

Abstract
There is considerable biologic plausibility to the hypothesis that genetic variability in pathways involved in insulin signaling and energy homeostasis may modulate dietary risk associated with colorectal cancer. We utilized data from 2 population-based case-control studies of colon (n = 1,574 cases, 1,970 controls) and rectal (n = 791 cases, 999 controls) cancer to evaluate genetic variation in candidate SNPs identified from 9 genes in a candidate pathway: PDK1, RP6KA1, RPS6KA2, RPS6KB1, RPS6KB2, PTEN, FRAP1 (mTOR), TSC1, TSC2, Akt1, PIK3CA, and PRKAG2 with dietary intake of total energy, carbohydrates, fat, and fiber. We employed SNP, haplotype, and multiple-gene analysis to evaluate associations. PDK1 interacted with dietary fat for both colon and rectal cancer and with dietary carbohydrates for colon cancer. Statistically significant interaction with dietary carbohydrates and rectal cancer was detected by haplotype analysis of PDK1. Evaluation of dietary interactions with multiple genes in this candidate pathway showed several interactions with pairs of genes: Akt1 and PDK1, PDK1 and PTEN, PDK1 and TSC1, and PRKAG2 and PTEN. Analyses show that genetic variation influences risk of colorectal cancer associated with diet and illustrate the importance of evaluating dietary interactions beyond the level of single SNPs or haplotypes when a biologically relevant candidate pathway is examined.
The association between diet and colon and rectal cancer remains to be fully elucidated. Attempts to resolve differences observed across studies through the incorporation of genetic susceptibility data has had limited success. Most studies have focused on single nutrients such as dietary folate, vitamin D, or calcium and have taken a candidate-gene or candidate-pathway approach but have often evaluated only single polymorphisms to determine interactions with diet (1-7). Pathways involved in metabolic signaling, insulin sensitivity, and cellular energy balance may influence colon and rectal cancer risk and may be modified by dietary behaviors. Some studies of candidate genes and polymorphisms support these mechanisms (8-12). Many other genes and many other relevant polymorphisms associated with metabolic signaling and insulin sensitivity have not been studied, despite their plausibility (13). Assessment of associations beyond the single polymorphism has not generally been attempted.
One pathway of importance has been outlined (13) and focuses on the convergence of 3 critical elements associated with colon and rectal cancer risk: inflammation, hormones (including both sex steroid and growth hormones), and energy-related factors. The hormone and energy-related factors may be influenced by dietary components that contribute to energy consumption and insulin signaling. Key elements identified as central to the pathway include phosphatase tensin homolog deleted on chromosome 10 (PTEN), which acts as a negative regulator of cell growth in the insulin/IGF pathway by modulating signaling via phosphatidylinositol 3-kinase (PI3K) and v-akt murine thymoma viral oncogene homolog 1 (Akt1, also known as protein kinase B or PKB). PI3K and Akt1 are downstream of the insulin receptor. Akt1 also regulates the functioning of the tuberous sclerosis complex (TSC1 and TSC2), 2 other components of the pathway. TSC1 and TSC2 have been shown to be involved in the regulation and proliferation of cell size and may have other roles given their links to insulin signaling.
In cells with altered energy homeostasis, adenosine monophosphate (AMP)-dependent kinase (PRKAG2) is involved through phosphorylation of TSC1 and TSC2 (14-17). FK506 binding protein 12-rapamycin associated protein 1 (FRAP1, alias mTOR) ultimately represses anabolic processes (ATP utilization) and enhances catabolic processes (ATP generation). PDK1, another important component of the pathway, mediates the cellular influence of growth factors and insulin and plays a role in the activation of PKB/Akt1 (18) in response to insulin levels. PDK1 also regulates both RPS6KA1 and A2 (RSK1 and RSK2) and RPS6KB1 and B2 (S6K1 and S6K2). RSK has been identified as a component in the regulation of gut inflammation and apoptosis (19,20), and it is weakly activated by insulin (21); whereas S6K is involved in cell growth and regulation through growth factors such as EGF, PDGF, and insulin as well as mTOR. PDK1 mediates the cellular influence of growth factors and insulin by activating both RSK and S6K and is essential for activation of PKB/Akt1 (18). Genetic variation in this pathway may work with dietary consumption to alter colon and rectal cancer risk. In our previous work, we have discussed how these genes and genetic variation of these genes influence colon and rectal cancer risk (22,23).
Given the importance of this candidate pathway in regulation of metabolic signaling and insulin sensitivity, it is reasonable to hypothesize that genetic variation in this pathway—in conjunction with dietary factors, especially those related to energy consumption and insulin—would influence colon and rectal cancer risk beyond that observed for genetic variation alone. In this study, we evaluated the interaction between total energy intake, dietary fat, dietary carbohydrate, and dietary fiber on the one hand and genetic variation in this candidate pathway on the other. We evaluated candidate SNPs, haplotypes, and multiple-gene analysis to characterize these associations more comprehensively.
METHODS
Data for the study come from 2 case-control studies conducted in Utah, the Northern California Kaiser Permanente Medical Care Program (KPMCP), and the Twin Cities Metropolitan area of Minnesota (colon cancer study only). Eligibility criteria included being between 30 and 79 yr of age at time of diagnosis, English speaking, mentally competent to complete the interview, no previous history of colorectal cancer, and no known (as indicated on the pathology report or by self-report) familial adenomatous polyposis, ulcerative colitis, or Crohn’s disease. Controls were frequency matched to cases by sex and 5-yr age groups. Controls were randomly selected from membership lists (KPMCP) or from a driver’s license list (Minnesota). In Utah, controls 65 or older were randomly selected from lists provided by the Centers for Medicare and Medicaid Services and controls younger than 65 were randomly selected from driver’s license lists. Study eligibility and recruitment details of the study have been published previously (24,25). Cooperation rates were 83% at KPMCP, 76% at Utah and 67% Minnesota (colon cases); 73% at KPMCP, 53% at Minnesota and 69% at Utah (colon controls); 75.4% at KPCMP and 69.7% at Utah (rectal cases) and 69.9% at KPMCP and 67.2% at Utah (rectal controls).
The current analysis is restricted to subjects who provided a blood sample. The colon cancer study population consists of non-Hispanic White cases (n = 1444) and controls (n = 1841), Hispanic or American Indian cases (n = 60) and controls (n = 75), and African American cases (n = 70) and controls (n = 54). The rectal cancer study population consists of non-Hispanic white cases (n = 657) and controls (n = 856), Hispanic or American Indian cases (n = 63) and controls (n = 69), African American cases (n = 31) and controls (n = 44), and Asian cases (n = 40) and controls (n = 30). Numbers vary slightly by data available for each genotype.
Trained and certified interviewers collected diet and lifestyle data as previously described (26,27) using the same data questionnaire and study procedures for both the colon and rectal studies. The referent year for the study was the calendar year approximately 2 yr prior to date of diagnosis (cases) or selection (controls). Information was collected on demographic factors such as age, sex, and study center, and on exposures including diet, physical activity, aspirin and non-steroidal drug use, body size, and other lifestyle factors including medical, family, and reproductive history. Dietary intake data were obtained using a detailed diet history questionnaire that captured frequency of consumption, serving size, and details on added fat and sugar (27,28).
DNA was extracted from blood drawn from study participants. Tagging SNPs were identified using HapMap and the Illumina Platform tagSNP database. We have previously described methods used for tagSNP selection and gene coverage (22,23). Based on previous analyses, we include those SNPs that appear to be most important for the genes of interest. Table 1 shows the candidate SNPs included in these analyses.
TABLE 1.
Description of candidate SNPs for proposed dietary associations
| Symbol | Location | SNP | SNP Base | MAF | Major/Minor Allele | FDR HWE | Colon
|
Rectal
|
||
|---|---|---|---|---|---|---|---|---|---|---|
| Heterozygote OR | Homozygote Rare OR | Heterozygote OR | Homozygote Rare OR | |||||||
| AKT1 | 14q32.32 | rs2494738 | 104317731 | 0.07 | G/A | 0.62 | 1.05 (0.87, 1.27)* | 1.02 (0.78, 1.33)* | ||
| PDK1 | 2q31.1 | rs11904366 | 173170476 | 0.15 | G/T | 1.00 | 1.04 (0.89, 1.21) | 1.10 (0.73, 1.67) | 0.87 (0.70, 1.09) | 0.69 (0.35, 1.35) |
| rs4972842 | 173137279 | 0.19 | T/A | 1.00 | 0.88 (0.76, 1.03) | 0.95 (0.66, 1.37) | 0.85 (0.69, 1.06) | 1.43 (0.88, 2.33) | ||
| PRKAG2 | 7q36.1 | rs6464156 | 150950488 | 0.5 | A/G | 0.85 | 0.84 (0.71, 0.98) | 0.82 (0.67, 0.99) | 1.14 (0.91, 1.43) | 0.97 (0.73, 1.27) |
| rs1881632 | 151147274 | 0.2 | C/T | 1.00 | 1.11 (0.96, 1.29) | 1.02 (0.71, 1.45) | 0.95 (0.77, 1.17) | 0.80 (0.48, 1.32) | ||
| rs9632641 | 151106857 | 0.2 | A/C | 0.79 | 1.15 (1.00, 1.33) | 1.28 (0.92, 1.78) | 0.92 (0.74, 1.14) | 0.72 (0.44, 1.17) | ||
| rs9648723 | 151026570 | 0.18 | A/C | 0.98 | 0.96 (0.82, 1.11) | 1.48 (1.07, 2.06) | 1.06 (0.86, 1.31) | 0.63 (0.40, 1.00) | ||
| rs1104897 | 151063507 | 0.14 | G/A | 1.00 | 1.19 (1.02, 1.39) | 1.46 (0.94, 2.26) | 1.22 (0.99, 1.52) | 1.88 (1.08, 3.27) | ||
| rs4725431 | 151104112 | 0.16 | T/C | 1.00 | 1.06 (0.91, 1.23) | 0.97 (0.63, 1.48) | 0.80 (0.64, 1.01) | 0.70 (0.38, 1.28) | ||
| rs6464170 | 151107793 | 0.22 | C/G | 0.96 | 1.06 (0.92, 1.22) | 0.75 (0.55, 1.01) | 1.09 (0.89, 1.34) | 1.03 (0.69, 1.53) | ||
| rs9648724 | 151133235 | 0.25 | G/A | 0.95 | 0.89 (0.77, 1.03) | 0.74 (0.55, 1.00) | 0.80 (0.65, 0.98) | 0.67 (0.43, 1.04) | ||
| PTEN | 10q23.3 | rs532678 | 89713322 | 0.34 | C/T | 1.00 | 0.92 (0.80, 1.06) | 0.92 (0.74, 1.15) | 1.25 (1.01, 1.53) | 1.25 (0.92, 1.69) |
| RPS6KA1 | 1p | rs12025634 | 26738092 | 0.15 | C/T | 0.57 | 1.00 (0.85, 1.16) | 1.74 (1.08, 2.79) | 0.86 (0.69, 1.08) | 0.91 (0.44, 1.88) |
| RPS6KA2 | 6q27 | rs16898963 | 166802654 | 0.12 | A/C | 0.39 | 1.01 (0.86, 1.19) | 2.28 (1.27, 4.09) | 0.95 (0.76, 1.20) | 0.94 (0.41, 2.17) |
| rs2072638 | 167108340 | 0.3 | T/C | 0.25 | 1.23 (1.06, 1.42) | 1.17 (0.94, 1.46) | 1.02 (0.83, 1.26) | 1.77 (1.28, 2.45) | ||
| TSC1 | 9q34 | rs11243940 | 134811193 | 0.23 | A/G | 1.00 | 1.05 (0.91, 1.21) | 1.26 (0.93, 1.70) | 0.99 (0.81, 1.21) | 0.91 (0.59, 1.40) |
| TSC2 | 16p13.3 | rs3087631 | 2079128 | 0.18 | A/T | 0.38 | 1.07 (0.92, 1.24) | 0.68 (0.48, 0.95) | 1.10 (0.89, 1.36) | 0.79 (0.50, 1.27) |
OR, odds ratio; FDR HWE = False Discovery Rate Hardy Weinberg Equilibrium.
Indicates dominant model used due to MAF < 0.1.
STATISTICAL ANALYSIS
Candidate SNPs previously identified as being associated with colon and rectal cancer were targeted as interacting with dietary factors (22,23). Where 2 or more candidate SNPs were from the same gene and had similar associations, only the SNP with the most significant association as determined by level of significance was carried on for further analysis with dietary factors. We evaluated interactions based on single SNPs, haplotypes, and by multiple-gene analysis based on allele copies from different genes in a similar manner as would be done for haplotype analysis. All statistical analysis was performed using SAS v. 9.2 (SAS Institute, Cary, NC). The odds ratios (ORs) and corresponding 95% confidence intervals (CIs) presented here were generated using multiple logistic regression models for colon and rectal separately. PROC HAPLOTYPE was used to estimate haplotypes and combined genotype frequencies using the EM algorithm and assuming linkage equilibrium.
Based upon biologically informed hypotheses, we evaluated interactions between dietary intake of fat, carbohydrates, fiber, and energy on the one hand and SNPs from the candidate genes on the other, using a chi-square goodness-of-fit test that compared the maximum likelihood ratios of a model with and without an interaction term. Based on initial assessment from these dietary factors, a high-risk and low-risk diet score was developed that considered high intake of fat and low intake of carbohydrates and fiber as high risk and the opposite characteristics low risk. SNPs for assessment were determined by examining hypothesized dietary interactions with SNPs and groups of SNPs based on known biologic pathways. All possible pairwise haplotypes and combined genotypes were examined with dietary factors and only the SNPs involved in haplotypes that were statistically significant at the 0.01 level are shown; supplemental data are available for other less significant associations online. Additional adjustment of BMI, cigarette smoking, dietary energy, fiber, calcium, physical activity, aspirin/NSAIDs, and family history of colorectal cancer did not appreciably alter the risk estimates; therefore, data are presented for the minimally adjusted model. We do not present data where the P for interaction is >0.01, given the number of comparisons made; see Supplemental Data online.
RESULTS
PDK1 rs11904366 interacted statistically significantly with dietary fat for both colon and rectal cancer: having a copy of the T allele and a low-fat intake was associated with a reduced risk of cancer (Table 2). For colon cancer, the increased risk from having a T allele and a high-fat intake was more evident than for rectal cancer. The PDK1 rs4972842 SNP also interacted with dietary carbohydrate and risk of colon cancer: The risk declined with increasing presence of the A allele at low carbohydrate but showed no difference at high carbohydrate intake, itself associated with reduced risk. A haplotype analysis of both of these PDK1 SNPs together, showed statistically significant interaction with both dietary fat and carbohydrate for colon cancer and dietary carbohydrate (data not shown in Table 2) for rectal cancer. Dietary fiber, although not reaching statistical significance at the 0.01 level, showed trends of importance for rectal cancer. Combining these dietary variables, dietary fat, and carbohydrates for colon cancer and dietary fat, carbohydrates, and fiber for rectal cancer into a high- and low-risk diet score showed even stronger results with the P interaction being 0.0002 for colon cancer and 0.008 for rectal cancer. These results are shown in Fig. 1, with the risk estimate per copy of the haplo-type. The risk of cancer increases with the high-risk diet and is most evident when the T-T haplotype is present. The interaction between dietary carbohydrate and individual SNPs was not detected.
TABLE 2.
Associations with dietary fat and carbohydrates with individual PDK1 SNPs
| Controls Cases
|
(95% CI) | Controls Cases
|
(95% CI) | Controls Cases
|
(95% CI) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | N | OR | N | N | OR | N | N | OR | ||||
| Colon | Tertile 1 | Tertile 2 | Tertile 1 | |||||||||
| PDK1 (rs11904366 G > T) | Dietary Fat | |||||||||||
| GG | 515 | 379 | 1.00 | 491 | 362 | 0.99 | (0.82, 1.19) | 393 | 352 | 1.18 | (0.96, 1.43) | |
| GT/TT | 226 | 131 | 0.78 | (0.60, 1.00) | 186 | 181 | 1.28 | (0.997, 1.63) | 145 | 151 | 1.35 | (1.03, 1.76) |
| P Interaction | 0.01 | |||||||||||
| PDK1 (rs4972842 T > A) | Dietary Carbohydrate | |||||||||||
| TT | 381 | 393 | 1.00 | 446 | 373 | 0.84 | (0.69, 1.03) | 471 | 312 | 0.68 | (0.55, 0.84) | |
| TA/AA | 226 | 159 | 0.70 | (0.54, 0.89) | 211 | 166 | 0.80 | (0.62, 1.02) | 221 | 153 | 0.73 | (0.56, 0.94) |
| P Interaction | 0.02 | |||||||||||
| Rectal Cancer | ||||||||||||
| PDK1 (rs11904366 G > T) | Dietary Fat | |||||||||||
| GG | 149 | 123 | 1.00 | 228 | 180 | 0.98 | (0.72, 1.34) | 307 | 258 | 1.04 | (0.78, 1.40) | |
| GT/TT | 70 | 35 | 0.61 | (0.380.98) | 91 | 56 | 0.76 | (0.50, 1.15) | 114 | 102 | 1.12 | (0.78, 1.61) |
| P Interaction | 0.02 | |||||||||||
Adjusted for age, center, race, and sex.
FIG. 1.
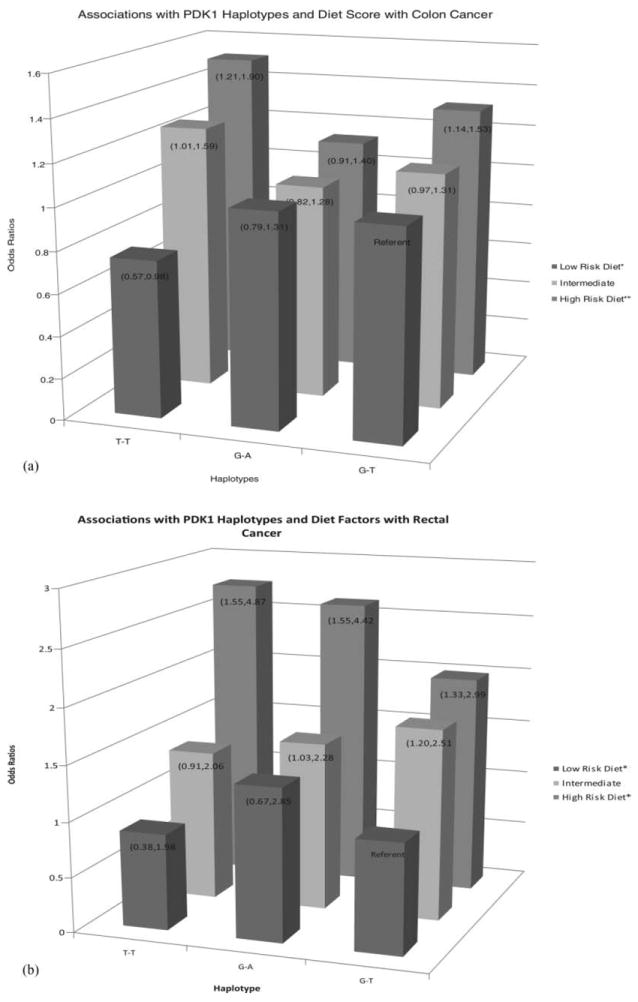
Associations among PDK1 haplotypes, high- and low-risk diet score and colon and rectal cancer risk.
Assessment of combined genes and dietary factors showed several important interactions with low- and high-risk diet and risk of colon cancer (Table 3). Associations are for a single copy of the combined alleles from the genes listed. The pattern of interactions in colon cancer shows that risk decreases (or is flat) with low-risk diet and increases (or is flat) with high-risk diet in the presence of variation in the following pairs of genes: Akt1/PDK1 rs11904366; PDK1/PTEN; PDK1/TSC1; and PRKAG2/PTEN. Additionally, energy in-take interacted significantly with PRKAG2 and PTEN (P interaction = 0.0017). Although we have only shown associations where the P interaction was 0.01 or less, associations with P interaction values between 0.05 and 0.01 were detected for several dietary factors and combined genes, including FRAP1 (rs1057079), PDK2 (rs1063647 and rs4794096), PIK3CA (rs7651265 and rs7640662), TSC1 (rs13295634 and rs11243940), and TSC2 (rs3087631). For rectal cancer (Table 4), the patterns are essentially similar. Other associations at the 0.05 to 0.01 level of significance that are not shown in the tables include all the SNPs in the list presented above for colon cancer.
TABLE 3.
Associations between low- and high-risk diet and genetic variation and risk of colon cancer
| Low Diet Risk*
|
Intermediate diet risk
|
High Diet Risk**
|
P interaction | ||||
|---|---|---|---|---|---|---|---|
| OR | (95% CI) | OR | (95% CI) | OR | (95% CI) | ||
| PDK1 rs11904366 (G > T) & PTEN rs532678 (C > T) | |||||||
| G-C | 1.00 | 1.04 | (0.88, 1.22) | 1.18 | (1.00, 1.39) | 0.0008 | |
| G-T | 0.82 | (0.66, 1.02) | 1.00 | (0.83, 1.22) | 1.20 | (0.99, 1.45) | |
| T-C | 0.73 | (0.53, 1.01) | 1.23 | (0.94, 1.61) | 1.41 | (1.07, 1.85) | |
| T-T | 0.64 | (0.41, 1.00) | 1.07 | (0.74, 1.55) | 1.42 | (1.02, 1.99) | |
| PRKAG2 rs1881632 (C > T) & PTEN rs532678 (C > T) | |||||||
| C-C | 1.00 | 1.13 | (0.96, 1.34) | 1.23 | (1.04, 1.45) | 0.0296 | |
| C-T | 0.85 | (0.68, 1.06) | 1.06 | (0.87, 1.29) | 1.28 | (1.06, 1.55) | |
| T-C | 1.03 | (0.77, 1.37) | 1.11 | (0.86, 1.44) | 1.51 | (1.18, 1.93) | |
| T-T | 0.78 | (0.51, 1.17) | 1.13 | (0.80, 1.58) | 1.42 | (1.02, 1.96) | |
| AKT1 rs2494738 (G > A) & PDK1 rs11904366 (G > T) | |||||||
| G-G | 1.00 | 1.09 | (0.95, 1.25) | 1.25 | (1.09, 1.43) | 0.0005 | |
| G-T | 0.76 | (0.58, 1.00) | 1.23 | (0.98, 1.55) | 1.53 | (1.22, 1.92) | |
| A-G | 0.85 | (0.57, 1.26) | 1.18 | (0.84, 1.65) | 1.52 | (1.09, 2.11) | |
| A-T | 0.51 | (0.20, 1.29) | 1.67 | (0.82, 3.40) | 1.55 | (0.75, 3.21) | |
| PDK1 rs11904366 (G > T) & TSC1 rs11243940 (A > G) | |||||||
| G-A | 1.00 | 1.08 | (0.92, 1.25) | 1.31 | (1.12, 1.52) | 0.0003 | |
| G-G | 1.13 | (0.89, 1.43) | 1.33 | (1.07, 1.65) | 1.30 | (1.05, 1.61) | |
| T-A | 0.80 | (0.59, 1.08) | 1.21 | (0.94, 1.56) | 1.63 | (1.27, 2.09) | |
| T-G | 0.69 | (0.41, 1.14) | 1.60 | (1.06, 2.44) | 1.32 | (0.86, 2.01) | |
| PRKAG2 rs4725431 (T > C) & PTEN rs532678 (C > T) | |||||||
| T-C | 1.00 | 1.15 | (0.98, 1.36) | 1.30 | (1.10, 1.53) | 0.0501 | |
| T-T | 0.82 | (0.66, 1.02) | 1.08 | (0.89, 1.31) | 1.27 | (1.05, 1.53) | |
| C-C | 1.10 | (0.80, 1.52) | 1.07 | (0.82, 1.40) | 1.24 | (0.95, 1.62) | |
| C-T | 0.95 | (0.63, 1.45) | 1.09 | (0.76, 1.56) | 1.56 | (1.12, 2.17) | |
Adjusted for age, center, race, and sex. OR indicates odds ratios.
Includes individuals in tertile 1 of dietary fat and tertile 3 of carbohydrates.
Includes individuals in tertile 3 of Fat and tertile 1 of Carbohydrates, tertile 3 of Fat and tertile 2 of Carbohydrates, tertile 2 of Fat and tertile 1 of Carbohydrate; intermediate risk includes all others.
TABLE 4.
Associations between low- and high-risk diet and genetic variation and risk of rectal cancer
| Low Diet Risk*
|
Intermediate Diet Risk
|
High Diet risk**
|
P interaction | ||||
|---|---|---|---|---|---|---|---|
| OR | (95% CI) | OR | (95% CI) | OR | (95% CI) | ||
| PRKAG2 rs4725431 (T > C) & TSC2 rs3087631 (A > T) | |||||||
| T-A | 1.00 | 1.61 | (1.13, 2.28) | 2.10 | (1.42, 3.10) | 0.01 | |
| T-T | 1.39 | (0.62, 3.12) | 1.51 | (1.02, 2.23) | 2.49 | (1.50, 4.16) | |
| C-A | 0.60 | (0.23, 1.53) | 1.45 | (0.96, 2.17) | 1.30 | (0.73, 2.33) | |
| C-T | 0.85 | (0.12, 6.09) | 1.01 | (0.57, 1.80) | 2.82 | (1.09, 7.27) | |
| PDK1 rs11904366 (G > T) & PTEN rs532678 (C > T) | |||||||
| G-C | 1.00 | 1.83 | (1.21, 2.76) | 2.26 | (1.44, 3.57) | 0.0004 | |
| G-T | 1.66 | (0.88, 3.13) | 2.03 | (1.33, 3.10) | 2.54 | (1.55, 4.15) | |
| T-C | 1.62 | (0.64, 4.07) | 1.36 | (0.84, 2.18) | 2.94 | (1.48, 5.83) | |
| T-T | 0.11 | (0.01, 1.88) | 1.86 | (1.11, 3.14) | 3.39 | (1.45, 7.91) | |
| PRKAG2 rs1881632 (C > T) & PTEN rs532678 (C > T) | |||||||
| C-C | 1.00 | 1.92 | (1.24, 2.96) | 2.51 | (1.56, 4.05) | 0.0219 | |
| C-T | 1.24 | (0.62, 2.49) | 2.26 | (1.45, 3.53) | 2.79 | (1.67, 4.66) | |
| T-C | 1.94 | (0.84, 4.44) | 1.74 | (1.08, 2.81) | 2.53 | (1.37, 4.66) | |
| T-T | 2.05 | (0.76, 5.53) | 1.75 | (1.05, 2.92) | 3.01 | (1.45, 6.26) | |
OR indicates odds ratio. Adjusted for age, center, race, and sex.
Includes individuals in tertile 1 of dietary fat and tertile 3 of both carbohydrates and fiber.
Includes individuals in tertile 3 of Fat and tertile 1 of both carbohydrates and fiber, tertile 3 of fat and tertile 2 of both carbohydrates and fiber, tertile 2 of fat and tertile 1 of both carbohydrates and fiber. Intermediate risk includes all others.
DISCUSSION
Our assessment of dietary intake with genetic variation in a proposed candidate pathway that involves metabolic signaling, insulin sensitivity, and cellular energy balance showed numerous associations. Although dietary factors were associated with only one gene when assessed with independent SNPs, assessment of genetic factors that include multiple SNPs within a gene and across multiple genes, showed numerous associations. The analysis supports the role of diet in this candidate pathway but also points to issues where the analysis involves the interpretation of gene–diet interaction.
There is a biologically plausible mechanism for the hypothesized and observed associations. PDK1 mediates the cellular influence of growth factors and insulin by activating both RSK and S6K and is essential for activation of PKB/Akt1 (18). PTEN acts as a metabolic regulator by modulating signalling via the PI3K and the Akt1 pathway, downstream of the insulin receptor. Akt1-dependent phosphorylation negatively regulates the functioning of TSC1 and TSC2 (29), 2 other factors involved in insulin signaling. FK506 binding protein 12-rapamycin associated protein 1 (FRAP1, alias mTOR) ultimately represses anabolic processes (ATP utilization) and enhances catabolic processes (ATP generation), restoring the system toward normal energy homeostasis (30). In cells with excess AMP due to altered energy homeostasis, phosphorylated AMP-dependent kinase (PRKAA1 and PRKAG2) (14-17) alters TSC1 and TSC2. These candidate genes were selected based on their known function within the pathway (13).
A challenge to the analysis of genetic data within a candidate pathway, when unknown functional polymorphisms have been identified, is to capture the representation of the genetic variation that is most meaningful. In this study, we used 3 methods to explore dietary factors associated with genetic variability that may influence energy and insulin signaling and ultimately colon and rectal cancer risk. We analyzed individual SNPs, haplotypes, and multiple genes. Each level of exploration contributed to our understanding of these associations and illustrated shortcomings of the other methods. A major assumption regarding the mechanisms of action of low-penetrance genes, such as those studied here, is that risk associated with such genes is influenced by exposures such as diet. As demonstrated in our analyses, associations would be missed if we limited our assessment to only those genes and SNPs where independent associations were detected. PDK1 illustrates these findings. In single-SNP analyses, we observed no association between PDK1 SNPs and either colon or rectal cancer. However, individual PDK1 SNPs interacted with several dietary components that comprise a high- and low-risk diet for both colon and rectal cancer. Similarly, haplotypes of multiple PDK1 SNPs, and the combined PDK1 SNPs in combination with other genes, influenced disease risk. The consistency of associations from the multiple levels of analysis adds to the likelihood of these being true associations.
Although our data support the utility of several approaches to analysis, differences in interpretation of associations between assessment of genotype and haplotype and across gene allele analysis exist. The association with a specific genotype is simply the risk associated with a given genotype. For both the multiple-gene-SNP analyses and the haplotype analyses, the interpretation is based on having one copy of the allele combination and assumes a dose effect. However, if a recessive genotype is most importantly related to the outcome of interest, an assumption of a linear association may be inappropriate. We have presented combinations of 2 SNPs in determining risk. However, combinations of 3 and 4 SNPs also were important in many instances, although not presented here because of the number of potential combinations and the increasing lack of precision with the increasing number of individual loci evaluated. From our 3- and 4-locus analyses, we selected SNPs that appeared to drive most of the associations and then focused on the 2-locus analyses. We have thus undoubtedly missed potentially important associations. Additionally, it is possible that other dietary factors are important and may impact the associations observed for fat and carbohydrates.
Although candidate pathways are often hypothesized as being important, appropriate analytic methods are often lacking to characterize the genetic variation in the pathway. Our analysis of haplotypes and multigenes helped encompass different components of the pathway, although methods to integrate all components are needed. Despite this limitation, our data illustrate the importance of dietary components to this pathway. Although many of these SNPs did not interact independently with dietary factors, haplotypes and/or gene combinations often gave clues to important interactions and to the genetic variability that influences dietary risk.
Our findings suggest that dietary factors associated with energy and insulin are influenced by a set of genes involved in insulin sensitivity. PDK1, in combination with dietary fat and carbohydrate, appears to be important for both colon and rectal cancer. PDK1 is a key regulator within the pathway and its importance is seen as it relates in combination with other genes and diet to alter risk. These findings need replication in other large studies. Our analysis emphasizes the complexity of diet and gene associations and the need of a more comprehensive analytical approach to understanding disease associations.
Acknowledgments
This study was funded by National Cancer Institute Grants CA48998 and CA61757. This research also was supported by the Utah Cancer Registry, which is funded by Contract #N01- PC-67000 from the National Cancer Institute, with additional support from the State of Utah Department of Health, the Northern California Cancer Registry, and the Sacramento Tumor Registry. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute. We would like to acknowledge the contributions of Sandra Edwards, Roger Edwards, Leslie Palmer, Donna Schaffer, Dr. Kristin Anderson, and Judy Morse for data management and collection
Contributor Information
Martha L. Slattery, Department of Medicine, University of Utah, Salt Lake City, Utah
Abbie Lundgreen, Department of Medicine, University of Utah, Salt Lake City, Utah.
Jennifer S. Herrick, Department of Medicine, University of Utah, Salt Lake City, Utah
Bette J. Caan, Kaiser Permanente Medical Research Program, Oakland, California
John D. Potter, Fred Hutchinson Cancer Research Institute, Seattle, Washington
Roger K. Wolff, Department of Medicine, University of Utah, Salt Lake City, Utah
References
- 1.Slattery ML, Potter JD, Samowitz W, Schaffer D, Leppert M. Methylenetetrahydrofolate reductase, diet, and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:513–518. [PubMed] [Google Scholar]
- 2.Giovannucci E, Rimm EB, Ascherio A, Stampfer MJ, Colditz GA, et al. Alcohol, low-methionine-low-folate diets, and risk of colon cancer in men. J Natl Cancer Inst. 1995;87:265–273. doi: 10.1093/jnci/87.4.265. [DOI] [PubMed] [Google Scholar]
- 3.Ulrich CM, Curtin K, Potter JD, Bigler J, Caan B, et al. Polymorphisms in the reduced folate carrier, thymidylate synthase, or methionine synthase and risk of colon cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2509–2516. doi: 10.1158/1055-9965.EPI-05-0261. [DOI] [PubMed] [Google Scholar]
- 4.Slattery ML, Sweeney C, Murtaugh M, Ma KN, Caan BJ, et al. Associations between vitamin D, vitamin D receptor gene and the androgen receptor gene with colon and rectal cancer. Int J Cancer. 2006;118:3140–3146. doi: 10.1002/ijc.21791. [DOI] [PubMed] [Google Scholar]
- 5.Garland C, Shekelle RB, Barrett-Connor E, Criqui MH, et al. Dietary vitamin D and calcium and risk of colorectal cancer: a 19-year prospective study in men. Lancet. 1985;1:307–309. doi: 10.1016/s0140-6736(85)91082-7. [DOI] [PubMed] [Google Scholar]
- 6.Huncharek M, Muscat J, Kupelnick B. Colorectal cancer risk and dietary intake of calcium, vitamin d, and dairy products: a meta-analysis of 26,335 cases from 60 observational studies. Nutr Cancer. 2009;61:47–69. doi: 10.1080/01635580802395733. [DOI] [PubMed] [Google Scholar]
- 7.Slattery ML, Wolff RK, Herrick JS, Caan BJ, Potter JD. IL6 genotypes and colon and rectal cancer. Cancer Causes Control. 2007;18:1095–1105. doi: 10.1007/s10552-007-9049-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takezaki T, Hamajima N, Matsuo K, Tanaka R, Hirai T, et al. Association of polymorphisms in the beta-2 and beta-3 adrenoceptor genes with risk of colorectal cancer in Japanese. Int J Clin Oncol. 2001;6:117–122. doi: 10.1007/pl00012092. [DOI] [PubMed] [Google Scholar]
- 9.Zou T, Fleisher AS, Kong D, Yin J, Souza RF, et al. Sequence alterations of insulin-like growth factor binding protein 3 in neoplastic and normal gastrointestinal tissues. Cancer Res. 1998;58:4802–4804. [PubMed] [Google Scholar]
- 10.Slattery ML, Samowitz W, Hoffman M, Ma KN, Levin TR, et al. Aspirin, NSAIDs, and colorectal cancer: possible involvement in an insulin-related pathway. Cancer Epidemiol Biomarkers Prev. 2004;13:538–545. [PubMed] [Google Scholar]
- 11.Murtaugh MA, Sweeney C, Ma KN, Potter JD, Caan BJ, et al. Vitamin D receptor gene polymorphisms, dietary promotion of insulin resistance, and colon and rectal cancer. Nutr Cancer. 2006;55:35–43. doi: 10.1207/s15327914nc5501_5. [DOI] [PubMed] [Google Scholar]
- 12.Slattery ML, Folsom AR, Wolff R, Herrick J, Caan BJ, et al. Transcription factor 7-like 2 polymorphism and colon cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:978–982. doi: 10.1158/1055-9965.EPI-07-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slattery ML, Fitzpatrick FA. Convergence of hormones, inflammation, and energy-related factors: a novel pathway of cancer etiology. Cancer Prev Res (Phila PA) 2009;2:922–930. doi: 10.1158/1940-6207.CAPR-08-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 15.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carling D. Ampk. Curr Biol. 2004;14:R220. doi: 10.1016/j.cub.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 17.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayascas JR. Dissecting the role of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) signalling pathways. Cell Cycle. 2008;7:2978–2982. doi: 10.4161/cc.7.19.6810. [DOI] [PubMed] [Google Scholar]
- 19.Fantini MC, Pallone F. Cytokines: from gut inflammation to colorectal cancer. Curr Drug Targets. 2008;9:375–380. doi: 10.2174/138945008784221206. [DOI] [PubMed] [Google Scholar]
- 20.Shant J, Cheng K, Marasa BS, Wang JY, Raufman JP. Akt-dependent NF-kappaB activation is required for bile acids to rescue colon cancer cells from stress-induced apoptosis. Exp Cell Res. 2009;315:432–450. doi: 10.1016/j.yexcr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Abe Y, Yoon SO, Kubota K, Mendoza MC, Gygi SP, et al. p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J Biol Chem. 2009;284:14939–14948. doi: 10.1074/jbc.M900097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slattery ML, Herrick JS, Lundgreen A, Fitzpatrick FA, Curtin K, et al. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis. 31:1604–1611. doi: 10.1093/carcin/bgq142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slattery ML, Lundgreen A, Herrick JS, Wolff RK. Genetic variation in RPS6KA1, RPS6KA2, PRS6KB1, RPS6KB2, PDK1 and risk of colon or rectal cancer. Mut Res. 2011;706:13–20. doi: 10.1016/j.mrfmmm.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slattery ML, Edwards S, Curtin K, Ma K, Edwards R, et al. Physical activity and colorectal cancer. Am J Epidemiol. 2003;158:214–224. doi: 10.1093/aje/kwg134. [DOI] [PubMed] [Google Scholar]
- 25.Slattery ML, Potter J, Caan B, Edwards S, Coates A, et al. Energy balance and colon cancer—beyond physical activity. Cancer Res. 1997;57:75–80. [PubMed] [Google Scholar]
- 26.Edwards S, Slattery ML, Mori M, Berry TD, Caan BJ, et al. Objective system for interviewer performance evaluation for use in epidemiologic studies. Am J Epidemiol. 1994;140:1020–1028. doi: 10.1093/oxfordjournals.aje.a117192. [DOI] [PubMed] [Google Scholar]
- 27.Slattery ML, Caan BJ, Duncan D, Berry TD, Coates A, et al. A computerized diet history questionnaire for epidemiologic studies. J Am Diet Assoc. 1994;94:761–766. doi: 10.1016/0002-8223(94)91944-5. [DOI] [PubMed] [Google Scholar]
- 28.McDonald A, Van Horn L, Slattery M, Hilner J, Bragg C, et al. The CARDIA dietary history: development, implementation, and evaluation. J Am Diet Assoc. 1991;91:1104–1112. [PubMed] [Google Scholar]
- 29.Agarwal A, Das K, Lerner N, Sathe S, Cicek M, et al. The AKT/I kappa B kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-kappa B and beta-catenin. Oncogene. 2005;24:1021–1031. doi: 10.1038/sj.onc.1208296. [DOI] [PubMed] [Google Scholar]
- 30.Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;27(Suppl 2):S43–S51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]