4-(4-Ethylpiperazin-1-yl)benzaldehyde (9c)
To a mixture of 4-bromobenzylaldehyde diethyl acetal (0.518 g, 2.0 mmol) and anhydrous toluene (4 mL) was added 1-ethylpiperazine (0.274 g, 2.4 mmol) followed by Pd2(dba)3 (0.018 g, 0.02 mmol), racemic BINAP (0.037 g, 0.06 mmol), and NaOtBu (0.326 g, 3.40 mmol). The reaction mixture was placed into an oil bath preheated to 100 °C, stirred at this temperature for 5 h under argon, and then allowed to cool to room temperature. Aqueous HCl (1 M, 10 mL) was added, the mixture was vigorously stirred for 2.5 h, and then the pH was adjusted to 13 with aqueous NaOH and extracted with ethyl acetate (3 × 50 mL). The combined organics were dried (Na2SO4) and concentrated in vacuo, and the residue was absorbed on silica gel and placed on a 20 g ISOLUTE column. Elution with a gradient of methanol (from 0 to 4%) in ethyl acetate/dichloromethane (v/v, 1:1) afforded the title compound as an oil (0.115 g, 26%). 1H NMR (500 MHz, DMSO-d6): 1.04 (t, J = 7.2 Hz, 3H, CH2CH3), 2.37 (q, J = 7.2 Hz, 2H, CH2CH3), 2.54 (m, 4H, piperazine C–H), 3.39 (t, J = 5.0 Hz, 4H, piperazine C–H), 7.05 (d, J = 8.9 Hz, 2H) and 7.71 (d, J = 8.9 Hz, 2H) (2,6-C6H4 and 3,5-C6H4), 9.72 (s, 1H, CHO). LC–MS (ESI, m/z): tR = 0.85 min; 219 (M + H)+.
3-((4-(6-Chloro-2-(4-(4-ethylpiperazin-1-yl)phenyl)-3H-imidazo[4,5-b]pyridin-7-yl)piperazin-1-yl)methyl)-5-methylisoxazole (10c)
To a mixture of 5-chloro-4-(4-((5-methylisoxazol-3-yl)methyl)piperazin-1-yl)-3-nitropyridin-2-amine32 (0.052 g, 0.15 mmol) and EtOH (7.0 mL) was added 4-(4-ethylpiperazin-1-yl)benzaldehyde (0.046 g, 0.21 mmol) followed by a freshly prepared aqueous solution of Na2S2O4 (1 M, 0.60 mL, 0.60 mmol). The reaction mixture was stirred at 80 °C for 18 h, allowed to cool to room temperature, and concentrated in vacuo. The residue was absorbed on silica gel and placed on a 10 g ISOLUTE silica column which was eluted first with a gradient of methanol (0 to 5%) in ethyl acetate/dichloromethane (v:v, 1:1) and then 6% methanol in chloroform. Fractions containing the product were combined and concentrated in vacuo, and the resulting solid residue was triturated with diethyl ether. The title compound was isolated by filtration as a white solid and washed with diethyl ether, water, and finally diethyl ether (0.012 g, 15%). 1H NMR (500 MHz, DMSO-d6): 1.04 (t, J = 6.8 Hz, 3H, CH2CH3), 2.37 (q, obscured by isoxazole 5-CH3 peak, CH2CH3), 2.40 (s, 3H, isoxazole 5-CH3), 2.62 (br s, 4H, piperazine C–H), 3.27 (br s, 4H, piperazine C–H), 3.59 (s, 2H, NCH2isoxazole), 3.67 (br s, 4H, piperazine C–H), 6.25 (s, 1H, isoxazole 4-H), 7.05 (d, J = 8.9 Hz, 2H) and 8.02 (d, J = 8.9 Hz, 2H) (2,6-C6H4 and 3,5-C6H4), 8.04 (s, 1H, imidazo[4,5-b]pyridine 5-H), 13.22 (br s, 1H, imidazo[4,5-b]pyridine N–H). LC–MS (ESI, m/z): tR = 1.70 min; 521, 523 (M + H)+, Cl isotopic pattern. HRMS: found 521.2532, calcd for C27H34ClN8O (M + H)+ 521.2539.
3-(4-Methylpiperazin-1-yl)benzaldehyde (19f)
To a solution of 3-bromobenzylaldehyde diethyl acetal (0.518 g, 2.0 mmol) and anhydrous toluene (4 mL) was added 1-methylpiperazine (0.240 g, 2.4 mmol) followed by Pd2(dba)3 (0.018 g, 0.02 mmol), racemic BINAP (0.037 g, 0.06 mmol), and NaOtBu (0.326 g, 3.4 mmol). The reaction mixture was placed into an oil bath preheated to 100 °C, stirred at this temperature for 18 h under argon, and then allowed to cool to room temperature. Aqueous HCl (1 M, 10 mL) was added, the mixture was vigorously stirred for 2.5 h, the pH was adjusted to 13 with 6 M aqueous NaOH, and the mixture was extracted with ethyl acetate (3 × 30 mL). The combined organics were dried (Na2SO4), and concentrated in vacuo, and the residue was absorbed on silica gel and placed on a 10 g ISOLUTE column. Elution with ethyl acetate/CH2Cl2 (v/v, 4:1) and then a gradient of methanol (from 3 to 7%) in ethyl acetate afforded the title compound as a yellow oil (0.170 g, 42%). 1H NMR (500 MHz, DMSO-d6): 2.22 (s, 3H, N-Me), 2.46 (t, J = 5.0 Hz, 4H, piperazine C–H), 3.21 (t, J = 5.1 Hz, 4H, piperazine C–H), 7.28 (m, 2H, PhH), 7.41 (m, 2H, PhH), 9.94 (s, 1H, CHO). LC–MS (ESI, m/z): tR = 0.86 min; 205 (M + H)+.
3-((4-(6-Chloro-2-(3-(4-methylpiperazin-1-yl)phenyl)-3H-imidazo[4,5-b]pyridin-7-yl)piperazin-1-yl)methyl)-5-methylisoxazole (20f)
To a mixture of 5-chloro-4-(4-((5-methylisoxazol-3-yl)methyl)piperazin-1-yl)-3-nitropyridin-2-amine32 (0.090 g, 0.26 mmol) and EtOH (20 mL) was added 3-(4-methylpiperazin-1-yl)benzaldehyde (0.057 g, 0.28 mmol) followed by a freshly prepared aqueous solution of Na2S2O4 (1 M, 0.76 mL, 0.76 mmol). The reaction mixture was stirred at 80 °C for 24 h, allowed to cool to room temperature, and concentrated in vacuo. The residue was partitioned between chloroform and 5% aqueous sodium hydrogen carbonate. The two layers were separated, and the organic layer was dried (Na2SO4) and concentrated in vacuo. The crude product was applied to an SCX ion-exchange column (5 g, 25 mL) which was eluted with 10% methanol in chloroform followed by 1 M ammonia in methanol. Fractions containing the pure product were combined and concentrated in vacuo. The title compound was obtained as a powder after trituration with diethyl ether (0.070 g, 54%). 1H NMR (500 MHz, DMSO-d6): 2.24 (s, 3H, N–CH3), 2.40 (s, 3H, isoxazole 5-CH3), 2.62 (br s, 4H, piperazine C–H), 3.24 (poorly resolved t, 4H, piperazine C–H), 3.59 (s, 2H, N–CH2-isoxazole), 3.69 (br s, 4H, piperazine C–H), 6.25 (s, 1H, 4-H isoxazole), 7.06 (dd, J = 1.8, 8.3 Hz, 1H, PhH), 7.35 (t, J = 8.3 Hz, 1H, PhH), 7.61 (d, J = 7.8 Hz, 1H, PhH), 7.72 (s, 1H, PhH), 8.10 (s, 1H, imidazo[4,5-b]pyridine 5-H), 13.40 (br s, 1H, imidazo[4,5-b]pyridine N–H). LC–MS (ESI, m/z): tR = 1.53 min; 507, 509 (M + H)+, Cl isotopic pattern. HRMS: found 507.2380, calcd for C26H32ClN8O (M + H)+ 507.2382.
3-((4-(6-Bromo-2-(1-methyl-1H-imidazol-5-yl)-3H-imidazo[4,5-b]pyridin-7-yl)piperazin-1-yl)methyl)-5-methylisoxazole (21a)
To a mixture of 5-bromo-4-(4-((5-methylisoxazol-3-yl)methyl)piperazin-1-yl)-3-nitropyridin-2-amine32 (0.1 g, 0.25 mmol) and EtOH (15 mL) was added 1-methyl-1H-imidazole-5-carbaldehyde (30 mg, 0.32 mmol) followed by a freshly prepared aqueous solution of Na2S2O4 (1 M, 1 mL, 1 mmol). The reaction mixture was heated at reflux for 24 h, allowed to cool to room temperature, and concentrated in vacuo. The residue was taken up in chloroform and a 10% aqueous bicarbonate solution. The aqueous layer was further extracted with dichloromethane, and the combined organic solutions were dried and concentrated. Ether was added to the residue and a pale white powder precipitated; this was filtered and dried (70 mg). The product was purified on an SCX ion-exchange column to provide the title compound as an off-white powder after trituration with ether (60 mg, 53%). 1H NMR (500 MHz, DMSO-d6): 2.39 (s, 3H, isoxazole 5-CH3), 2.63 (br s, 4H, piperazine NCH2), 3.59 (s, 2H, N-CH2-isoxazole), 3.65 (br s, 4H, piperazine NCH2), 4.05 (s, 3H, N–CH3), 6.23 (s, 1H, 4-H isoxazole), 7.73 (s, 1H imidazole-CH), 7.86 (s, 1H, imidazole-CH), 8.21 (s, 1H, imidazo[4,5-b]pyridine 5-H), 13.30 (br s, 1H, imidazo[4,5-b]pyridine N–H). LC–MS (ESI, m/z): tR = 1.72 min; 457, 459 (M + H)+, Br isotopic pattern. HRMS: found 457.1094, calcd for C19H22BrN8O (M + H)+ 457.1091.
3-((4-(6-Chloro-2-(1-methyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridin-7-yl)piperazin-1-yl)methyl)-5-methylisoxazole (21i)
To a mixture of 5-chloro-4-(4-((5-methylisoxazol-3-yl)methyl)piperazin-1-yl)-3-nitropyridin-2-amine (0.06 g, 0.17 mmol) and EtOH (10 mL) was added 1-methyl-1H-pyrazole-4-carbaldehyde (0.023 g, 0.21 mmol) followed by a freshly prepared aqueous solution of Na2S2O4 (1 M, 0.45 mL, 0.45 mmol). The reaction mixture was heated at reflux for 24 h, allowed to cool to room temperature, and concentrated in vacuo. The residue was taken up in chloroform and 10% bicarbonate. The aqueous layer was further extracted with dichloromethane, and the combined organic solutions were dried and concentrated in vacuo. The crude product was purified by silica column chromatography eluting with 2–10% methanol in dichloromethane. The pure fractions provided 38 mg of title compound (54%). 1H NMR (500 MHz, DMSO-d6): 2.40 (s, 3H, isoxazole 5-CH3), 2.61 (br s, 4H, piperazine NCH2), 3.59 (s, 2H, N–CH2-isoxazole), 3.62 (br s, 4H, piperazine NCH2), 3.92 (s, 3H, NCH3), 6.24 (s, 1H, 4-H isoxazole), 8.03 (s, 1H, pyrazole-H), 8.04 (s, 1H, H pyrazole-H), 8.32 (s, 1H, imidazo[4,5-b]pyridine 5-H), 13.20 (br s, 1H, imidazo[4,5-b]pyridine N–H). LC–MS (ESI, m/z): tR = 2.35 min; 413, 415 (M + H)+, Cl isotopic pattern. HRMS: found 413.1599, calcd for C19H22ClN8O (M + H)+ 413.1599.
6-Chloro-2-(1,3-dimethylpyrazol-4-yl)-7-(4-(5-methylisoxazol-3-ylmethyl)piperazin-1-yl)-3H-imidazo[4,5-b]pyridine (22d)
To a mixture of 1,3-dimethylpyrazole-4-aldehyde (27.3 mg, 0.20 mmol) and 2-amino-5-chloro-4-(4-(5-methylisoxazol-3-yl)methylpiperazin-1-yl)-3-nitropyridine (70.5 mg, 0.20 mmol) in ethanol (1.4 mL) was added freshly prepared 1 M aqueous Na2S2O4 solution (0.7 mL, 0.7 mmol). The reaction was stirred and heated at 75 °C for 19.5 h. The reaction was cooled, 5 M ammonia solution (0.4 mL) was added, and the mixture was stirred for 15 min. Most of the ethanol was evaporated and water (2 mL) was added. The product was extracted into ethyl acetate (8, 4, 4 mL), and the combined extracts were washed with brine, dried (Na2SO4), and evaporated to leave a cream solid. This was triturated with ether (2 mL) and the solid further washed with ether (1 mL) to leave the product (58 mg, 67%). 1H NMR (500 MHz, DMSO-d6): 2.41 (s, 3H, CH3), 2.52 (s, 3H, CH3), 2.63 (m, 4H, piperazine C–H), 3.59 (s, 2H, CH2), 3.69 (m, 4H, piperazine C–H), 3.85 (s, 3H, N–CH3), 6.24 (s, 1H, heteroaryl H), 8.03 (s, 1H, heteroaryl H), 8.19 (s, 1H, heteroaryl H), 12.98 (s, 1H, NH). LC–MS (ESI, m/z): tR = 1.78 min; 427, 429 (M + H)+, Cl isotope pattern. HRMS: found 427.1756, calcd for C20H24ClN8O (M + H)+ 427.1756.
5-Chloro-4-(4-(4-chlorobenzyl)piperazin-1-yl)-3-nitropyridin-2-amine (26e)
To a mixture of 2-amino-4,5-dichloro-3-nitropyridine (0.152 g, 0.73 mmol) and 2-propanol (22 mL) was added 1-(4-chlorobenzyl)piperazine (0.165 g, 0.78 mmol) followed by diisopropylethylamine (0.17 mL, 0.97 mmol). The reaction mixture was heated at 45 °C for 18 h, allowed to cool to room temperature, and diluted with 2-propanol (5 mL). The precipitate was collected by filtration, washed with 2-propanol, and diethyl ether. The title compound was thus obtained as a yellow solid (0.215 g, 77%). 1H NMR (500 MHz, DMSO-d6): 2.48 (br s, obscured by DMSO peak, 4H, piperazine C–H), 3.06 (br t, J = 4.3 Hz, 4H, piperazine C–H), 3.52 (s, 2H, NCH2C6H4Cl), 6.95 (s, 2H, NH2), 7.35 (d, J = 8.5 Hz, 2H) and 7.38 (d, J = 8.5 Hz, 2H) (3,5-ArH and 2,6-ArH), 8.06 (s, 1H, 6-H). LC–MS (ESI, m/z): tR = 1.70 min; 382, 384, 386 (M + H)+, Cl2 isotopic pattern.
6-Chloro-7-(4-(4-chlorobenzyl)piperazin-1-yl)-2-(1,3-dimethyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine (27e)
To a mixture of 5-chloro-4-(4-(4-chlorobenzyl)piperazin-1-yl)-3-nitropyridin-2-amine (0.076 g, 0.20 mmol) and EtOH (4.0 mL) was added 1,3-dimethyl-1H-pyrazole-4-carbaldehyde (0.027 g, 0.22 mmol) followed by a freshly prepared aqueous solution of Na2S2O4 (1 M, 0.85 mL, 0.85 mmol). The reaction mixture was stirred at 80 °C for 24 h, allowed to cool to room temperature, and concentrated in vacuo, and the residue was absorbed on silica gel and placed on a 10 g ISOLUTE silica column. Elution with ethyl acetate/dichloromethane (v/v, 1:1) and then 4% methanol in ethyl acetate/dichloromethane (v/v; 1:1) afforded the title compound as a white solid after trituration with diethyl ether (0.023 g, 25%). 1H NMR (500 MHz, DMSO-d6): 2.51 (s, obscured by solvent peak, pyrazole 3-CH3), 2.57 (br s, 4H, piperazine C–H), 3.54 (s, 2H, N-CH2C6H4Cl), 3.68 (br s, 4H, piperazine C–H), 3.84 (s, 3H, pyrazole N-Me), 7.37 (d, J = 8.5 Hz, 2H) and 7.40 (d, J = 8.5 Hz, 2H) (C6H4Cl), 8.02 (s, 1H), and 8.18 (s, 1H) (pyrazole 5-H, and imidazo[4,5-b]pyridine 5-H), 12.95 (br s, 1H, imidazo[4,5-b]pyridine N–H). LC–MS (ESI, m/z): tR = 1.97 min; 456, 458, 460 (M + H)+, Cl2 isotopic pattern. HRMS: found 456.1457, calcd for C22H24Cl2N7 (M + H)+ 456.1465.
Acknowledgments
This work was supported by Cancer Research UK [CUK] grant numbers C309/A8274 and C309/A11566. A.S.M. was supported by Cancer Research UK (Grant number C1178/A10294) and in part by a New Investigator Scholarship awarded by the Haematology Society of Australia and New Zealand. S.L. is also supported by Breakthrough Breast Cancer. R.B. is a Royal Society University Research Fellow and acknowledges the support of Breakthrough Breast Cancer Project Grant AURA 05/06 and infrastructural support for Structural Biology at the ICR from Cancer Research UK. We acknowledge NHS funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and the Royal Marsden NHS Foundation Trust. We also thank Dr. Amin Mirza, Mr. Meirion Richards, and Dr. Maggie Liu for their assistance with NMR, mass spectrometry, and HPLC.
Glossary
Abbreviations Used:
- AML
acute myeloid leukemia
- AUC
area under the curve
- Boc
tert-butoxycarbonyl
- DIPEA
N,N-diisopropylethylamine
- ESI
electrospray ionization
- FLT3
FMS-like tyrosine kinase 3
- hERG
the human ether-a-go-go related gene
- HPLC
high-performance liquid chromatography
- HLM
human liver microsomes
- HRMS
high-resolution mass spectrometry
- inh
inhibition
- LC
liquid chromatography
- MLM
mouse liver microsomes
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- Pd2(dba)3
tris(dibenzylideneacetone)dipalladium(0)
- PPB
plasma protein binding
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid.
Supporting Information Available
Experimental procedures for compounds 11–13, 9b,d,e, 10a–e, 17, 19a,b, 20a-e, 21b–h, 22a–c,e, 24b,d,f,g, 25b,d,f,g, 26a–d, 26f,g, 27a–d, 27f,g; summary of crystallographic analysis of compound 21a (Table S1); and kinase selectivity profiling of compound 27e (Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Atomic coordinates and structure factors for the crystal structure of Aurora-A with compound 21a can be accessed using PDB code 4B0G.
The authors declare the following competing financial interest(s): Please note that all authors who are, or have been, employed by The Institute of Cancer Research are subject to a “Rewards to Inventors Scheme” which may reward contributors to a programme that is subsequently licensed. Paul Workman was a founder and shareholder in Piramed Pharma and is a founder and shareholder in Chroma Therapeutics. Paul Workman is a former employee of AstraZeneca. Julian Blagg is former employee and shareholder of Pfizer Inc.
Author Present Address
∥ Queensland Children’s Medical Research Institute, Royal Children’s Hospital, Herston Rd, Herston, QLD 4029, Australia.
Author Present Address
⊥ University of Leicester, Department of Biochemistry, Lancaster Road, Leicester LE1 9HN, United Kingdom.
Supplementary Material
References
- Carmena M; Earnshaw W. C. The Cellular Geography of Aurora Kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [DOI] [PubMed] [Google Scholar]
- Ducat D.; Zheng Y. Aurora Kinases in Spindle Assembly and Chromosome Segregation. Exp. Cell Res. 2004, 301, 60–67. [DOI] [PubMed] [Google Scholar]
- Marumoto T.; Zhang D.; Saya H. Aurora-A, a Guardian of Poles. Nat. Rev. Cancer 2005, 5, 42–50. [DOI] [PubMed] [Google Scholar]
- Barr A. R.; Gergely F. Aurora-A: The Maker and Breaker of Spindle Poles. J. Cell Sci. 2007, 120, 2987–2996. [DOI] [PubMed] [Google Scholar]
- Bayliss R; Sardon T.; Vernos I.; Conti E. Structural Basis of Aurora-A Activation by TPX2 at the Mitotic Spindle. Mol. Cell 2003, 12, 851–862. [DOI] [PubMed] [Google Scholar]
- Giet R.; Glover D. M. Drosophila Aurora B Kinase Is Required for Histone H3 Phosphorylation and Condensin Recruitment During Chromosome Condensation and to Organise the Central Spindle During Cytokinesis. J. Cell Biol. 2001, 152, 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann R.; Carvalho A.; Henzing A. J.; Ruchaud S.; Hudson D. F.; Honda R.; Nigg E. A.; Gerloff D. L.; Earnshaw W. C. Borealin: A Novel Chromosomal Passenger Required for Stability of the Bipolar Mitotic Spinde. J. Cell Biol. 2004, 166, 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa F.; Mapelli M.; Ciferri C.; Tarricone C.; Areces L. B.; Schneider T. R.; Stukenberg P. T.; Musacchio A. Mechanism of Aurora B Activation by INCENP and Inhibition by Hesperadin. Mol. Cell 2005, 18, 379–391. [DOI] [PubMed] [Google Scholar]
- Bishop J. D.; Schumacher J. M. Phosphorylation of the Carboxyl Terminus of Inner Centromere Protein (INCENP) by Aurora B Kinase Stimulates Aurora B Kinase Activity. J. Biol. Chem. 2002, 277, 27577–27580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T.; Kimura M.; Matsunaga K.; Fukada D.; Mori H.; Okano Y. Centrosomal Kinase AIK1 Is Overexpressed in Invasive Ductal Carcinoma of the Breast. Cancer Res. 1999, 59, 2041–2044. [PubMed] [Google Scholar]
- Bischoff J. R.; Anderson L.; Zhu Y.; Mossie K.; Ng L.; Souza B.; Schryver B.; Flanagan P.; Clairvoyant F.; Ginther C.; Chan C. S. M.; Novotny M.; Slamon D. J.; Plowman G. D. A Homologue of Drosophila Aurora Kinase is Oncogenic and Amplified in Human Colorectal Cancers. EMBO J. 1998, 17, 3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsko T. M.; Coppola D.; Paciga J. E.; Yang L.; Sun M.; Shelley S. A.; Fiorica J. V.; Nicosia S. V.; Cheng J. Q. Activation and Overexpression of Centrosome Kinase BTAK/Aurora-A in Human Ovarian Cancer. Clin. Cancer. Res. 2003, 9, 1420–1426. [PubMed] [Google Scholar]
- Reichardt W.; Jung V.; Brunner C.; Klein A.; Wemmert S.; Romeike B. F. M.; Zang K. D.; Urbschat S. The Putative Serine/Threonine Kinase Gene STK15 on Chromosome 20q13.2 is Amplified in Human Gliomas. Oncol. Rep. 2003, 10, 1275–1279. [PubMed] [Google Scholar]
- Chieffi P.; Troncone G.; Caleo A.; Libertini S.; Linardopoulos S.; Tramontano D.; Portella G. Aurora B Expression in Normal Testis and Seminonas. J. Endocrinol. 2004, 181, 263–270. [DOI] [PubMed] [Google Scholar]
- Araki K.; Nozaki K.; Ueba T.; Tatsuka M.; Hashimoto N. High Expression of Aurora-B/Aurora and Ipl1I-like Midbody-Associated Protein (AIM-1) in Astrocytomas. J.Neurooncol. 2004, 67, 53–64. [DOI] [PubMed] [Google Scholar]
- Sorrentino R.; Libertini S.; Pallante P. L.; Troncone G.; Palombini L.; Bavetsias V.; Cernia D. S.; Laccetti P.; Linardopoulos S.; Chieffi P.; Fusco A.; Portella G. Aurora B Overexpression Associates with the Thyroid Carcinoma Undifferentiated Phenotype and is Required for Thyroid Carcinoma Cell Proliferation. J. Clin. Endocrinol. Metab. 2005, 90, 928–935. [DOI] [PubMed] [Google Scholar]
- Kimura M.; Matsuda Y.; Yoshioka T.; Okano Y. Cell Cycle-dependent Expression and Centrosome Localisation of a Third Human Aurora/Ipl1-related Protein Kinase, AIK3. J. Biol. Chem. 1999, 274, 7334–7340. [DOI] [PubMed] [Google Scholar]
- Pollard J. R.; Mortimore M. Discovery and Development of Aurora kinase inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 2629–2651. [DOI] [PubMed] [Google Scholar]
- Moore A. S.; Blagg J.; Linardopoulos S.; Pearson A. D. J. Aurora Kinase Inhibitors: Novel Small Molecules With Promising Activity in Acute Myeloid and Philadelphia– Positive Leukemias. Leukemia 2010, 24, 671–678. [DOI] [PubMed] [Google Scholar]
- Gautschi O.; Heighway J.; Mack P. C.; Purnell P. R.; Lara P. N. Jr.; Gandara D. R. Aurora Kinases as Anticancer Drug Targets. Clin. Cancer Res. 2008, 14, 1639–1648. [DOI] [PubMed] [Google Scholar]
- Ikezoe T.; Yang J.; Nishioka C.; Tasaka T.; Taniguchi A.; Kuwayama Y.; Komatsu N.; Bandobashi K.; Togitani K.; Koeffler H. P.; Taguchi H. A Novel Treatment Strategy Targeting Aurora Kinases in Myelogenous Leukemia. Mol. Cancer Ther. 2007, 6, 1851–1857. [DOI] [PubMed] [Google Scholar]
- Ochi T.; Fujiwara H.; Suemori K.; Azuma T.; Yakushijin Y.; Hato T.; Kuzushima K.; Yasukawa M. Aurora-A Kinase: A Novel Target of Cellular Immunotherapy for Leukemia. Blood 2009, 113, 66–74. [DOI] [PubMed] [Google Scholar]
- Huang X.-F.; Luo S.-K.; Xu J.; Li J.; Xu D.-R.; Wang L.-H.; Yan M.; Wang X.-R.; Wan X.-B.; Zheng F.-M.; Zeng Y.-X.; Liu Q. Aurora Kinase inhibitory VX-680 Increases Bax/Bcl-2 Ratio and Induces Apoptosis in Aurora-A-High Acute Myeloid Leukemia. Blood 2008, 111, 2854–2865. [DOI] [PubMed] [Google Scholar]
- Walsby E.; Walsh V.; Pepper C.; Burnett A.; Mills K. Effects of the Aurora Kinase Inhibitors AZD1152-HQPA and ZM447439 on Growth Arrest and Polyploidy in Acute Myeloid Leukemia Cell Lines and Primary Blasts. Haematologica 2008, 93, 662–669. [DOI] [PubMed] [Google Scholar]
- Green M. R.; Woolery J. E.; Mahadevan D. Update on Aurora Kinase Targeted Therapeutics in Oncology. Expert Opin. Drug Discovery 2011, 6, 291–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C. H. A.; Coumar M. S.; Chang J.-Y.; Hsieh H.-P. Aurora Kinase Inhibitor Patents and Agents in Clinical Testing: An Update (2009–10). Expert Opin. Ther. Patents 2011, 21, 857–884. [DOI] [PubMed] [Google Scholar]
- Harrington E. A.; Bebbington D.; Moore J.; Rasmussen R. K.; Ajose-Adeogun A. O.; Nakayama T.; Graham J. A.; Demur C.; Hercend T.; Diu-Hercend A.; Su M.; Golec J. M. C.; Miller K. M. VX-680, a Potent and Selective Small-Molecule Inhibitor of the Aurora Kinases, Suppresses Tumor Growth. In Vivo Nat. Med. 2004, 10, 262–267. [DOI] [PubMed] [Google Scholar]
- Mortlock A. A.; Foote K. M.; Heron N. M.; Jung F. H.; Pasquet G.; Lohmann J-J. M.; Warin N.; Renaud F.; De Savi C.; Roberts N. J.; Johnson T.; Dousson C. B.; Hill G. B.; Perkins D.; Hatter G.; Wilkinson R. W.; Wedge S. R.; Heaton S. P.; Odedra R.; Keen N. J.; Crafter C.; Brown E.; Thompson K.; Brightwell S.; Khatri L.; Brady M. C.; Kearney S.; McKillop D.; Rhead S.; Parry T.; Green S. Discovery, Synthesis, and in Vivo Activity of a New Class of Pyrazoloquinazolines as Selective Inhibitors of Aurora B Kinase. J. Med. Chem. 2007, 50, 2213–2224. [DOI] [PubMed] [Google Scholar]
- Fancelli D.; Moll J.; Varasi M.; Bravo R.; Artico R.; Berta D.; Bindi S.; Cameron A.; Candiani I.; Cappella P.; Carpinelli P.; Croci W.; Forte B.; Giorgini M. L.; Klapwijk J.; Marsiglio A.; Pesenti E.; Rocchetti M.; Roletto F.; Severino D.; Soncini C.; Storici P.; Tonani R.; Zugnoni P.; Vianello P. 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazoles: Identification of a Potent Aurora Kinase Inhibitor With a Favorable Antitumor Kinase Inhibition Profile. J. Med. Chem. 2006, 49, 7247–7251. [DOI] [PubMed] [Google Scholar]
- Caprinelli. P.; Ceruti R.; Giorgini M. L.; Cappella P.; Gianellini L.; Croci V.; Degrassi A.; Texido G.; Rocchetti M.; Vianello P.; Rusconi L.; Storici P.; Zugnoni P.; Arrigoni C.; Soncini C.; Alli C.; Patton V.; Marsiglio A.; Ballinari D.; Pesenti E.; Fancelli D.; Moll J. PHA-739358, a Potent Inhibitor of Aurora Kinases With a Selective Target Inhibition Profile Relevant to Cancer. Mol. Cancer Ther. 2007, 6, 3158–3168. [DOI] [PubMed] [Google Scholar]
- Payton M.; Bush T. L.; Chung G.; Ziegler B.; Eden P.; McElroy P.; Ross S.; Cee V. J.; Deak H. L.; Hodous B. L.; Nguyen H. N.; Olivieri P. R.; Romero K.; Schenkel L. B.; Bak A.; Stanton M.; Dussault I.; Patel V. F.; Geuns-Meyer S.; Radinsky R.; Kendall R. L. Preclinical Evaluation of AMG900, a Novel Potent and Highly Selective Pan-Aurora Kinase Inhibitor with Activity in Taxane-Resistant Tumor Cell Lines. Cancer Res. 2010, 70, 9846–9854. [DOI] [PubMed] [Google Scholar]
- Bavetsias V.; Large J. M.; Sun C.; Bouloc N.; Kosmopoulou M.; Matteucci M.; Wilsher N. E.; Martins V.; Reynisson J.; Atrash B.; Faisal A.; Urban F.; Valenti M.; de Haven Brandon A; Box G.; Raynaud F. I.; Workman P.; Eccles S. A.; Bayliss R.; Blagg J.; Linardopoulos S.; McDonald E. Imidazo[4,5-b]pyridine Derivatives as Inhibitors of Aurora kinases: Lead Optimization Studies Toward the Identification of an Orally Bioavailable Preclinical Development Candidate. J. Med. Chem. 2010, 53, 5213–5228. [DOI] [PubMed] [Google Scholar]
- Yang D.; Fokas D.; Li J.; Yu L.; Baldino C. M. A Versatile Method for the Synthesis of Benzimidazoles from o-Nitroanilines and Aldehydes in One Step via a Reductive Cyclization. Synthesis 2005, 47–56. [Google Scholar]
- Nielsen S. F.; Larsen M.; Boesen T.; Schonning K; Kromann H. Cationic Chalcone Antibiotics. Design, Synthesis, and Mechanism of Action. J. Med. Chem. 2005, 48, 2667–2677. [DOI] [PubMed] [Google Scholar]
- Bavetsias V.; Sun C.; Bouloc N.; Reynisson J.; Workman P.; Linardopoulos S.; McDonald E. Hit Generation and Exploration: Imidazo[4,5-b]pyridine Derivatives as Inhibitors of Aurora Kinases. Bioorg. Med. Chem. Lett. 2007, 17, 6567–6571. [DOI] [PubMed] [Google Scholar]
- Free S. M. Jr.; Wilson J. W. A Mathematical Contribution to Structure–Activity Studies. J. Med. Chem. 1964, 7, 395–399. [DOI] [PubMed] [Google Scholar]
- log D and pKa measurements were performed by Pharmorphix Solid State Services, Member of the Sigma-Aldrich Group, Cambridge, UK.
- Calculated using Moka 1.1.0, by Molecular Discovery Ltd.
- Milletti F.; Storchi L.; Sforna G.; Cruciani G. New and Original pKa Prediction Method Using Grid Molecular Interaction Fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [DOI] [PubMed] [Google Scholar]
- Zachariae U.; Giordanetto F.; Leach A. G. Side Chain Flexibilities in the Human Ether-a-go-go Related Gene Potassium Channel (hERG) Together with Matched-Pair Binding Studies Suggest a New Binding Mode for Channel Blockers. J. Med. Chem. 2009, 52, 4266–4276. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal Chemistry of hERG Optimizations: Highlights and Hang-Ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Aronov A. M. Ligand Structural Aspects of hERG Channel Blockade. Curr. Top. Med. Chem. 2008, 8, 1113–1127. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Johnstone C. A Quantitative Assessment of hERG Liability as a Function of Lipophilicity. Bioorg. Med. Chem. Lett. 2007, 17, 1759–1764. [DOI] [PubMed] [Google Scholar]
- Whitlock G. A.; Blagg J.; Fish P. V. 1-(2-Phenoxypheny)methanamines: SAR for Dual Serotonin/Noradrenaline Reuptake Inhibition, Metabolic Stability and hERG Affinity. Bioorg. Med. Chem. Lett. 2008, 18, 596–599. [DOI] [PubMed] [Google Scholar]
- Van de Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. Property-Based Design: Optimization of Drug Absorption and Pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [DOI] [PubMed] [Google Scholar]
- clogP was calculated using ChemBioDraw Ultra 12 by CambridgeSoft (www.cambridgesoft.com).
- The measured (see ref (37)) log D7.4 for compound 27e is 3.84.
- Kinase profiling using the KINOMEScan technology: www.kinomescan.com.
- Fabian M. A.; Biggs W. H. III; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J.-M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A Small Molecule-Kinase Interaction Map for Clinical Kinase Inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A Quantitative Analysis of Kinase Inhibitor Selectivity. Nat. Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Moore A. S.; Faisal A.; Gonzalez de Castro D.; Bavetsias V.; Sun C.; Atrash B.; Valenti M.; de Haven Brandon A.; Avery S.; Mair D.; Mirabella F.; Swanbury J.; Pearson A. D. J.; Workman P.; Blagg J.; Raynaud F. I.; Eccles S. A.; Linardopoulos S. Selective FLT3 Inhibition of FLT3-ITD+ Acute Myeloid Leukaemia Resulting in Secondary D835Y Mutation: A Model for Emerging Clinical Resistance Patterns. Leukemia 2012, 26, 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirewalt D. L.; Radich J. P. The Role of FLT3 in Haematopoietic Malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [DOI] [PubMed] [Google Scholar]
- Kindler T.; Lipka D. B.; Fischer T. FLT3 as a Therapeutic Target in AML: Still Challenging After All These Years. Blood 2010, 116, 5089–5102. [DOI] [PubMed] [Google Scholar]
- Meshinchi S.; Appelbaum F. R. Structural and Functional Alterations of FLT3 in Acute Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 4263–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshinchi S.; Alonzo T. A.; Stirewalt D. L.; Zwaan M.; Zimmerman M.; Reinhardt D.; Kaspers G. J. L.; Heerema N. A.; Gerbing R.; Lange B. J.; Radich J. P. Clinical Implications of FLT3 Mutations in Pediatric AML. Blood 2006, 108, 3654–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis M. J. Will Newer Tyrosine Kinase Inhibitors Have an Impact in AML?. Best Pract. Res. Clin. Haematol. 2010, 23, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi M. G.; Ecsedy J. A.; Meetze K. A.; Balani S. K.; Burenkova O.; Chen W.; Galvin K. M.; Hoar K. M.; Huck J. J.; LeRoy P. J.; Ray E. T.; Sells T. B.; Stringer B.; Stroud S. G.; Vos T. J.; Weatherhead G. S.; Wysong D. R.; Zhang M.; Bolen J. B.; Claiborne C. F. Antitumour Activity of MLN8054, an Orally Active Small-Molecule Inhibitor of Aurora A kinase. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 4106–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloc N.; Large J. M.; Kosmopoulou M.; Sun C.; Faisal A.; Matteucci M.; Reynisson J.; Brown N.; Atrash B.; Blagg J.; McDonald E; Linardopoulos S.; Bayliss R.; Bavetsias V. Structure-based Design of Imidazo[1,2-a]pyrazine Derivatives as Selective Inhibitors of Aurora-A Kinase in Cells. Bioorg. Med. Chem. Lett. 2010, 20, 5988–5993. [DOI] [PubMed] [Google Scholar]
- Mizuki M.; Fenski R.; Halfter H.; Matsumura I.; Schmidt R.; Muller C.; Gruning W.; Kratz-Albers K.; Serve S.; Steur C.; Buchner T.; Kienast J.; Kanakura Y.; Berdel W. E.; Serve H. Flt3 Mutations from Patients with Acute Myeloid Leukemia Induce Transformation of 32D Cells Mediated by Ras and STAT5 Pathways. Blood 2000, 96, 3907–3914. [PubMed] [Google Scholar]
- Choudhary C.; Brandts C.; Schwable J.; Tickenbrock L.; Sargin B.; Ueker A.; Bohmer F.-D.; Berdel W. E.; Muller-Tidow C.; Serve H. Activation Mechanisms of STAT5 by Oncogenic Flt3-ITD. Blood 2007, 110, 370–374. [DOI] [PubMed] [Google Scholar]
- Moore A.; Faisal A.; Bavetsias V.; Gonzalez de Castro D.; Sun C.; Atrash B.; Valenti M.; de Haven Brandon A.; Avery S.; Pearson A.; Workman P.; Blagg J.; Raynaud F.; Eccles S.; Linardopoulos S.. The Dual FLT3-Aurora Inhibitor CCT241736 Overcomes Resistance to Selective FLT3 Inhibition Driven by FLT3 Ligand and FLT3 Point Mutations in Acute Myeloid Leukemia. Mol Cancer Ther, 2011, 10, Issue 11, Supplement 1, doi 10.1158/1535-7163.TARG-11-B74; Abstracts of AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Nov 12–16, 2011, San Francisco, CA. [DOI] [Google Scholar]
- Chan F.; Sun C.; Perumal M.; Nguyen Q.-D.; Bavetsias V.; McDonald E.; Martins V.; Wilsher N.; Valenti M.; Eccles S.; te Poele R.; Workman P.; Aboagye E. O.; Linardopoulos S. Mechanism of Action And in Vivo Quantification of Biological Activity of the Aurora Kinase Inhibitor CCT129202. Mol. Cancer Ther. 2007, 6, 3147–3157. [DOI] [PubMed] [Google Scholar]
- The CCP4 (Collaborative Computational Project Number 4) Suite: Programmes for Protein Crystallography. Acta Crystallogr. Sect. D 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr., Section D 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Moreno-Farre J.; Workman P.; Raynaud F. I. Analysis of Potential Drug–Drug Interactions for Anticancer Agents in Human Liver Microsomes by High Throughput Liquid Chromatography/Mass Spectrometry Assay. Aust.-Asian J. Cancer 2007, 6, 55–68. [Google Scholar]
- Ion Channel Cardiac Profiler; Millipore: Billerica, MA; http://www.millipore.com/life_sciences/flx4/ld_ion [Google Scholar]
- hERG Safety Assay; Cyprotex plc, Cheshire, UK; www.cyprotex.com
- Workman P; Aboagye E. O.; Balkwill F.; Balmain A.; Bruder G.; Chaplin D. J.; Double J. A.; Everitt J.; Farningham D.; Glennie M. J; Kelland L. R.; Robinson V.; Stratford I. J.; Tozer G. M.; Watson S.; Wedge S. R.; Eccles S. A. Guidelines for the Welfare and Use of Animals in Cancer Research. Br. J. Cancer 2010, 102, 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.