

Abstract
Cross-species genomic analyses have proven useful for identifying common genomic alterations that occur in human cancers and mouse models designed to recapitulate human tumor development. High-throughput molecular analyses provide a valuable tool for identifying particular animal models that may represent aspects of specific subtypes of human cancers. Corresponding alterations in gene copy number and expression in tumors from mouse and human suggest that these conserved changes may be mechanistically essential for cancer development and progression, and therefore, they may be critical targets for therapeutic intervention. Using a cross-species analysis approach, mouse models in which the functions of p53, Rb, and BRCA1 have been disrupted demonstrate molecular features of human, triple-negative (ER-, PR-, and ERBB2-), basal-type breast cancer. Using mouse tumor models based on the targeted abrogation of p53 and Rb function, we identified a large, integrated genetic network that correlates to poor outcome in several human epithelial cancers. This gene signature is highly enriched for genes involved in DNA replication and repair, chromosome maintenance, cell cycle regulation, and apoptosis. Current studies are determining whether inactivation of specific members within this signature, using drugs or siRNA, will identify potentially important new targets to inhibit triple-negative, basal-type breast cancer for which no targeted therapies currently exist.
Keywords: genomics, mouse models, mammary cancer, preclinical testing
Introduction
Despite enormous efforts, there have been relatively few promising new therapies for the treatment of breast cancer over the past decade. Although new agents have been developed, testing in a clinical setting takes many years and is limited by tremendous expense. Additionally, testing of combination therapies adds another level of complexity to the relatively limited numbers of patients who will participate in protocols. Model systems for preclinical testing, including cell lines and human xenografts implanted into mice, have been used to test whether new agents interfere with predicted molecular targets and inhibit tumor growth, but these systems have had limited success in predicting response in human patients (Talmadge et al. 2007).
Such disparities between the results of preclinical testing and outcome in clinical trials have impeded progress in treating cancer. However, the application of high-throughput genomic approaches to the study of cancer, including gene expression profiling and array comparative genomic hybridization (CGH), has provided more detailed molecular insights into tumor biology. Gene expression profiling identifies alterations in gene expression that constitute the cancer transcriptome, whereas array CGH reveals copy number alterations in the genome of the tumor. Importantly, comparison of gene expression data with array CGH data enhances our understanding of how cancer evolves and what is required for survival of the cancer cell. For instance, overexpression of particular genes may be attributed to whole chromosome gains, regions of chromosomal amplification, whereas chromosomal deletions may indicate the selective loss of tumor suppressor genes.
Gene expression profiling analyses of genetically engineered mouse (GEM) models of mammary cancer have begun to identify models that represent particular molecular subtypes of human breast cancer (Deeb et al. 2007; Herschkowitz et al. 2007). These data provide strong rationales for choosing particular GEM models in preclinical testing regimens and thus may better represent human breast tumor response to therapy. Future comparisons using array CGH and miRNA analyses will likely further define in what ways the models recapitulate specific molecular features of human breast cancer. This symposium report will highlight and update the advances of cross-species genomic analyses on understanding the biology of breast cancer for improved therapy testing (Bennett and Green 2008).
Defining Breast Cancer Subtypes Based on the Tumor Transcriptome
Breast cancer diagnosis and therapy regimens are, in part, determined based on expression of several genetic markers, including estrogen receptor (ER), progesterone receptor (PR), and ERBB2 (Her2, Neu) (Figure 1). Although these markers provide a basis for selecting particular drugs as part of the treatment of patients, - ER+ (estrogen antagonist), amplified ERBB2 (tratuzumab; ERBB2 humanized monoclonal antibody), tumors lacking these markers (ER-, PR-, ERBB2-) currently can only be treated using a nontargeted therapy approach, primarily conventional chemotherapy. Moreover, many tumors expressing ER or ERBB2 do not respond as predicted, suggesting that the molecular phenotypes of these tumors are more complex and additional predictors and targets will need to be identified. In fact, analyses of breast tumor RNA by gene expression profiling has determined that breast tumors may be categorized into at least 5 major molecular categories based on distinct genetic signatures: luminal A, luminal B, basal, ERBB2+, and normal-like (Sorlie et al. 2001). Importantly, these transcriptome-based classifiers better predict overall prognosis than previous methods (van ‘t Veer et al. 2002; van de Vijver et al. 2002). Based on these findings, microarray chips, containing a subset of the predictor genes (Buyse et al. 2006; Glas et al. 2006), are actively being tested in clinical trials for utility as personalized prognostic biomarkers for overall risk and therapy response (Dowsett and Dunbier 2008).
Figure 1.
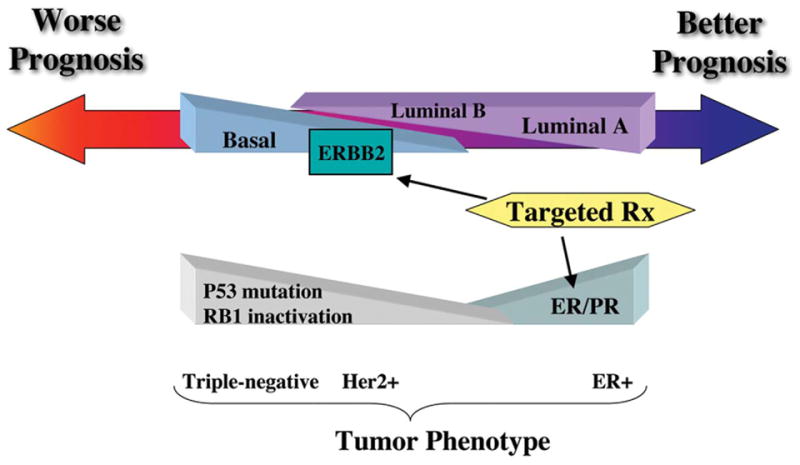
Patient outcome based on categorization of human breast tumors by molecular subtype.
Breast cancer survival appears to be defined, in part, by the apparent luminal or basal lineage phenotype of the tumor. Luminal A tumors tend to have the best prognosis, whereas basal subtype and ERBB2-positive tumors generally have worse outcomes (Figure 1). Differences in survival are also attributed to the availability of targeted therapies for luminal A and ERBB2-amplified tumors and the absence of a responsive molecular target for basal subtype tumors. A subset of basal-type breast tumors responds well to standard chemotherapy, yet a significant portion of patients do not respond or develop recurrence within a few years (Cleator et al. 2007). Therefore, treatments against basal TN (triple-negative) breast cancer are urgently needed.
Classifying breast tumors by molecular subtype has also provided a new means to assess relevance of particular GEM models of mammary cancer. Our lab and others have used cross-species gene expression profiling to delineate which GEM models genetically represent subtypes of breast cancer (Deeb et al. 2007; Dourdin et al. 2008; Herschkowitz et al. 2007; Schade et al. 2009). Although there are many challenges to comparing large genomic data sets between species, defined bio-informatic protocols have eased many of these concerns and yielded datasets of high statistical confidence (Hoenerhoff et al. 2009). This research approach will undoubtedly lead to better choice of models for preclinical testing of targeted therapies.
Several GEM models of mammary cancer have been designated as luminal based on a gene expression signature that is conserved across species, including GATA3, keratin 8, keratin 18, and Xbp1. Genetically engineered mouse tumors with some luminal features include MMTV-Neu, MMTV-PyMT, WAP-Myc, WAP-Int3, and K14-cre; ApcCK°/+ GEM models (Herschkowitz et al. 2007; Kuraguchi et al. 2009). Interestingly, very few GEM models of mammary cancer develop tumors that would be considered ER positive by most human pathologists. Estrogen receptor expression is closely associated with human luminal A–type breast cancer, but it is markedly decreased or not expressed in tumors falling into the luminal B category. Why most mammary tumor mouse models lack ER expression is unclear. The reason may be, in part, the manner in which the genetic lesions have been modeled in the mice. Most transgenic models have used the MMTV LTR to drive expression, which may preferentially be transcribed in progenitor tumor cells that do not express ER. This may possibly be the reason why MMTV-Neu and MMTV-PyMT tumors express luminal markers rather than clustering with human ErbB2+ tumors. It is quite possible that knockout models with basal features may target progenitor cells that develop tumors with basal features.
Some GEM models of mammary cancer develop tumors that represent more than one subtype, which suggests that the genetic aberrations may affect a multipotent progenitor cell in the mammary gland. Alternatively, it remains possible that transformed cells may dedifferentiate, altering their phenotype. For instance, cross-species gene expression analyses of mammary tumors from the p53 fp/fp WAP-Cre conditional knockout (Michalowski, A.M., unpublished data) and MMTV-Metmut GEM (Ponzo et al. 2009) models revealed that individual tumors may express either luminal or the basal-type phenotype. These models may prove to be especially useful in testing agents that target differentiation in the mammary gland, such as histone deacetylase inhibitors in combination with other agents (Botrugno et al. 2009). Interestingly, the MMTV-Wnt1 model develops tumors that have basal (K5+), luminal (K8/18+), and epithelial regions absent for either marker (Herschkowitz et al. 2007). This heterogeneous tumor population has been suggested to be an accessible model for isolation of cancer stem cells (Cho et al. 2008) and may prove to be important for testing therapies that target these progenitor populations.
Genetically engineered mouse mammary tumor models, including C3(1)Tag, Brca1+/−; p53+/−; IR, WAP-Tag, WAP-T121 and Brca1c°/c°; MMTV-Cre; p53+/− have been classified as falling into the basal subtype (Deeb et al. 2007; Herschkowitz et al. 2007). Mouse mammary tumors that develop in these models lack expression for ER, PR, and ErbB2, and they express the basal marker keratin 5 and/or a genetic signature enriched for genes associated with cellular proliferation and functional loss of DNA repair. These molecular features are consistent with the human basal subtype of breast cancer and are associated with poor prognosis (Deeb et al. 2007; Herschkowitz et al. 2007).
Cross-species gene expression comparisons between mouse models of mammary cancer and human breast cancers have identified a new subgroup within human basal subtype breast cancer, designated claudin-low (Herschkowitz et al. 2007). The subtype is characterized by low gene expression of occluding e-cadherin claudins 3, 4, and 7, as well as loss of ER, PR, and ERBB2 expression. Genetically engineered mouse mammary tumors included in this category often have spindle cell morphology, including the C3(1)TAg model, Brca1 c°/c°, MMTV-Cre; p53+/− model, p53 null transplant model, and DMBA-induced mammary tumors. Recently, genetic expression analysis of a cohort of metaplastic breast cancer samples revealed that these tumors, which are generally genetically characterized as basal-like and phenotypically described as mesenchymal/sarcomatoid and/or squamous, have a genetic phenotype that is distinguishable from other basal-subtype tumors and most closely related to the claudin-low subtype (Hennessy et al. 2009). Identification of this small population of breast tumors as a distinct subset may have been overlooked had a cross-species approach not been considered. However, whether this signature distinguishes therapeutic response or overall prognosis between basal subtype tumors and the claudin-low population has still not been reported.
Genomic Changes Related to the Subtype of Breast Cancer
Choice of therapy may also be influenced by the changes in chromosome copy number and rearrangements that are identified in breast tumor samples. Standard molecular cytogenetic techniques involve in vitro culturing of tumor cells and fluorescence in situ hybridization (FISH)–labeled probes during the metaphase of the cell cycle. This technique uses probes that span regions containing known oncogenes and tumor suppressor genes to identify the “usual suspects” involved in tumorigenesis. Another method of chromosome analysis, array CGH, offers another approach for detecting genomic changes. Array CGH uses a large number of smaller probes spanning the entire genome to identify copy number changes. The density of these arrays has significantly increased, allowing for more precise localization of copy number changes in very small regions (Gouas et al. 2008).
Several studies have assessed genomic changes in relation to the molecular signatures that define the subtypes of human breast cancer and found that there are characteristic genomic alterations. Luminal A tumors are associated with amplifications within chromosomes 1 and 16, as well as 11q13-14 and 8p11-12 (Adelaide et al. 2007; Bergamaschi et al. 2006). Luminal B tumors have significant amplifications within chromosomes 8 and 20 (Bergamaschi et al. 2006), and 62% of tumors have Rb loss of heterozygosity (Herschkowitz et al. 2008). The ERBB2 locus in chromosome 17 is amplified in the ERBB2 subtype of breast tumors.
The basal subtype of breast cancer has been reported to have more genomic gains and losses than other subtypes, including amplifications in chromosomes 1, 6, 7,10, 17, and 21 and losses in chromosomes 4 and 14 (Bergamaschi et al. 2006). As also observed in luminal B tumors, Rb loss of heterozygosity occurs frequently, in nearly 72% of tumors (Herschkowitz et al. 2008). K-Ras overexpression has also been noted in a subset of human basal subtype breast cancers (Herschkowitz et al. 2008). The potential role of K-Ras in basal-type tumors was actually first identified in the C3(1)Tag mammary cancer model (Liu et al. 2001) and designed into the MMTV-KRAS(G12D) model (Klinakis et al. 2009).
Several recent studies suggest that what has been referred to as the basal subtype may be composed of even more specific subcategories with the particular genomic alterations that may correlate with different rates of survival (Hennessy et al. 2009; Vincent-Salomon et al. 2007). For instance, medullary breast carcinomas, which are classified as basal subtype but have a better prognosis (Rakha et al. 2009), have greater expression of keratin 5/6; a higher rate of chromosomal gains including 9p, 10p, 16q; and loss of 4p, as well as amplification of other chromosome regions than the general basal subtype population (Vincent-Salomon et al. 2007). Identifying such differences within the heterogeneous basal subtype population will clarify how patterns of genomic changes are associated with therapy response. For instance, array CGH analyses revealed that metaplastic breast tumors are distinguishable from claudin-low and other basal subtype tumors owing to their increased frequency of genomic mutations associated with PIK3A, PTEN, and KRAS and may represent the “basal” tumors that do not respond to chemotherapy (Hennessy et al. 2009; Vincent-Salomon et al. 2007). Such stratification of hormone-negative “basal” subtype tumors into additional subtypes will undoubtedly better define the nuances of basal tumors and identify populations that respond to standard chemotherapy and more targeted drugs.
Although many GEM models of mammary tumorigenesis do not acquire as many genomic alterations as observed in human breast cancer, several GEM models, including the BRCA-deficient, MMTV-Myc, MMTV-PyVmT mice, have genomic changes that are conserved in human breast tumors (Bennett and Green 2008; Borowsky et al. 2005; Montagna et al. 2003; Weaver et al. 2002, 1999). We have begun to assess a number of GEM models of mammary cancer for chromosomal changes associated with the initiating genetic alteration using array CGH and gene expression microarray analyses. These studies will determine how the primary genetic mutation influences accumulation of secondary mutations to affect tumor development. This work will complement earlier work using CGH based on hybridizations to chromosomal spreads (Barkan et al. 2004). Our preliminary data suggest that there are particular genomic changes associated with the initiating genetic mutation within some of the models (M. Zhu, unpublished data). Understanding this progression, and whether it is similar in human breast tumors, may provide a means to determine treatment options based on tumor stage.
Another means to assess how accumulation of genomic changes in GEM models affects mammary tumorigenesis has been through genetic alterations of specific GEM models. For example, the MMTV-Cre; BRCAc°/11-GEM model, in which one BRCA allele is lost in the mammary gland, develops mammary tumors after an extended latency (Xu et al. 1999). However, incorporation of a p53-null allele into the model reduces tumor latency (Brodie et al. 2001). Since p53 loss is almost always found in BRCA1 human breast cancers, this genetic combination in the mouse is an excellent model of human BRCA1 breast tumors that shares other features with the human disease: ER negativity, genomic instability, and activation of other oncogenes. Recent studies have reported that genomic loss of PTEN in GEM models of ERBB2-induced mammary tumors influences tumor progression and tumor subtype (Dourdin et al. 2008; Schade et al. 2009). Comparison of such models in drug studies will likely reveal how drug response shifts in relation to acquiring genomic mutations, an issue that may account for acquiring drug resistance or changes in drug efficacy with time.
Global Approaches to Identify Novel Therapeutic Targets
Global genomic approaches to characterize mouse mammary and human breast tumors have aided in identifying potential therapeutic targets and provided a strong rationale for using specific GEM models to test therapies relevant to particular subtypes of breast cancer (Figure 2). The next question becomes “Can these GEM models, and use of global approaches in general, assist in identifying novel targets for therapy?”
Figure 2.
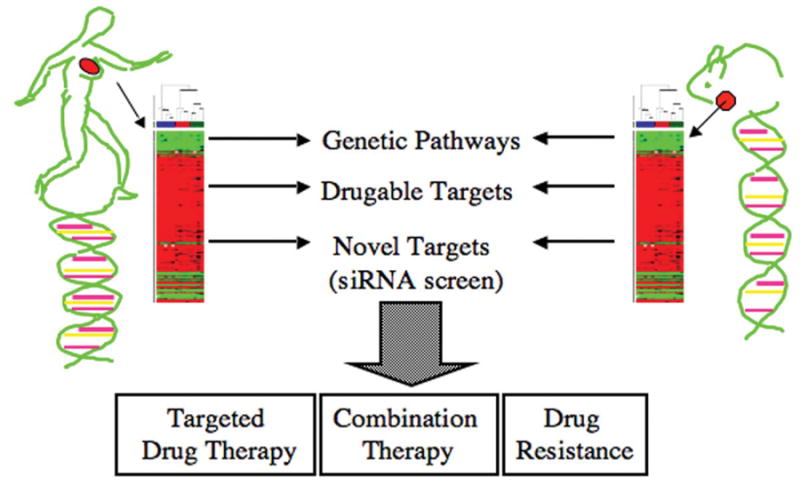
Use of genetically engineered mouse models to identify genetic pathways, drugable targets, and novel targets that are critical for tumor growth and survival. Mouse models are amenable to testing of targeted drug therapies and novel drug combinations, and understanding mechanisms of drug resistance that cannot be easily accomplished in clinical trials.
Our lab has begun to answer this question by testing whether novel therapeutic targets for basal subtype breast cancer can be extracted from a gene expression signature related to the C3(1)T/t-antigen mammary tumor model. The signature was initially defined by identifying genes that were similarly dysregulated in three SV40-T/t-antigen GEM models of cancer (mammary, prostate, and lung) in which p53 and Rb functions are abrogated (Figure 3) (Deeb et al. 2007). Deeb et al. reported that the expression of about 150 genes was similarly altered in all three models. The majority of the genes were associated with genetic pathways involved in proliferation and DNA repair. This finding suggested that the genes within this signature are critical for tumor survival. Importantly, we found this signature to be highly represented in human basal-type breast cancer, metastatic prostate cancer, and aggressive lung cancer and that the signature is predictive of poor outcome.
Figure 3.

Intrinsic biological network associated with SV40-T/t-antigen inactivation of p53 and Rb. Individual genetic nodes are highlighted. Genes in red exhibit increased expression; genes in green exhibit decreased expression. Adapted from Deeb et al. 2007.
We are now functionally testing which genes in this signature are necessary for tumor cell maintenance and survival. Some of these targets can be inhibited with existing pharmaceutical agents, whereas most are not drug targets. Therefore, we have begun to test, in vitro and in vivo, the response of basal-type tumor cells from mouse and human to existing drugs and experimental compounds, both alone and in combination. Additionally, we have performed a focused siRNA screen to determine which of the up-regulated genes in the signature may be critical for tumor cell proliferation or the inhibition of apoptosis. Based on the results of the initial screens, secondary synthetic lethal screens can also be performed. This experimental approach will identify drug targets and effective combinations without relying on readily available agents (Turner et al. 2008; Zhang et al. 2009). Moreover, identification of novel genetic combinations that enhance efficacy of standard chemo- or radiotherapies will guide development of novel drug targets.
Another means to identify novel chemopreventative targets is through comparison of gene expression signatures induced by drug therapy in a particular GEM model. The genetic profile that is conserved in mammary tissue responsive to effective drug treatment may identify the signaling pathways important for prevention of tumor development and growth. For example, such a signature was determined by identifying the common genes affected in the p53-null mammary epithelial GEM model treated with three different chemopreventive agents using SAGE analysis (Abba et al. 2009; Medina et al. 2009). Abba et al. reported that the expression of about 34 genes was similarly altered by two effective compounds, bexarotene and gefitinib, but not by an ineffective compound, celocoxib (Abba et al. 2009). The genes were associated with genetic pathways involved in transcriptional regulation, cell cycle/proliferation, cytoskeleton organization, and protein metabolism. These data suggest that control of tumor progression is dependent on more than just regulation of cellular proliferation. Other cellular pathways, including those associated with the extracellular matrix and cellular metabolism, also may need to be tightly regulated in combination. Functional assays will now have to be pursued to identify the novel genetic targets and effective combinations of drugs that target these pathways as a means to prevent tumor progression.
Toward a Better Model System for Preclinical Testing
The advances in our understanding of the global genetic changes that are conserved between human breast tumors and mouse mammary tumors provide tangible justification for choosing particular models in preclinical testing regimens. As highlighted, a growing number of GEM mammary tumor models have been characterized using cross-species genomic analyses and now are being used for targeted preclinical studies.
Genetically engineered mouse mammary models provide the opportunity to test targeted agents on mammary tissue and tumors with known genetic mutations. Several recent reports have used GEM models of mammary cancer for testing targeted agents. Strecker et al. (2009) determined that treatment initiation and duration of lapatinib, an EGFR and ErbB2 inhibitor, affects tumor growth in the Her2 subtype mammary tumor model MMTV-ErbB2. Klinakis et al (2009). identified that spontaneous upregulation of IGF1r in invasive carcinomas of MMTV-KRAS(G12D) contributes to poor prognosis unless treated with a specific IGF1r inhibitor, picropodophyllin, which dramatically decreased tumor mass. Several reports have identified PARP inhibitor AZD2281 as an effective inhibitor of BRCA mutant basal subtype tumors using BRCA1-associated and BRCA2/p53-deleted GEM models (Hay et al. 2009; Rottenberg et al. 2008). Another recent report determined that inhibition of Aurora kinase using the inhibitor PHA-68032, causes tumor stasis in the MMTV-v-HA-ras mouse model (Carpinelli et al. 2007). These examples highlight how understanding the genetics of GEM models provides relevance to testing targeted drugs in an in vivo system. Indeed, GEM models are not available for every drug target, but by capitalizing on the data derived from gene expression assays and cgh arrays, GEM models can be used for testing a number of genetic targets.
Preclinical testing provides the opportunity to test combination therapies based on genetic targets. For example, recent reports found that PARP inhibitors increase overall survival of BRCA1-associated and BRCA2/p53 mutant GEM models when given in combination with the DNA damage agents cisplatin and carboplatin (Hay et al. 2009; Rottenberg et al. 2008). As seen with the advances in PARP inhibitor drugs for treatment of BRCA mutant basal subtype tumors, successes in such studies will provide more educated rationales for drug combinations used in clinical trials and direct small patient populations into relevant therapeutic modalities.
Genetically engineered mouse models of mammary cancer are also amenable for studying drug resistance. Cancer cell lines and mouse models have been developed with resistance to particular drugs. Drug screens can be performed to test whether drug sensitivity can be restored or whether another agent is now more effective. Several studies have shown that inhibitors of the drug efflux transporter ATP-binding cassette Bi/p glycoprotein (P-gp) efflux pumps, like tariquidar, restore drug sensitivity (Pajic et al. 2009; Rottenberg et al. 2008), but such positive outcomes have not been replicated in the clinic (Fox and Bates 2007). Others suggest that clonal evolution and increases in resident stem cell populations contributes to chemoresistance (Shafee et al. 2008). Further assessment of various GEM models of mammary cancer that develop drug-resistant tumors will aid in identifying paradigms that prolong tumor sensitivity and undoubtedly aid in understanding how tumors evolve to evade therapy.
Preclinical studies focusing on tumor response rather than prevention can expedite experiment duration by using mammary transplants. This adapted method of preclinical testing distributes dissociated primary tumor cells into large numbers of immunocompromised or syngeneic hosts at the same time, resulting in a tumor-staged and highly reproducible experimental model (Martinez et al. 2006; Varticovski et al. 2007). Studies in our lab have used this methodology to screen drug candidates in C3(1)Tag mammary transplants prior to testing in the GEM model. In this fashion, only drug candidates that show efficacy in the transplant study will be tested in the GEM model that spontaneously develops tumors.
Conclusion
In conclusion, high-throughput genomic technologies are proving to be effective tools for better defining the etiology of human breast cancer, as well as the various GEM models of mammary tumorigenesis. It is through this comparison between mouse and man that conserved features of mammary tumors have been identified and genetic pathways essential for tumor survival will continue to be revealed. Importantly, GEM models expressing genetic features similar to human subtypes of breast cancer can be used for testing targeted therapies and combination therapies and for assessing mechanisms of drug resistance in a preclinical setting. Ideally, preclinical testing will accelerate drug evaluation and stratify agents based on their in vivo efficacy prior to entering the clinic. In this way, molecularly identified patient populations will be treated with the most relevant therapy modalities approaching the goal of personalized medicine. Although there is much work to be done, many significant contributions have been made in clinical and basic breast cancer research within the past decade. However, this summary highlights only a small fraction of these advances.
Acknowledgments
This work was supported by the Intramural Program, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD 20892.
Footnotes
For reprints and permissions queries, please visit SAGE’s Web site at http://www.sagepub.com/journalsPermissions.nav.
References
- Abba MC, Hu Y, Levy CC, Gaddis S, Kittrell FS, Hill J, Bissonnette RP, Brown PH, Medina D, Aldaz CM. Identification of modulated genes by three classes of chemopreventive agents at preneoplastic stages in a p53-null mouse mammary tumor model. Cancer Prev Res (Phila Pa) 2009;2:175–84. doi: 10.1158/1940-6207.CAPR-08-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelaide J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F, Charafe-Jauffret E, Cervera N, Desplans J, Parzy D, Schoenmakers E, Viens P, Jacquemier J, Birnbaum D, Bertucci F, Chaffanet M. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007;67:11565–75. doi: 10.1158/0008-5472.CAN-07-2536. [DOI] [PubMed] [Google Scholar]
- Barkan D, Montagna C, Reid T, Green JE. Mammay Gland Cancer. In: Holland EC, editor. Mouse Models of Human Cancer. John Wiley & Sons, Inc; Hoboken, NJ: 2004. pp. 103–31. [Google Scholar]
- Bennett CN, Green JE. Unlocking the power of cross-species genomic analyses: Identification of evolutionarily conserved breast cancer networks and validation of preclinical models. Breast Cancer Res. 2008;10:213. doi: 10.1186/bcr2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi A, Kim YH, Wang P, Sorlie T, Hernandez-Boussard T, Lonning PE, Tibshirani R, Borresen-Dale AL, Pollack JR. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006;45:1033–40. doi: 10.1002/gcc.20366. [DOI] [PubMed] [Google Scholar]
- Borowsky AD, Namba R, Young LJ, Hunter KW, Hodgson JG, Tepper CG, McGoldrick ET, Muller WJ, Cardiff RD, Gregg JP. Syngeneic mouse mammary carcinoma cell lines: Two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis. 2005;22:47–59. doi: 10.1007/s10585-005-2908-5. [DOI] [PubMed] [Google Scholar]
- Botrugno OA, Santoro F, Minucci S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 2009;280:134–44. doi: 10.1016/j.canlet.2009.02.027. [DOI] [PubMed] [Google Scholar]
- Brodie SG, Xu X, Qiao W, Li WM, Cao L, Deng CX. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001;20:7514–23. doi: 10.1038/sj.onc.1204929. [DOI] [PubMed] [Google Scholar]
- Buyse M, Loi S, van’t Veer L, Viale G, Delorenzi M, Glas AM, d’Assignies MS, Bergh J, Lidereau R, Ellis P, Harris A, Bogaerts J, Therasse P, Floore A, Amakrane M, Piette F, Rutgers E, Sotiriou C, Cardoso F, Piccart MJ. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183–92. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, Arrigoni C, Soncini C, Alli C, Patton V, Marsiglio A, Ballinari D, Pesenti E, Fancelli D, Moll J. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6:3158–68. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- Cho RW, Wang X, Diehn M, Shedden K, Chen GY, Sherlock G, Gurney A, Lewicki J, Clarke MF. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells. 2008;26:364–71. doi: 10.1634/stemcells.2007-0440. [DOI] [PubMed] [Google Scholar]
- Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007;8:235–44. doi: 10.1016/S1470-2045(07)70074-8. [DOI] [PubMed] [Google Scholar]
- Deeb KK, Michalowska AM, Yoon CY, Krummey SM, Hoenerhoff MJ, Kavanaugh C, Li MC, Demayo FJ, Linnoila I, Deng CX, Lee EY, Medina D, Shih JH, Green JE. Identification of an integrated SV40 T/t-antigen cancer signature in aggressive human breast, prostate, and lung carcinomas with poor prognosis. Cancer Res. 2007;67:8065–80. doi: 10.1158/0008-5472.CAN-07-1515. [DOI] [PubMed] [Google Scholar]
- Dourdin N, Schade B, Lesurf R, Hallett M, Munn RJ, Cardiff RD, Muller WJ. Phosphatase and tensin homologue deleted on chromosome 10 deficiency accelerates tumor induction in a mouse model of ErbB-2 mammary tumorigenesis. Cancer Res. 2008;68:2122–31. doi: 10.1158/0008-5472.CAN-07-5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowsett M, Dunbier AK. Emerging biomarkers and new understanding of traditional markers in personalized therapy for breast cancer. Clin Cancer Res. 2008;14:8019–26. doi: 10.1158/1078-0432.CCR-08-0974. [DOI] [PubMed] [Google Scholar]
- Fox E, Bates SE. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–59. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- Glas AM, Floore A, Delahaye LJ, Witteveen AT, Pover RC, Bakx N, Lahti-Domenici JS, Bruinsma TJ, Warmoes MO, Bernards R, Wessels LF, Van’t Veer LJ. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 2006;7:278. doi: 10.1186/1471-2164-7-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouas L, Goumy C, Veronese L, Tchirkov A, Vago P. Gene dosage methods as diagnostic tools for the identification of chromosome abnormalities. Pathol Biol (Paris) 2008;56:345–53. doi: 10.1016/j.patbio.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Hay T, Matthews JR, Pietzka L, Lau A, Cranston A, Nygren AO, Douglas-Jones A, Smith GC, Martin NM, O’Connor M, Clarke AR. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009;69:3850–55. doi: 10.1158/0008-5472.CAN-08-2388. [DOI] [PubMed] [Google Scholar]
- Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, He X, Weigman V, Fan C, Palazzo J, Hortobagyi GN, Nolden LK, Wang NJ, Valero V, Gray JW, Perou CM, Mills GB. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69:4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10:R75. doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, Bastein R, Quackenbush J, Glazer RI, Brown PH, Green JE, Kopelovich L, Furth PA, Palazzo JP, Olopade OI, Bernard PS, Churchill GA, Van Dyke T, Perou CM. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenerhoff MJ, Michalowski AM, Qiu TH, Green JE. Bioinformatics in Cancer and Cancer Therapy. Humana Press; Totowa, NJ: 2009. Chapter 4: Bioinformatics Approaches to the Analysis of the Transcriptome of Animal Models of Cancer. [Google Scholar]
- Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, Efstratiadis A. Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci U S A. 2009;106:2359–64. doi: 10.1073/pnas.0810221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127:1041–55. doi: 10.1016/j.cell.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraguchi M, Ohene-Baah NY, Sonkin D, Bronson RT, Kucherlapati R. Genetic mechanisms in Apc-mediated mammary tumorigenesis. PLoS Genet. 2009;5:e1000367. doi: 10.1371/journal.pgen.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ML, Shibata MA, Von Lintig FC, Wang W, Cassenaer S, Boss GR, Green JE. Haploid loss of Kiras delays mammary tumor progression in C3 (1)/SV40 Tag transgenic mice. Oncogene. 2001;20:2044–49. doi: 10.1038/sj.onc.1204280. [DOI] [PubMed] [Google Scholar]
- Martinez JM, Sali T, Okazaki R, Anna C, Hollingshead M, Hose C, Monks A, Walker NJ, Baek SJ, Eling TE. Drug-induced expression of nonsteroidal anti-inflammatory drug-activated gene/ macrophage inhibitory cytokine-1/prostate-derived factor, a putative tumor suppressor, inhibits tumor growth. J Pharmacol Exp Ther. 2006;318:899–906. doi: 10.1124/jpet.105.100081. [DOI] [PubMed] [Google Scholar]
- Medina D, Kittrell F, Hill J, Zhang Y, Hilsenbeck SG, Bissonette R, Brown PH. Prevention of tumorigenesis in p53-null mammary epithelium by rexinoid bexarotene, tyrosine kinase inhibitor gefitinib, and celecoxib. Cancer Prev Res (Phila PA) 2009;2:168–74. doi: 10.1158/1940-6207.CAPR-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagna C, Lyu MS, Hunter K, Lukes L, Lowther W, Reppert T, Hissong B, Weaver Z, Ried T. The Septin 9 (MSF) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Res. 2003;63:2179–87. [PubMed] [Google Scholar]
- Pajic M, Iyer JK, Kersbergen A, van der Burg E, Nygren AO, Jonkers J, Borst P, Rottenberg S. Moderate increase in Mdr1a/1b expression causes in vivo resistance to doxorubicin in a mouse model for hereditary breast cancer. Cancer Res. 2009;69:6396–6404. doi: 10.1158/0008-5472.CAN-09-0041. [DOI] [PubMed] [Google Scholar]
- Ponzo MG, Lesurf R, Petkiewicz S, O’Malley FP, Pinnaduwage D, Andrulis IL, Bull SB, Chughtai N, Zuo D, Souleimanova M, Germain D, Omeroglu A, Cardiff RD, Hallett M, Park M. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci U S A. 2009;106:12903–8. doi: 10.1073/pnas.0810402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakha EA, Aleskandarany M, El-Sayed ME, Blamey RW, Elston CW, Ellis IO, Lee AH. The prognostic significance of inflammation and medullary histological type in invasive carcinoma of the breast. Eur J Cancer. 2009;45:1780–87. doi: 10.1016/j.ejca.2009.02.014. [DOI] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NM, Borst P, Jonkers J. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade B, Rao T, Dourdin N, Lesurf R, Hallett M, Cardiff RD, Muller WJ. PTEN Deficiency in a Luminal ErbB-2 Mouse Model Results in Dramatic Acceleration of Mammary Tumorigenesis and Metastasis. J Biol Chem. 2009;284:19018–26. doi: 10.1074/jbc.M109.018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, Stanbridge EJ, Lee EY. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68:3243–50. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker TE, Shen Q, Zhang Y, Hill JL, Li Y, Wang C, Kim HT, Gilmer TM, Sexton KR, Hilsenbeck SG, Osborne CK, Brown PH. Effect of lapatinib on the development of estrogen receptor-negative mammary tumors in mice. J Natl Cancer Inst. 2009;101:107–13. doi: 10.1093/jnci/djn436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007;170:793–804. doi: 10.2353/ajpath.2007.060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–77. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–36. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Varticovski L, Hollingshead MG, Robles AI, Wu X, Cherry J, Munroe DJ, Lukes L, Anver MR, Carter JP, Borgel SD, Stotler H, Bonomi CA, Nunez NP, Hursting SD, Qiao W, Deng CX, Green JE, Hunter KW, Merlino G, Steeg PS, Wakefield LM, Barrett JC. Accelerated preclinical testing using transplanted tumors from genetically engineered mouse breast cancer models. Clin Cancer Res. 2007;13:2168–77. doi: 10.1158/1078-0432.CCR-06-0918. [DOI] [PubMed] [Google Scholar]
- Vincent-Salomon A, Gruel N, Lucchesi C, MacGrogan G, Dendale R, Sigal-Zafrani B, Longy M, Raynal V, Pierron G, de Mascarel I, Taris C, Stoppa-Lyonnet D, Pierga JY, Salmon R, Sastre-Garau X, Fourquet A, Delattre O, de Cremoux P, Aurias A. Identification of typical medullary breast carcinoma as a genomic subgroup of basal-like carcinomas, a heterogeneous new molecular entity. Breast Cancer Res. 2007;9:R24. doi: 10.1186/bcr1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver Z, Montagna C, Xu X, Howard T, Gadina M, Brodie SG, Deng CX, Ried T. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene. 2002;21:5097–5107. doi: 10.1038/sj.onc.1205636. [DOI] [PubMed] [Google Scholar]
- Weaver ZA, McCormack SJ, Liyanage M, du Manoir S, Coleman A, Schrock E, Dickson RB, Ried T. A recurring pattern of chromosomal aberrations in mammary gland tumors of MMTV-cmyc transgenic mice. Genes Chromosomes Cancer. 1999;25:251–60. [PubMed] [Google Scholar]
- Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Jones TL, Martin SE, Caplen NJ, Pommier Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J Biol Chem. 2009;284:18085–95. doi: 10.1074/jbc.M109.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]