

Abstract
Genomic applications of DNA-binding molecules require an unbiased knowledge of their high affinity sites. We report the high-throughput analysis of pyrrole-imidazole polyamide DNA-binding specificity in a 1012-member DNA sequence library using affinity purification coupled with massively parallel sequencing. We find that even within this broad context, the canonical pairing rules are remarkably predictive of polyamide DNA-binding specificity. However, this approach also allows identification of unanticipated high affinity DNA-binding sites in the reverse orientation for polyamides containing β/Im pairs. These insights allow the redesign of hairpin polyamides with different turn units capable of distinguishing 5′-WCGCGW-3′ from 5′-WGCGCW-3′. Overall, this study displays the power of high-throughput methods to aid the optimal targeting of sequence-specific minor groove binding molecules, an essential underpinning for biological and nanotechnological applications.
Introduction
Defining the recognition motifs of DNA-binding small molecules in genome-sized sequence space is critical for applications in biology and nanotechnology. Py-Im polyamides are DNA-binding synthetic oligomers composed of analogues of N-methylpyrrole.1 Aromatic amino acids combined as unsymmetrical ring pairs can be used to read the minor groove of DNA according to well-defined pairing rules (Figure 1). Side-by-side stacked N-methylimidazole (Im) and N-methylpyrrole (Py) carboxamides (Im/Py pairs) distinguish G·C from C·G base pairs,2 whereas N-methyl-3-hydroxypyrrole (Hp)/Py shows specificity for T·A over A·T.3 Finally, Py/Py pairs specify for both A·T and T·A.4 The γ-aminobutyric acid (GABA) turn unit functions to keep rings unambiguously paired when folded in a hairpin configuration. The turn unit also functions in the hairpin as an A·T/T·A selective element (Figure 1A). Hairpin polyamides can potentially bind in two orientations: aligned 5′-3′ on the DNA with respect to the N-C terminus of the polyamide (referred to as the forward orientation) or 3′-5′ on DNA with respect to the N-C terminus (reverse orientation). Hairpin polyamides incorporating an unsubstituted GABA turn unit show a modest energetic preference for the forward orientation.5 This preference is increased when a chiral, α-substituted (R)-amino-GABA turn is introduced (>100-fold greater binding of forward versus reverse sites).6 More recently, “second generation” hairpin polyamides with β-amino-GABA turn elements showed increased binding affinity for some hairpin polyamides. These β-turn hairpins increase polyamide biological activity and nuclear uptake in several cases.7−9 The effect of the β-amino GABA turn on polyamide orientation and sequence-specificity are less well characterized.10 Here we apply massively parallel sequencing to assay polyamide-DNA binding allowing unanticipated binding motifs to be discovered. This unbiased sequencing assay facilitates iterative polyamide design, guiding the reprogramming of polyamide specificity and allowing us to codify general design principles critical to minor groove recognition.
Figure 1.

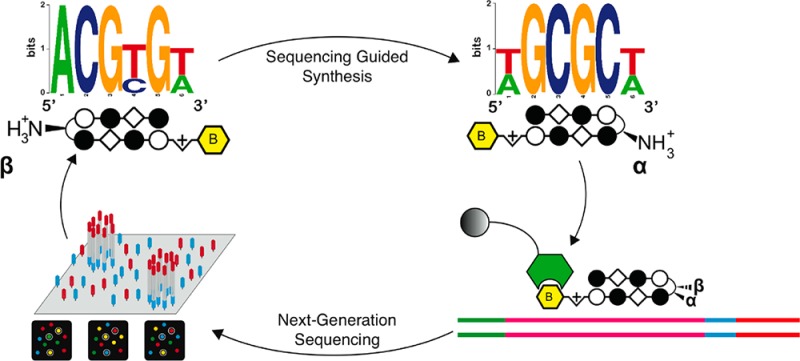
(A) Hairpin Py-Im polyamide recognition of the DNA minor groove. Aromatic amino acid ring pairs recognize distinct DNA base pairs. Complete structures for each hairpin polyamide-biotin conjugate used can be found in Supporting Information. (B) Scheme for Bind-n-Seq analysis of DNA binding polyamides. Double-stranded DNA containing a degenerate, 21-bp segment is enriched, purified, and analyzed via high-throughput sequencing. Commonly bound DNA consensus sequences are identified via motif searching.
Historically the design of sequence-specific DNA-binding molecules has been guided by pivotal advances in analytical screening techniques. For an eight-ring hairpin that binds 6 bp, there are formally 2080 different 6mer duplex DNA sites that are potential targets.11 Footprinting and affinity cleavage supported development of the pairing rules by defining the sequence preferences and orientation of Py-Im hairpin eight-ring oligomers in the context of a 150–250 bp DNA restriction fragment, effectively a library of a few hundred potential binding sites, each 6 bp in size.12−15 Quantitative DNase footprinting titrations added critical information regarding the energetics and penalties for single base mismatch binding.16,17 However, applications of Py-Im polyamides in biology and nanotechnology will require knowledge of sequence-specificity in larger sequence contexts and have necessitated the development of screening techniques that analyze sequence space in a higher throughput, less labor intensive manner than gel-based footprinting. The fluorescence intercalator displacement assay was applied to rank polyamide binding to all unique 5 bp sequences but is qualitative in nature and has limitations in its ability to address larger binding sites in the microplate format.18,19 A SELEX-like approach developed by Van Dyke and co-workers was used to determine the binding preferences to two hairpin polyamides within a library of 1.3 × 108 DNA sequences but required multiple rounds of selection and was limited to qualitative identification of the highest affinity polyamide binding sites.20 Ansari and co-workers applied a microarray platform to analyze the binding of Cy3-labeled polyamides in the context of all distinct 10mers (5.24 × 105 unique sequences).21−23 When calibrated with DNase I footprinting data, this method allows the determination of quantitative equilibrium association constant (Ka) values for polyamide binding to all possible match and single base-pair mismatch sites.22 However, routine application of microarray methods are hindered by the need for custom synthesis and analysis of arrays, presenting a significant technical obstacle as core genomics facilities transition to high-throughput sequencing based platforms.24
Recently, several methods have been developed to characterize protein–DNA interactions using massively parallel sequencing.25−28 One such technique, termed Bind-n-Seq, uses affinity-tagged transcription factors to enrich a pool of oligonucleotides containing random 21mers (>2 × 1012 unique sequences) in a single round of selection (Figure 1B) .27 Sequencing and data analysis allows identification of high affinity sequences and correlates well with solution-phase measurements of binding affinity. Each Bind-n-Seq reaction contains 2.5 × 107 copies of each possible 10mer, compared to 4 copies of each 10mer sampled by microarray methods.21 This ensures sampling of short sequences embedded in a diverse 21mer sequence, providing a context-averaged picture of binding. Combined with the ability to query longer sequences and deep sequence multiple binding reactions simultaneously, this approach could represent a useful alternative to array-based methods and provide important insights into polyamide-DNA binding in genome-sized sequence space.
Results
Validation of a Sequencing-Based Platform for Analyzing Polyamide-DNA Binding
As an initial test of the ability of this platform to comprehensively interrogate small molecule–DNA interactions, we synthesized a small panel of biotinylated polyamides (Figure 2, 1–6). Each of these molecules contains a heterocyclic Py-Im core that has been explored in cellular contexts for transcriptional inhibitory activity (1–3, 5, 6)29−32 and/or nuclear uptake (4),9 making their genome-wide specificities biologically relevant (complete structures are provided in Supporting Information). Although it was presumed the molecules would not violate the pairing rules, the introduction of the β-substituted turn coupled with β/Im pairs suggested this would be a good test of the Bind-n-Seq methodology. Each Py-Im polyamide-biotin conjugate (50 nM) was allowed to equilibrate with the mixed 21mer oligonucleotide, and the bound and unbound sequences were separated via affinity purification using streptavidin beads (Figure 1). Following elution, polyamide-enriched sequences were PCR amplified, purified, and subjected to massively parallel sequencing analysis. Current next-generation sequencing instruments can determine one hundred million or more short sequences per run. In contrast, a 6-bp binding polyamide flanked by 2-bp on each side represents only 524,800 potential sequences. This redundant sampling capacity allowed the analysis of multiple compounds and/or conditions in a single Illumina sequencing lane, with each binding reaction indexed by a unique, 3-bp barcode. Following sequencing, each unique binding reaction was analyzed by (i) recovering a clean data set of high fidelity enriched sequences for each bar-coded binding condition and (ii) counting the occurrence of unique DNA sequences using a sliding window of length “k”. These analyses were performed using MERMADE, a new pipeline for Bind-n-Seq analysis available at http://korflab.ucdavis.edu/Datasets/BindNSeq.27 Replicate measurements show the enrichment of DNA sequences by polyamide 1 is highly reproducible (R2 = 0.979 for two separate binding and enrichment experiments), establishing the reproducibility of this approach (Supplementary Figure S1).
Figure 2.

Structures of polyamides 1–6 and primary motifs identified for each via high-throughput analysis.
In order to validate our high-throughput approach and calibrate its dynamic range, we compared data generated via the Bind-n-Seq platform with previously determined solution phase polyamide binding affinities. Quantitative data for Bind-n-Seq enrichments, including comparison to quantitative footprinting-derived Ka values and E-values for motif analysis can be found in the Supporting Information. Hairpin polyamides containing the heterocyclic Py-Im cores of 1–3 and 5-6, have been previously analyzed by quantitative DNase footprint titration, while Cy3-labeled analogues of 1-2 and 5 have been studied by microarray.16,21−23,33,34 Assuming the number of times a given DNA is sequenced is proportional to the fractional occupancy of the polyamide at that oligonucleotide, one would expect to see a linear relationship between Ka and sequence counts. Indeed, we find good correlation (R2 = 0.99) for the number of times a DNA sequence was counted in a binding reaction enriched by polyamide 1 and previously reported DNase footprinting-derived Ka values (Supplementary Figure S2).16,21,33
Similar results were found for polyamides 2, 3 and 5, 6 and hold both for comparisons to DNase footprinting-derived Ka values as well as CSI microarray intensities (R2 ≥ 0.89; Supplementary Figures S2–S6). Closer inspection of the data shows that Bind-n-Seq sequence counts and footprinting-derived Ka values are best correlated for high affinity polyamide-DNA binding sites, over a Ka range of approximately 10-fold. A striking example is polyamide 6, in which sequencing-based analysis rank orders a number of polyamide-DNA match sites that differ only on the basis of their flanking sequences (Supplementary Figure S6). However, in contrast to microarray methods, we find this method is less useful in studying polyamide binding to single base pair mismatch sites. For example, DNA sequences bound by polyamide 1 with a Ka ≤ 3.8 × 109 M–1 all cluster around ∼1000 counts and are not clearly discriminated by Bind-n-Seq (Supplementary Figure S2). This limitation could be due to the known bias of enrichment-based approaches such as SELEX to discover high affinity binding events20,35 or factors specific to the binding conditions used in this study, such as the large number of all possible 6mers (the preferred binding site size for polyamides 1–6) found in each binding reaction or the presence of the same primer sequence in each randomized DNA sample. Thus, for comprehensive determination of binding affinities to all possible sequences (match and mismatch), non-enrichment based optical platforms such as CSI microarray and the more recently developed HiTS-FLIP remain the methods of choice.21,28 However, in terms of speed and simplicity, Bind-n-Seq provides a facile assay for the unbiased identification and quantitative rank ordering of very high affinity ligand-DNA binding sites that does not require custom array fabrication or specialized flow cell chemistry, thereby filling a valuable middle ground that can rapidly inform iterative cycles of molecular design as demonstrated below.
Motif Analysis
In order to graphically depict high affinity polyamide-DNA binding events we generated sequence logos for 1–6 using the motif finding program DREME.36 DREME was specially developed to discover short (4–8 bp) response elements typically bound by eukaryotic transcription factors from chromatin immunoprecipitation sequencing (ChIP-Seq) data, which typically span a read length of 30–50 bp.24 This approach similarly provides an optimal discovery tool for motifs bound by the relatively short, 6 bp DNA-binding polyamides 1–6 examined in this study. The strongest motifs generated from DREME analysis of raw enriched sequences for polyamides 1–6 are depicted in Figure 2. In general, the highest information content for each polyamide is found at a site width of six, verifying the binding site size expected when 1–6 are bound in a fully ring-paired, hairpin configuration. Motifs generated by 1, 5, and 6 are indicative of polyamide-DNA binding consistent with the Py-Im pairing rules in the forward orientation. Notably, polyamides 1 and 5 also generated strong secondary motifs (Supplementary Figure S7), which constitute formal match sites but are expected to be of relatively lower affinity due to variations in the minor groove width and sequence-specific microstructure of DNA.37 This reflects the ability of the method to parse subtle differences in high affinity polyamide-DNA binding events.
In addition to the expected motifs, DREME analysis also revealed several unanticipated binding contexts for the investigated molecules. For example, compounds 2 and 3 target the sequences 5′-WTWCGW-3′ and 5′-WGWCGW-3′, respectively, according to the pairing rules. However, each shows a formal degeneracy in the fourth Im/Py ring pair adjacent to the GABA turn, binding G or A rather than the expected G (Figure 2). Notably, evidence for the nonspecificity of the Im/Py pair of 2 in this particular sequence context had previously been observed using CSI microarray, providing another validation of the Bind-n-Seq approach.22 The most surprising result was the unanticipated 5′-WCGYGW-3′ (Y = C/T) consensus motif generated from DNA-enriched by polyamide 4 (Figure 2). Structurally, 4 is differentiated from the other polyamides by its incorporation of two Im/β-alanine (Im/β) pairs, which are required to reset the curvature and register of the internal Im amino acids and allow high affinity DNA binding.38 The simplest interpretation for the observed 5′-WCGYGW-3′ motif of polyamide 4 arises from a reversed binding mode,39 in which the N-to-C terminus of 4 is oriented in the 3′-5′ direction with regards to DNA (Figure 3), along with C/T binding by one of the Im/β pairs. To verify the high-throughput findings, we applied two solution-phase assays. First we analyzed the melting temperature of a 5′-WCGYGW-3′ oligonucleotide in the presence and absence of polyamide 4, as the DNA duplex stabilization (ΔTm) induced by a polyamide provides a measure of its overall binding affinity. Indeed, we find 4 productively binds the 5′-WCGCGW-3′sequence (ΔTm = 14.1 °C). To more quantitatively analyze the orientation preferences of polyamide 4, we measured its dissociation rate constant (koff) using oligonucleotides containing either the forward (5′-WGCGCW-3′) and reverse (5′-WGCGCW-3′) orientation binding sites by a fluorescence assay (Supporting Information). This approach is based on previous studies, which have shown the dissociation rate of a polyamide for different DNA sequences is the primary determinant of its binding specificity.40 In the case of 4, kinetic analysis reveals an ∼10× slower dissociation rate for oligonucleotides containing the reverse orientation binding site relative to the forward orientation (Table 1A, Supplementary Table S1). These findings confirm and highlight the ability of the Bind-n-Seq platform to reveal preferred binding sites.
Figure 3.
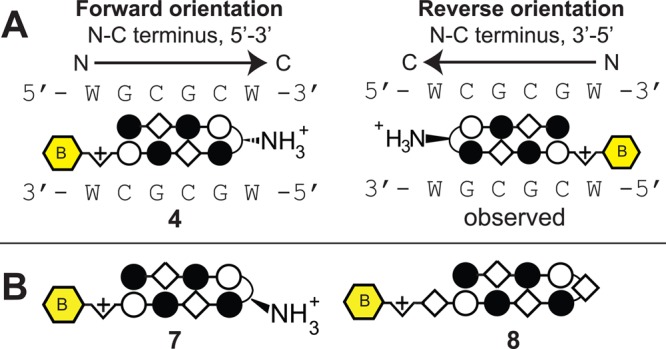
(A) Polyamide 4 with β/Im pairs as found in the forward (left) and reverse (right) binding orientations. (B) Second generation molecules used to probe the effect of turn modification (7, 8) on polyamide-DNA binding preferences.
Table 1. Melting Temperatures and Dissociation Kinetics for Polyamides with DNA Duplexesa.
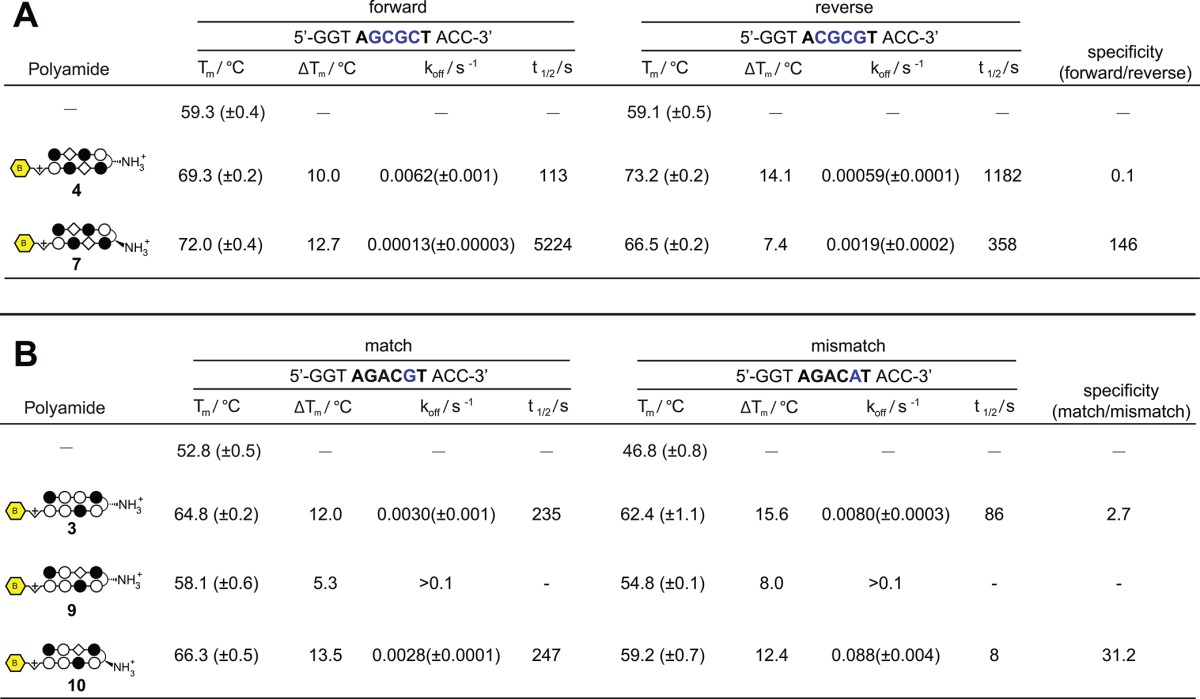
(A) Melting temperatures and dissociation kinetics for polyamides with DNA duplexes containing forward and reverse orientation binding sites for polyamide 4. Kinetic assays were measured using fluorescent analogues of polyamides 4 and 7, as described in Supporting Information. Error represents the standard deviation of replicate measurements. (B) Melting temperatures and dissociation kinetics for polyamides with DNA duplexes containing match and mismatch orientation binding sites for polyamide 3. Kinetic assays were measured using fluorescent analogues of polyamides 3, 9, and 10 as described in Supporting Information. Error represents the standard deviation of replicate measurements.
High-Throughput Sequencing Guided Redesign of Py-Im Polyamides: from 5′-CGCG-3′ to 5′-GCGC-3′
The reverse binding preference displayed by polyamide 4 for 5′-WCGCGW-3′ was unanticipated but is not without precedent. A previous study from our laboratory had observed a preference for reverse, 3′-5′ binding by hairpin polyamides containing a flexible β/β pair and an unsubstituted GABA turn unit.39 The reverse binding preference observed for β/Im containing polyamide 4 implicates conformational flexibility, rather than the β/β pair itself as the primary determinant of reverse binding. In contrast to α-amino GABA turn, which has been shown to enforce forward orientation binding even for conformationally flexible hairpins,39 the β-amino GABA turn unit is sterically compatible with reverse binding. Understanding the relationship between polyamide flexibility, turn unit, and orientation preference is important as these reverse-binding modes, if understood in a predictive sense, may be exploited for new sequence targeting applications. For example, a computational survey of the expected human genomic match sites of polyamides 1–6 shows the 5′-WCGCGW-3′ recognition site of 4 correlates most highly with genic features and also has the fewest genomic match sites of any of these polyamides (Supplementary Figure S8). Such qualities may be ideal for biological activity and specificity.
In addition to the opportunity to utilize reverse binding modes, polyamide 4 also provides an ideal case study to test our ability to utilize Bind-n-Seq in concert with synthetic chemistry to establish structure-motif relationships. In the case of 4, replacement of the β-amino-GABA turn with an α-amino-GABA turn unit is expected to restore the forward 5′-WGCGCW-3′ binding orientation, due to increased steric interaction of the α-substituted GABA substituent with the minor groove floor.6,41,42 Accordingly, we synthesized polyamide 7 and compared its ability to stabilize duplex DNA containing the forward (5′-WGCGCW-3′) and reverse (5′-WCGCGW-3′) binding orientations with compound 4. As can be seen in Table 1A, shifting the position of the chiral amine from the β to the α position on the turn unit substantially reduces the ability of 7 to stabilize the reverse orientation oligonucleotide relative to compound 4 (ΔTm4 = 14.1 °C, ΔTm7 = 7.4 °C) while showing an increase in melting temperature for the forward orientation (ΔTm4 = 10.0 °C, ΔTm7 = 12.7 °C). Analysis of the dissociation kinetics of an analogue of 7 suggest this effect reflects a substantial decrease in off-rate (koff) for DNA containing the forward orientation binding site coupled with a substantial increase in koff for duplex DNA containing the reverse orientation binding site. To further validate this redesign, we subjected 7 to Bind-n-Seq analysis, returning the expected 5′-WGCGCW-3′ motif (Figure 4). The observed motif suggests that enforcement of the 5′→3′ orientation by the α-amino GABA turn has the additional and unexpected benefit of improving alignment of the Im/β pairs with the central 5′-CG-3′ step.
Figure 4.

High-throughput sequencing guided redesign of polyamide 4.
As a further demonstration of the sensitivity of polyamide binding to modification of the turn linkage, we replaced the α-amino-GABA turn unit of 7 with a shorter β-alanine linker to afford polyamide 8. This molecule has been extensively studied (structure, affinity, and orientation) for binding of the sequence 5′-AAAGAGAAGAG-3′ as a 1:1 complex.43−45 The hairpin core of 8 differs from 7 only via a single amino-methylene unit in the linker (Figure 4). However, Bind-n-Seq analysis of 8 confirms this minor structural difference is sufficient to favor binding as an extended 1:1 complex, in the 3′-5′ reverse orientation with regards to the G-rich strand. Removal of the one amino-methylene unit of the GABA linker serves to stabilize the 1:1 motif and disfavor formation of the ring paired hairpin.46 This is consistent with earlier studies, which suggest the β linker is too short to make the hairpin turn without bending the flanking amide bonds out of the ring plane of the two heterocyclic Py-Im subunits.47,48 Thus, the choice of turn linkage (β-amino GABA, α-amino-GABA, β-alanine) redirects polyamide binding to three distinct DNA recognition motifs (Figure 4).
High-Throughput Sequencing Guided Redesign of Py-Im Polyamides: 5′-GWCG-3′
Next we analyzed whether the Bind-n-Seq platform could be used to guide iterative cycles of polyamide design. In both polyamides 2 and 3, the Im/Py pair in the position adjacent to the turn unit binds G/A instead of the expected G (Figure 2). Microarray analysis has previously confirmed this finding for polyamide 2. Thermal denaturation analysis validates this observation for polyamide 3 as well, which similarly stabilizes duplexes incorporating either a G·C or A·T base pair at this position (Table 1B). This represents a deviation from the G·C specificity predicted for the Im/Py pair by the pairing rules. One explanation is the Im residue of this Im/Py pair is not optimally aligned to productively bind with the exocyclic amine of guanine, possibly due to overcurvature of the N-terminal, Im-rich polyamide subunit with respect to DNA.38 Applying a design principle that has proven useful in the past,49 we synthesized polyamide 9, replacing the Py/Im pair of 3 with a β/Im pair in an attempt to relax polyamide curvature and restore sequence-specific binding. However, polyamide 9 exhibited overall low affinity binding and showed no substantial preference for match or mismatch sites in the forward orientation by either melting temperature or kinetic assays (Table 1B). This was unexpected, as previous studies from our group have shown the β/Im pair can serve as a functional surrogate for the Py/Im pair within eight-ring hairpin polyamides and bind DNA with high affinity and specificity.38
To understand this, we analyzed compound 9 by Bind-n-Seq, and as with polyamide 4, we observe a reverse (3′-5′) binding orientation (Figure 5). Polyamide 9 shows relatively little binding of its cognate forward orientation site (Table 1B). Using the same rationale as before, we replaced the β-amino GABA turn of 9 with an α-amino-GABA turn. The product of this effort, 10, shows greater stabilization of forward match (ΔTm = 13.5 °C) duplex DNA binding sites in contrast to a mismatch site (ΔTm = 12.4 °C) (Table 1B). Kinetic analysis suggests this represents a >10-fold increase in specificity relative to parent polyamide 3, which binds both sequences equally well. High-throughput analysis of 10 followed by motif analysis confirms the increased preference for a 5′-WGWCGW-3′ match site (Figure 5). Also interesting was the observation that polyamide 9 demonstrates no sequence preference for the C-terminal triamine residue, which usually codes for W presumably for steric reasons. Polyamide 10 similarly shows a reduced sequence preference at this position (Figure 5). It is possible that inclusion of the conformationally flexible Im/β pair favors a C-terminus orientation that does not interact with the minor groove.
Figure 5.

High-throughput sequencing guided redesign of turn unit for polyamide 3 with a single β/Im pair.
Having arrived at a molecule with the desired specificity, we correlated our stepwise structure-motif relationship for polyamide 3 with literature data to see if any general design principles arose. We found several examples where the Im heterocycle of an Im/Py pair must be preceded by a β (replacing Py) in order to optimize polyamide affinity and/or specificity.34,49 This appears most important when the Im/Py pair is preceded by two or more rigid Im or Py subunits (counting from the N- to C-terminus of the polyamide; Supplementary Figure S9). Remarkably, sequence context-dependent rules for the proper use of β within an eight-ring hairpin polyamide have not been previously elucidated. The weak binding of polyamide 9 compared to 10 (Table 1B) also supports an additional conclusion: the ability of β/Im to function as a surrogate for Py/Im is dependent on a forward orientation. Therefore, incorporation of the flexible β subunit should be paired with use of a chiral α-amino-GABA turn to maintain high affinity binding. A synopsis of footprinting-derived Ka values consistent with these design principles can be found in the Supporting Information (Figure S9).
Discussion
Implications for the Design of Minor Groove Binding Molecules
Despite progress, the ability to target dsDNA in any sequence context using synthetic molecules remains a challenging task. Variable sequence-dependent DNA structural features such as minor groove width, flexibility, and intrinsic helix curvature may reduce polyamide binding at specific sites. However, the results presented here reveal several insights that should inform future polyamide design:
-
(i)
Fully ring-paired Py-Im polyamides (1–3, 5, 6) prefer a 5′-3′ forward orientation, as previously reported, regardless of turn modificaton.5
-
(ii)
Conformationally flexible, β-containing polyamides with a β-amino-GABA linker (i.e., 4, 8, 9) prefer a reverse orientation, in which the N-terminal Im is aligned with the 3′ end of the binding site.44
-
(iii)
Conformationally flexible, β-containing polyamides with a α-amino-GABA linker (i.e., 7, 10) prefer a forward orientation, in which the N-terminal Im is aligned with the 5′ end of the polyamide binding site.39
-
(iv)
Polyamide binding in the reverse orientation may cause misalignment of internal Im/Py ring pairs (as in 4 and 8). Restoration of forward orientation can restore sequence-specific binding by these ring pairs (as in 7 and 10).
-
(v)
For optimal binding affinity and specificity, internal β residues should be used as follows: Counting from the polyamide N-terminus, Im heterocycles that are found following two or more ring pairs (Im or Py) should be preceded by a flexible β if possible to restore alignment with the GC base pair (i.e., compare 3 and 10). Similar observations have been previously reported but never formally codified.34,38,49
While polyamides incorporating β/β pairs had been previously found to allow or prefer reverse binding, a similar preference for the staggered β/Im pairs of 4 was unexpected.39 The current study suggests reverse binding may be a general characteristic of conformationally relaxed β-incorporating hairpin polyamides dictated by choice of turn unit, possibly reflecting a more favorable alignment between Im-N3 and G-NH2 due to the propeller twist of G·C base pairs. While the abilities of the GABA turn unit to enforce ring pairing and selectively bind A/T base pairs in hairpin polyamides are well-established, our findings suggest a third potential function: to program forward or reverse orientation via rational pairing of chiral GABA turn units with β/Im pairs. Interestingly, several biologically active polyamides incorporating multiple β residues have been reported in the literature, although these molecules utilize an unsubstituted GABA linker and target substantially larger binding sites.50,51 None of these molecules has been examined via an unbiased assay as presented here. Future questions that will help advance the field are (i) do these larger polyamides exhibit a similar ambiguity in binding orientation, (ii) how is this mediated by the unsubstituted GABA turn, and (iii) how does polyamide size (polyvalency) and architechture (hairpin, cyclic, head-to-tail dimers) affect sequence-specificity when examined in an unbiased context?
Conclusion
We have applied a high-throughput sequencing platform in combination with single-step affinity purification to globally benchmark the binding preferences of six biologically active polyamides. This platform allows rapid, quantitative identification of high affinity polyamide binding sites, correlates well with solution-phase and microarray platforms, and can be used to guide the refinement of general polyamide design principles. In the future we envision extending this platform to analyze a library of hairpins and alternative polyamide architectures, providing a database of polyamide-DNA interactions that will greatly benefit the molecular recognition field. A better understanding of polyamide-DNA recognition and difficult to target DNA sequences will be essential for applications of polyamides in vitro, such as DNA nanotechnology.52−55 Whether the Bind-n-Seq method provides an ideal platform for optimizing pulldown and labeling chemistries that will be required to generate high-resolution maps of small molecule-DNA binding in cellular environments, analogous to transcription factor ChIP-Seq assays, remains to be seen.24,56 Such methods will be essential to understanding how polyamides and other DNA-binding molecules interact with DNA in its native, chromatin context.
Experimental Section
Bind-n-Seq: Equilibration Reactions
Oligonucleotide and primer sequences, as well as polyamide structures, are provided in the Supporting Information. Template oligonucleotides were made double-stranded by primer extension in a 25 μL reaction containing Bind-n-Seq 93mer (3 μM, 75 nmols, 4.5 × 1013 molecules), Primer 1 (9 μM), and 1x TaqPro complete (2.0 mM Mg2+). Reactions were heated to 95 °C (2 min), 63 °C (1 min), 72 °C (4 min), and then 4 °C using a thermocycler. To initiate equilibrium binding reactions, a 25 μL of a solution containing polyamide (100 nM), Tris-HCl pH 7.5 (30 mM), KCl (20 mM), MgCl2 (20 mM), and CaCl2 (10 mM) was added directly to the primer extension reaction to give a final volume of 50 μL. The mixed and diluted reactions were allowed to equilibrate for 16 h at room temperature prior to affinity purification.
Bind-n-Seq: Enrichment Reactions
Streptavidin M-280 Dynabeads (650 μL, 6.5 mg) were prewashed with 0.5 mL of BSA blocking buffer (3.5 mg/mL BSA, 1x PBS, 2 × 10 min), 0.5 mL of calf thymus DNA blocking buffer (0.5 mg/mL, 1 × 90 min), and 0.5 mL of binding and washing buffer (10 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl, 3 × 1 min). Beads were isolated on a magnet for 8 min prior to removal of the supernatant in between each step. Aliquots of M-280 Dynabeads (50 μL, 0.5 mg) were then added to each equilibrium binding reaction and incubated at room temperature for 1 h with gentle mixing every 10 min. Reactions were washed with 0.5 mL of binding and washing buffer (recipe above, 1 × 5 min) and 0.5 mL of TKMC (10 mM Tris-HCl pH 7.0, 10 mM KCl, 10 mM MgCl2, 5 mM CaCl2, 2 × 10 min), with beads isolated for 8 min in between each step prior to removal of the supernatant. Enriched DNA was then isolated by addition of 100 μL of elution buffer (2% SDS, 100 mM NaHCO3, 3 mM biotin) followed by gentle shaking at 65 °C for 4–12 h. Beads were briefly centrifuged and isolated via magnet, and the supernatant was removed and saved.
Bind-n-Seq: Amplification and Sequencing
Recovered DNA was diluted 1:25 and analyzed by quantitative PCR (qPCR) to assess enrichment. Each qPCR reaction (20 μL) contained 5 μL of enriched DNA (1:25 dilution), 2.5 μL of Primer 2 (3.6 μM), 2.5 μL of Primer 3 (3.6 μM), and 10 μL of Roche qPCR Master Mix. Reactions were analyzed for the number of cycles required to achieve a saturated fluorescence signal. This number of cycles was then recorded and used to guide a subsequent touchdown PCR amplification reaction in order to prepare sufficient DNA for Illumina sequencing. Each touchdown PCR reaction (50 μL) contained 2.5 μL of enriched DNA (1:25 dilution), 2.5 μL of Primer 2 (3.6 μM), 2.5 μL of Primer 3 (3.6 μM), and 25 μL of TaqPro Complete (2.0 mM Mg2+). Amplification reactions were initiated by heating to 95 °C (4 min), followed by 10 cycles of heating to 60 °C (0.5 min), 72 °C (4 min), and then 95 °C (0.5 min), with the temperature of the 60 °C step being progressively decreased in 0.5 °C increments with each cycle. Depending on qPCR analysis of enrichment, reactions were subjected to another 5–10 cycles of heating to 45 °C (0.5 min), 72 °C (4 min), and then 95 °C (0.5 min), followed by cooling to 4 °C. Amplified DNA was purified using a QIAquick PCR purification kit (Qiagen), using the in-buffer pH indicator to adjust the pH using sodium acetate if necessary. Recovered DNA was quantified by Qubit dsDNA high sensitivity assay kit, and 100 ng of DNA from each enrichment reaction was pooled and reduced to ∼50 μL by SpeedVac. All samples were processed as single read sequencing runs at the California Institute of Technology Millard and Muriel Jacobs Genetics and Genomics Laboratory on an Illumina HiSeq 2000 Genome Analyzer.
Bind-n-Seq: Data Analysis
Sequencing reads were filtered and sorted using custom Perl scripts found in the MERMADE package, an updated version of the Bind-n-Seq data analysis pipeline. MERMADE is freely available with user documentation at http://korflab.ucdavis.edu/Datasets/BindNSeq. Briefly, high quality reads (composed only of A, C, T, or G, with a valid constant region [“AA”] and unique random region) were retained and split into separate files based on their unique 3-nt barcode (MERMADE scripts: sequence_converter.pl, debarcode.pl). For motif analysis of polyamides 1–7 and 9, 10, a random 10% of the sequences from each reaction condition was extracted, converted to FASTA format, and analyzed by DREME (http://meme.sdsc.edu/meme/cgi-bin/dreme.cgi) using the default settings. For motif analysis of polyamide 8, recovered sequences were analyzed relative to a file of unenriched, background 21mer sequences using a sliding window of 10 bp (MERMADE scripts: kmer_counter.pl, kmer_selector.pl). Sequences showing ≥2-fold enrichment relative to background were then analyzed by MERMADE using an iterative motif searching approach (MERMADE scripts: mermade.pl, motif_expander.pl). Graphical representations of all sequence motifs were rendered using Weblogo.
Acknowledgments
This work is supported by the National Institutes of Health (GM27681). J.L.M. is supported by a postdoctoral grant from the American Cancer Society (PF-10-015-01-CDD). We thank Adam Urbach for the gift of a precursor to compound 8. We also thank Adam Urbach (Trinity University) and Sarah Lockwood (UC Davis) for helpful discussions.
Supporting Information Available
Complete synthetic procedures, analytical data for 1–10, chemical structures, procedures for kinetic and melting temperature analyses, correlation with footprinting and CSI data, and supplemental figures and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dervan P. B.; Edelson B. S. Curr. Opin. Struct. Biol. 2003, 13, 284–99. [DOI] [PubMed] [Google Scholar]
- Wade W. S.; Mrksich M. L.; Dervan P. B. J. Am. Chem. Soc. 1992, 114, 8783–94. [Google Scholar]
- White S.; Szewczyk J. W.; Turner J. M.; Baird E. E.; Dervan P. B. Nature 1998, 391, 468–71. [DOI] [PubMed] [Google Scholar]
- Pelton J. G.; Wemmer D. E. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 5723–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1997, 119, 8756–65. [Google Scholar]
- Herman D. M.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1998, 120, 1382–1391. [Google Scholar]
- Dose C.; Farkas M. E.; Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2008, 130, 6859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth D. M.; Harki D. A.; Phillips J. W.; Dose C.; Dervan P. B. J. Am. Chem. Soc. 2009, 131, 7182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J. L.; Montgomery D. C.; Dervan P. B. Nucleic Acids Res. 2012, 40, 2345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas M. E.; Li B. C.; Dose C.; Dervan P. B. Bioorg. Med. Chem. Lett. 2009, 19, 3919–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dervan P. B. Science 1986, 232, 464–71. [DOI] [PubMed] [Google Scholar]
- Galas D. J.; Schmitz A. Nucleic Acids Res. 1978, 5, 3157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke M. W.; Hertzberg R. P.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 5470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz P. G.; S. T. J.; Dervan P. B. J. Am. Chem. Soc. 1982, 104, 6861–3. [Google Scholar]
- Mrksich M.; Wade W. S.; Dwyer T. J.; Geierstanger B. H.; Wemmer D. E.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 7586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauger J. W.; Baird E. E.; Dervan P. B. Nature 1996, 382, 559–61. [DOI] [PubMed] [Google Scholar]
- Trauger J. W.; Dervan P. B. Methods Enzymol. 2001, 340, 450–66. [DOI] [PubMed] [Google Scholar]
- Tse W. C.; Boger D. L. Acc. Chem. Res. 2004, 37, 61–9. [DOI] [PubMed] [Google Scholar]
- Tse W. C.; Ishii T.; Boger D. L. Bioorg. Med. Chem. 2003, 11, 4479–86. [DOI] [PubMed] [Google Scholar]
- Vashisht Gopal Y. N.; Van Dyke M. W. Biochemistry 2003, 42, 6891–903. [DOI] [PubMed] [Google Scholar]
- Warren C. L.; Kratochvil N. C.; Hauschild K. E.; Foister S.; Brezinski M. L.; Dervan P. B.; Phillips G. N. Jr.; Ansari A. Z. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett J. W.; Muzikar K. A.; Tietjen J.; Warren C. L.; Ansari A. Z.; Dervan P. B. J. Am. Chem. Soc. 2007, 129, 12310–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson C. D.; Warren C. L.; Hauschild K. E.; Ozers M. S.; Qadir N.; Bhimsaria D.; Lee Y.; Cerrina F.; Ansari A. Z. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P. J. Nat. Rev. Genet. 2009, 10, 669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Granas D.; Stormo G. D. PLoS Comput. Biol. 2009, 5, e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulet E.; Busso S.; Camargo A. A.; Simpson A. J.; Mermod N.; Bucher P. Nat. Biotechnol. 2002, 20, 831–5. [DOI] [PubMed] [Google Scholar]
- Zykovich A.; Korf I.; Segal D. J. Nucleic Acids Res. 2009, 37, e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutiu R.; Friedman R. C.; Luo S.; Khrebtukova I.; Silva D.; Li R.; Zhang L.; Schroth G. P.; Burge C. B. Nat. Biotechnol. 2011, 29, 659–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols N. G.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols N. G.; Jacobs C. S.; Farkas M. E.; Dervan P. B. ACS Chem. Biol. 2007, 2, 561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzikar K. A.; Nickols N. G.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskatov J. A.; Meier J. L.; Puckett J. W.; Yang F.; Ramakrishnan P.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 1023–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foister S.; Marques M. A.; Doss R. M.; Dervan P. B. Bioorg. Med. Chem. 2003, 11, 4333–40. [DOI] [PubMed] [Google Scholar]
- Hsu C. F.; Phillips J. W.; Trauger J. W.; Farkas M. E.; Belitsky J. M.; Heckel A.; Olenyuk B. Z.; Puckett J. W.; Wang C. C.; Dervan P. B. Tetrahedron 2007, 63, 6146–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo G. D.; Zhao Y. Nat. Rev. Genet. 2010, 11, 751–60. [DOI] [PubMed] [Google Scholar]
- Bailey T. L. Bioinformatics 2011, 27, 1653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R.; West S. M.; Sosinsky A.; Liu P.; Mann R. S.; Honig B. Nature 2009, 461, 1248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J. M.; Swalley S. E.; Baird E. E.; Dervan P. B. J. Am. Chem. Soc. 1998, 120, 6219–26. [Google Scholar]
- Rucker V. C.; Melander C. M.; Dervan P. B. Helv. Chim. Acta 2003, 87, 1839–51. [Google Scholar]
- Baliga R.; Baird E. E.; Herman D. M.; Melander C.; Dervan P. B.; Crothers D. M. Biochemistry 2001, 40, 3–8. [DOI] [PubMed] [Google Scholar]
- Chenoweth D. M.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2010, 132, 14521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen S.; Durussel T.; Laemmli U. K. Mol. Cell 2000, 6, 999–1011. [DOI] [PubMed] [Google Scholar]
- Urbach A. R.; Dervan P. B. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 4343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach A. R.; Love J. J.; Ross S. A.; Dervan P. B. J. Mol. Biol. 2002, 320, 55–71. [DOI] [PubMed] [Google Scholar]
- Urbach A. R. Ph.D. Thesis, California Institute of Technology, 2002. [Google Scholar]
- Mrksich M. L.; Parks M. E.; Dervan P. B. J. Am. Chem. Soc. 1994, 116, 7983–8. [Google Scholar]
- deClairac R. P. L.; Geierstanger B. H.; Mrksich M. L.; Wemmer D. E.; Dervan P. B. J. Am. Chem. Soc. 1997, 119, 7906–16. [Google Scholar]
- Wang C. C.; Ellervik U.; Dervan P. B. Bioorg. Med. Chem. 2001, 9, 653–7. [DOI] [PubMed] [Google Scholar]
- Matsuda H.; Fukuda N.; Ueno T.; Tahira Y.; Ayame H.; Zhang W.; Bando T.; Sugiyama H.; Saito S.; Matsumoto K.; Mugishima H.; Serie K. J. Am. Soc. Nephrol. 2006, 17, 422–32. [DOI] [PubMed] [Google Scholar]
- Edwards T. G.; Koeller K. J.; Slomczynska U.; Fok K.; Helmus M.; Bashkin J. K.; Fisher C. Antiviral Res. 2011, 91, 177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh I.; Stains C. I.; Ooi A. T.; Segal D. J. Mol. Biosyst. 2006, 2, 551–60. [DOI] [PubMed] [Google Scholar]
- Rucker V. C.; Foister S.; Melander C.; Dervan P. B. J. Am. Chem. Soc. 2003, 125, 1195–202. [DOI] [PubMed] [Google Scholar]
- Krpetić Z.; Singh I.; Su W.; Guerrini L.; Faulds K.; Burley G. A.; Graham D. J. Am. Chem. Soc. 2012, 134, 8356–9. [DOI] [PubMed] [Google Scholar]
- Schmidt T. L.; Heckel A. Small 2009, 5, 1517–20. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Mortazavi A.; Myers R. M.; Wold B. Science 2007, 316, 1497–502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.