

Noncentrosomal microtubules recruit myosin II to the cell cortex in order to engage adherens junctions and increase tight junction formation, resulting in an increase in mechanical integrity of cell sheets.
Abstract
During differentiation, many cells reorganize their microtubule cytoskeleton into noncentrosomal arrays. Although these microtubules are likely organized to meet the physiological roles of their tissues, their functions in most cell types remain unexplored. In the epidermis, differentiation induces the reorganization of microtubules to cell–cell junctions in a desmosome-dependent manner. Here, we recapitulate the reorganization of microtubules in cultured epidermal cells. Using this reorganization assay, we show that cortical microtubules recruit myosin II to the cell cortex in order to engage adherens junctions, resulting in an increase in mechanical integrity of the cell sheets. Cortical microtubules and engaged adherens junctions, in turn, increase tight junction function. In vivo, disruption of microtubules or loss of myosin IIA and B resulted in loss of tight junction–mediated barrier activity. We propose that noncentrosomal microtubules act through myosin II recruitment to potentiate cell adhesion in the differentiating epidermis, thus forming a robust mechanical and chemical barrier against the external environment.
Introduction
Although centrosomal microtubule arrays have been well characterized in cultured cells, differentiated cells in situ adopt diverse noncentrosomal arrays of microtubules that are likely matched to the tissue’s physiology. For example, cortical microtubules in plants direct the synthesis of cell wall components through association with cellulose synthase, while apical-basal arrays of microtubules in simple epithelia play roles in polarity and directed trafficking of cellular components (Müsch, 2004; Paradez et al., 2006). In other cell types the function of noncentrosomal microtubule arrays has not been studied in detail—in fact, the organization of microtubule arrays in many differentiated cells has not been adequately described.
In the epidermis, microtubules undergo a stereotypical reorganization upon terminal differentiation, accumulating at the cell cortex (Lechler and Fuchs, 2007). This reorganization requires desmosomes, cell adhesion structures that are especially prominent in differentiated epidermis. Desmosomal components recruit a subset of centrosomal proteins, including ninein, Lis1, and Nde1/Ndel1, to the cortex (Lechler and Fuchs, 2007; Sumigray et al., 2011). These proteins are implicated in microtubule organization in a number of cell types, and ninein and Ndel1 are required for microtubule anchoring at the centrosome (Delgehyr et al., 2005; Guo et al., 2006). Loss of Lis1 in the epidermis results in loss of cortical microtubules, a phenotype that mirrors loss of the desmosomal protein desmoplakin (Lechler and Fuchs, 2007; Sumigray et al., 2011). Due to pleiotropic effects resulting from genetic ablation of these genes, it has not been possible to assign specific functions to cortical microtubules in the epidermis.
The major functions of the epidermis are to serve as a barrier against mechanical and chemical assaults and to prevent dehydration. This requires the presence of robust cell adhesions (desmosomes, adherens junctions, and tight junctions) in the terminally differentiating cells—the same cells with robust cortical microtubules. Here, we demonstrate that noncentrosomal microtubules increase both the mechanical strength and the impermeability of epithelial sheets. These effects are mediated by myosin II–induced tension, which stabilizes adherens junctions and increases tight junction activity. Therefore, cortical microtubules coordinate cytoskeletal and cell adhesion structures to generate a robust barrier in the differentiated layers of the skin.
Results
Microtubule stabilization promotes their cortical accumulation
To study the function of cortical microtubules, we attempted to develop an assay in cultured keratinocytes. However, the cortical localization of the centrosomal proteins ninein and Lis1 was not sufficient for cortical microtubules to robustly form in cultured cells. When calcium was added to keratinocytes to induce desmosome formation, these proteins were recruited to cell junctions within hours (Fig. 1 A and unpublished data). Despite this, microtubules did not become strongly reorganized to the cell cortex (Fig. 1 B), though low levels of cortical microtubules were seen in some cells. Similarly, in the first layer of cells committing to differentiation in the epidermis, ninein and Lis1 were cortically localized, but microtubules were largely cytoplasmic (Lechler and Fuchs, 2007; Sumigray et al., 2011).
Figure 1.

Stabilization of microtubules promotes their cortical reorganization. (A) Mouse keratinocytes grown in 1.2 mM Ca2+-containing media for 24 h have Lis1 at cell junctions. Microtubules, however, remain cytoplasmic (B). (C) Immunofluorescence analysis of Tau (red) in E17.5 mouse embryo epidermis. Hoechst labels DNA (blue) and the dotted line marks the basement membrane. (D) Tau levels in lysates prepared from basal and suprabasal (differentiated) cells of the epidermis. (E–G) Organization of microtubules in wild-type keratinocytes that were transfected with GFP-tagged Map2b (E), Map4 (F), or Tau-24 (G). Transfected cells were identified by GFP fluorescence and are marked with an asterisk. (H–J) Organization of microtubules in DP-null cells that were transfected with Map2b (H), Map4 (I), or Tau-24 (J). Microtubule organization in WT control cells (K) and in cells treated with 10 µM taxol for 1 h (L). (M) Microtubule organization in a small colony of WT keratinocytes. Note that microtubules accumulate at cell–cell junctions, but not free leading edges (arrows). Microtubule organization in α-catenin–null (N), p120-catenin–null (O), and DP-null cells (P). (Q) Transmission electron micrograph of a taxol-treated keratinocyte. Some of the microtubules are highlighted in red adjacent to the electron-dense desmosomes. All bars are 10 µm except Q, which is 0.5 µm.
A number of genomic studies have reported increased expression of microtubule-associated proteins in the differentiated and/or granular layers of the mouse or human epidermis. These include MAP2, MAP4, MAP7, and Tau (Fabre-Jonca et al., 1999; Patel et al., 2006; Raymond et al., 2008; Mattiuzzo et al., 2011). We have verified the increased expression of Tau in differentiating epidermis by both immunofluorescence and biochemical analyses (Fig. 1, C and D). In E17.5 mouse skin, Tau was clearly up-regulated in the differentiated cells of the granular layers of the epidermis. Little staining was seen in basal/spinous cells (Fig. 1 C). When isolated epidermis was separated into basal (proliferative) and suprabasal (differentiated) fractions, Tau levels were significantly higher in the differentiated cell compartment (Fig. 1 D). In cultured cells, Tau levels were undetectable and did not significantly increase within the first 48 h of calcium shift (not depicted). We similarly saw increased MAP4 levels in the upper differentiated region of the epidermis (Fig. S1 A).
To determine whether the expression of Tau and/or other MAPs could promote the accumulation of cortical microtubules in cultured cells, we exogenously expressed MAP2b, MAP4, or Tau in cultured keratinocytes. 24 h after the addition of calcium (to induce desmosome formation), we analyzed microtubule organization in the cells. Each of these MAPs was sufficient to induce the reorganization of microtubules to the cell cortex (Fig. 1, E–G). In vivo, the desmosomal protein desmoplakin is required for cortical microtubules to form (Lechler and Fuchs, 2007). This is also true in culture as none of the MAPs were able to promote cortical microtubule reorganization in desmoplakin-null cells, though they were able to bundle microtubules (Fig. 1, H–J).
Most MAPs can promote microtubule stabilization (Dehmelt and Halpain, 2005). To determine whether stabilization was sufficient to induce reorganization, we treated keratinocytes with taxol, a small molecule that stabilizes microtubules (Orr et al., 2003). In taxol, robust cortical microtubules formed in keratinocytes (Fig. 1, compare K with L). This occurred at doses as low as 500 nM (not depicted), a concentration at which taxol stabilizes microtubules but does not substantially lead to their bundling (Derry et al., 1995; Orr et al., 2003). Cortical microtubules did not form at the free “leading edge” of colonies of cells grown in high calcium or in cells grown in low calcium-containing media, where cell adhesion structures do not form (Fig. 1 M; Fig. S1 B). These data demonstrate that both cell contact and adhesion are required for the local recruitment of cortical microtubules. Taxol-induced reorganization of microtubules also required the desmosomal protein desmoplakin, but not the adherens junction components α-catenin or p120-catenin (Fig. 1, N–P). These requirements are identical to those found for cortical microtubules in intact epidermis (Lechler and Fuchs, 2007). Cell surface E-cadherin levels are very low in p120-catenin–null cells (Perez-Moreno et al., 2006), demonstrating that robust adherens junctions are not required for cortical microtubule reorganization. Therefore, although not absolute, microtubule stabilization induces a shift from cytoplasmic arrays to cortical arrays—allowing a more robust reorganization than occurs in untreated keratinocytes.
The strong recruitment of microtubules to cell junctions in taxol-treated cells allowed us to visualize them ultrastructurally. By electron microscopy we saw clear enrichment of microtubules near the cell cortex in taxol-treated cells (Fig. 1 Q; Fig. S1, E and F). Microtubules came within 50 nm of the desmosomes and were highly enriched in the area 50–200 nm away. They were aligned roughly parallel to the plasma membrane. We did not note any specific contact points mediating direct association between the desmosome and the microtubules. In control cells, cortical microtubules were not easily identifiable by ultrastructural analysis (Fig. S1 G).
Cortical microtubules increase epithelial sheet integrity
The establishment of a cell culture model that robustly recapitulates the microtubule reorganization seen in vivo allowed us to study the functional consequences of this reorganization. A primary role of the epidermis is to act as a mechanical and chemical barrier against the environment. To determine whether cortical microtubules are important for the mechanical properties of keratinocytes, we tested the integrity of isolated epidermal sheets. Confluent, adherent sheets of cells were released from the underlying substrate by treatment with dispase. The sheets were then subjected to mechanical stress, resulting in fragmentation. Quantitation of the number of fragments was used to determine the mechanical and cell-adhesive properties of the sheets. Upon taxol treatment, epidermal sheets became significantly more resistant to mechanical disruption, whereas disruption of microtubules with nocodazole treatment had little effect (Fig. 2, A and B; Fig. S1, H and I). These data demonstrate that cortical microtubules promote the integrity of epidermal sheets, whereas the cytoplasmic arrays make little contribution to mechanical strength.
Figure 2.

Cortical microtubules increase epidermal sheet integrity. (A) Epidermal sheet fragments resulting from mechanical disruption of untreated or taxol-treated keratinocytes. Bar, 1 cm. (B) Quantitation of sheet fragments resulting from mechanical disruption of keratinocyte monolayers. P = 0.0017 for control vs. taxol treatment; n = 5 for control, 7 for taxol treatment. (C) Quantitation of epidermal sheet stability in cells treated with normal rat serum (NRS) in the presence or absence of taxol and treated with the E-cadherin inhibitory antibodies, DECMA, in the presence or absence of taxol. P = 0.02. (D) Quantitation of epidermal sheet stability of α-catenin–null cells with and without taxol treatment. (E) Quantitation of epidermal sheet stability after treatment with the myosin II inhibitor (blebbistatin) with or without taxol (compare to control in B). (F) Epidermal sheet integrity in myosin IIA floxed cells, with or without Cre recombinase and taxol, as indicated. P < 0.0001. Error bars are SEM.
Cortical microtubules cause tension-induced stabilization of adherens junctions
Tissue integrity is mediated by cell–cell adhesions and their connection to the underlying cytoskeleton. Adherens junctions are essential adhesion structures in the skin that interact with the actomyosin cytoskeleton (Tunggal et al., 2005; Tinkle et al., 2008; Cavey and Lecuit, 2009; Smutny et al., 2010). Immunofluorescence analysis of β-catenin, a core component of the adherens junction, revealed increased cortical intensity after taxol treatment (Fig. 3 A). Quantitation of the cortical/cytoplasmic ratio of fluorescence intensity confirmed a significant increase in the cortical pool of β-catenin (Fig. 3 B). We did not detect significant increases in either E-cadherin or α-catenin at cell junctions by immunofluorescence analysis (Fig. S2, A and B). In addition, both total and surface pools of E-cadherin, as measured by biotinylation, were unchanged by taxol treatment (Fig. 3 C). This is also true of the total levels of adherens junction components such as α- and β-catenin. However, when β-catenin was immunoprecipitated from taxol-treated cells, an increase in E-cadherin association was noted as compared with control cells, again supporting a change in adherens junctions upon microtubule reorganization (Fig. 3 D). The changes in both β-catenin localization and E-cadherin association required α-catenin (Fig. S2, D and E). In addition, we noted increased association of the actin-binding protein vinculin with β-catenin complexes (Fig. 3 D). There was not a significant increase in cortical localization of vinculin (Fig. S2 C). This demonstrates a change in engagement of vinculin with the junction, rather than its recruitment.
Figure 3.

Cortical microtubules induce engagement of adherens junctions. (A) Immunofluorescence analysis of β-catenin localization in control and taxol-treated cells, as indicated. (B) Quantitation of the cortical/cytoplasmic intensity measurements of β-catenin in control and taxol-treated cells. P = 0.014, n = 200 cells. (C) Western blots of adherens junction components in control and taxol-treated cell extracts, as indicated. The bottom panel represents exposed E-cadherin as identified by cell surface biotinylation. (D) Analysis of β-catenin immunoprecipitates from control and taxol-treated cultured cells. Proteins were subjected to Western blot analysis with E-cadherin, β-catenin, α-catenin, and vinculin antibodies. (E) Analysis of β-catenin immunoprecipitates from control and nocodazole-treated epidermal tissue extracts. Proteins were subjected to Western blot analysis with β-catenin and vinculin antibodies. (F) FRAP analysis of α-catenin-GFP in control and taxol-treated cells. Shown is a kymograph over 1 min. The red arrow indicates the time of photobleaching. Bar, 2 µm. (G–J) Quantitation of the mobile fraction of indicated adherens junction proteins in control and taxol-treated cells. All are in wild-type cells, except H, which is in desmoplakin-null cells. Boxes are 25–75% marks, whiskers are 5–95% marks. For G, P = 0.004; n = 8 for control and 11 for taxol treatment. (K) Immunofluorescence analysis of wild-type cells with the α18 antibody, which recognizes a tension-dependent conformation of α-catenin. Control cells, taxol-treated cells, and taxol plus blebbistatin-treated cells are shown, as indicated. (L) Immunofluorescence analysis of α18 in control and myosin IIA–null cells in the presence and absence of taxol, as indicated. Bars: (A, K, and L) 10 µm.
In the granular cells of the epidermis, most microtubules are cortical (as they are after taxol treatment of cultured cells). In extracts from intact epidermis, we detected association of vinculin with β-catenin (Fig. 3 E). When epidermis was pretreated with nocodazole to disrupt microtubules, this association was decreased. Therefore, in cultured cells, a shift toward cortical microtubules resulted in increased vinculin association with the adherens junction and, in intact epidermis, loss of microtubules resulted in a decrease in the association of vinculin with the adherens junction.
We next tested whether the dynamics and stability of adherens junctions were also altered in the presence of cortical microtubules. We performed fluorescence recovery after photobleaching (FRAP) on control and taxol-treated cells expressing α-catenin-GFP. Although α-catenin turnover occurred quickly in control cells, this was reduced in taxol-treated cells (Fig. 3 F). Quantitation of the mobile fraction revealed a significant decrease after taxol treatment (Fig. 3 G). This change in α-catenin turnover is dependent upon desmoplakin, as no increase in α-catenin stability was found in desmoplakin-null cells, though these cells have a slightly increased mobile fraction under normal conditions (Fig. 3 H). Therefore, stabilization of microtubules, in the absence of their cortical localization, does not impact α-catenin dynamics. β-Catenin also trended toward increased stability; however, this did not reach statistical significance (Fig. 3 I). Finally, E-cadherin mobile fractions were unchanged (Fig. 3 J). Therefore, both the composition and dynamics of adherens junctions are altered in the presence of cortical microtubules.
The lack of change in surface E-cadherin and the selective stabilization of α-catenin could be explained by increased engagement of α-catenin to the underlying cytoskeleton, which would strengthen adherens junctions. Recent work has demonstrated that adherens junctions function as mechanosensors and responders (le Duc et al., 2010; Liu et al., 2010; Yonemura et al., 2010; Taguchi et al., 2011). This has been proposed to involve conformational changes in α-catenin, allowing increased interaction with vinculin and/or other actin-binding proteins (Yonemura et al., 2010). The changes we observed in β-catenin immunoprecipitates (especially the association with vinculin) and in α-catenin dynamics were consistent with engaged adherens junctions. A monoclonal antibody to α-catenin, α18, recognizes a conformation of the protein that is induced by tension (Yonemura et al., 2010). In wild-type keratinocytes, there was sparse labeling of cell contacts with the α18 antibody (Fig. 3 K). Upon taxol treatment, staining of α18 at junctions became robust, suggesting a conformational change of α-catenin or an alteration in its association with other proteins. Because the α18 epitope is thought to be tension sensitive, we tested whether blebbistatin, an inhibitor of nonmuscle myosin II, prevented its appearance (Straight et al., 2003). When cells were treated with both taxol and blebbistatin, no increase in α18 was detected (Fig. 3 K). To test myosin II function genetically, we cultured myosin IIA fl/fl keratinocytes in which Cre-recombination leads to loss of myosin IIA. Although control cells (both myosin IIA fl/fl that were not treated with adeno-Cre and wild-type cells treated with adeno-Cre) exhibited an increase in α18 staining upon taxol treatment, cells depleted of myosin IIA did not (Fig. 3 L and unpublished data). Therefore, we conclude that myosin II activity is required for cortical microtubules to exert their effect on adherens junctions. Despite this, we did not detect any kinetic delays in the cortical localization of E-cadherin or α-catenin after calcium switch in myosin IIA–null cells (Fig. S3). This suggests that assembly and engagement of adherens junctions are molecularly distinct.
Cortical microtubules act through adherens junctions to increase epithelial sheet integrity
Although cortical microtubules induced changes in adherens junctions, it was not clear whether those changes accounted for the differences in epithelial sheet integrity observed in Fig. 2 B. We therefore exposed control and taxol-treated keratinocytes to inhibitory E-cadherin antibodies (DECMA) to perturb adherens junctions. When treated with taxol, DECMA-treated cells did not show an increase in mechanical strength, whereas control cells did (Fig. 2 C). Under these conditions, E-cadherin inhibitory antibodies had negligible effects on cell sheet integrity. In addition, α-catenin–null cells showed no increase in mechanical integrity upon taxol treatment (Fig. 2 D). These experiments clearly show that cortical microtubules do not have a direct stabilizing role on their own, as taxol-treated α-catenin–null cells and E-cadherin inhibitory antibody-treated cells have robust cortical microtubules yet show no change in mechanical integrity (Fig. 1 N and unpublished data). In total, these data demonstrate that cortical microtubules promote tissue strength through engagement of adherens junctions. The lack of significant effect on sheet integrity after DECMA treatment or loss of α-catenin suggests that the majority of the mechanical integrity of keratinocytes is provided by desmosomes. Although adherens junction perturbation does not significantly decrease integrity, their engagement can increase integrity.
To test the functional significance of contractility on cortical microtubule-induced epithelial sheet strengthening, we treated epidermal sheets with a myosin II inhibitor. Blebbistatin treatment caused both increased fragility of wild-type cells and loss of the stabilizing effect of cortical microtubules (Fig. 2, compare E with B). The loss of response to taxol was not secondary to defects in the microtubule cytoskeleton, as cortical microtubules were present after treatment (not depicted). Because blebbistatin caused cell sheet fragility on its own, we also examined the integrity of myosin IIA–depleted cells. Although control cells showed clear strengthening in response to taxol, the myosin IIA–depleted cells did not (Fig. 2 F). Wild-type cells treated with adeno-Cre responded normally, indicating that this was not a secondary effect of infection (not depicted). Thus, cortical microtubules act through a myosin II–dependent pathway to engage adherens junctions, increasing epidermal strength.
Cortical microtubules promote tight junction function
In intact epidermis, cortical microtubules are robust in granular layer cells where functional tight junctions also form (Lechler and Fuchs, 2007). Because adherens junction components are known to be essential for proper tight junction assembly (Gumbiner et al., 1988; Tunggal et al., 2005), we analyzed the effect of taxol treatment on tight junction function in cultured cells, as measured by transepithelial resistance (TER). TER was measured in both control cells and taxol-treated cells. As has been previously reported, TER increased within 24 h of calcium addition, and the reported measurements were taken at 48 h when TER had stabilized (Helfrich et al., 2007). In taxol-treated cells, TER values were double those in control cells (Fig. 4 A). Therefore, cortical microtubules promote an increase in tight junction barrier function. No changes in tight junction protein staining or ZO1 dynamics were observed (Fig. S4, A and B). Importantly, this also demonstrates that cortical microtubules do not globally alter turnover rates of peripheral membrane proteins. In support of this, there was not a significant change in fluorescence recovery of actin-GFP at the cell junctions after taxol treatment (Fig. S4, C and D).
Figure 4.

Tight junction barrier activity is increased by cortical microtubules. (A) Transepithelial resistance (TER) measurements were taken of wild-type cells, cells treated with taxol, cells treated with E-cadherin inhibitory DECMA antibodies (with and without taxol), and in blebbistatin-treated cells (with and without taxol). P = 0.0037 for control vs. taxol treatment; n = 9. (B) TER levels in myosin IIA WT and null cells (with and without taxol). P = 0.007. (C and D) In vivo epidermal barrier assay. DMSO or nocodazole was injected subcutaneously with a biotin tracer. After 30 min the skin was removed, embedded, and sectioned for analysis. Streptavidin-FITC (green) allowed visualization of the diffusion of the biotin, and occludin (red) puncta mark the tight junctions in the granular layer of the epidermis. Bar, 10 µm (5 µm for insets).
Cortical microtubules could act directly on tight junctions or they could exert their effect through adherens junctions, which are necessary for tight junction formation. To distinguish between these possibilities, we determined whether contractility and adherens junctions were required for the cortical microtubule-induced increase in the tight junction barrier activity. Treatment of cells with anti–E-cadherin inhibitory antibodies decreased basal TER levels and prevented the increase in TER that is normally induced by cortical microtubules. Furthermore, we did not observe an increase in TER in taxol plus blebbistatin-treated cells (Fig. 4 A). In addition, loss of myosin IIA in keratinocytes prevented the increase in TER normally seen after taxol treatment while having only minor effects on basal TER. Cells untreated with adeno-Cre or wild-type cells treated with the virus showed normal increase in TER after taxol treatment (Fig. 4 B and unpublished data). These data demonstrate that myosin II is also required for the increase in the tight junction barrier. Although we cannot rule out direct effects of the cortical microtubules on tight junctions, our data are consistent with the engagement of adherens junctions regulating tight junction activity.
If cortical microtubules normally act to enhance tight junctions, a decrease in tight junction activity would be expected upon loss of the microtubules in vivo. To test this, we subcutaneously injected nocodazole in newborn wild-type mice to disrupt microtubules, which are largely cortical in the granular cells. Loss of microtubules under these conditions was validated with EMTB-GFP–expressing mice, in which the microtubule cytoskeleton can be more easily visualized (unpublished data). However, experiments were performed in wild-type mice so that we could examine both tight junction localization and function concurrently. We injected biotin tracer, which normally diffuses through the epidermis up to the granular layer where occludin-containing tight junctions block its movement. In the presence of nocodazole, we saw increased diffusion of the biotin past occludin puncta, which was not seen in the control sample (Fig. 4 C). These data are consistent with the conclusion that cortical microtubules regulate tight junction function in vivo. However, genetic models to specifically perturb cortical microtubules (rather than pharmacologic methods that disrupt all microtubules) will be required to verify this in intact tissue.
Cortical microtubules recruit myosin II to the cell cortex
Consistent with our findings that myosin II is required for the engagement of adherens junctions, we noted an increase in cortical localization of myosin IIA and IIB after taxol treatment (Fig. 5, A and B; Fig. S5, A and B). Although not as efficient, expression of Tau-GFP was also able to induce the cortical localization of myosin IIA (Fig. 5 D). This occurred in 22% of Tau-GFP–transfected cell doublets versus 6% of GFP-transfected cell doublets (n = 50 cells). This change in localization was not seen in desmoplakin-null cells, though increased levels of myosin IIA were detected (Fig. 5, E and F). The increase in myosin II at the cell cortex did not require myosin motor activity, as pharmacologic inhibition of myosin II or its upstream activator Rho-kinase did not perturb cortical localization (Fig. 5, G–J). Similar results were found for myosin IIB (Fig. S5, C–F). This demonstrates a noncanonical recruitment of myosin II to cell junctions. We did not note any significant changes in the total levels of myosin light chain phosphorylated on residues 19 or 18/19, again suggesting these changes were not downstream of Rho/Rock or MLCK signaling (Fig. S5 G). Therefore, downstream effects of myosin II required its activity (as demonstrated by pharmacologic inhibition), but junctional localization was independent of activity.
Figure 5.
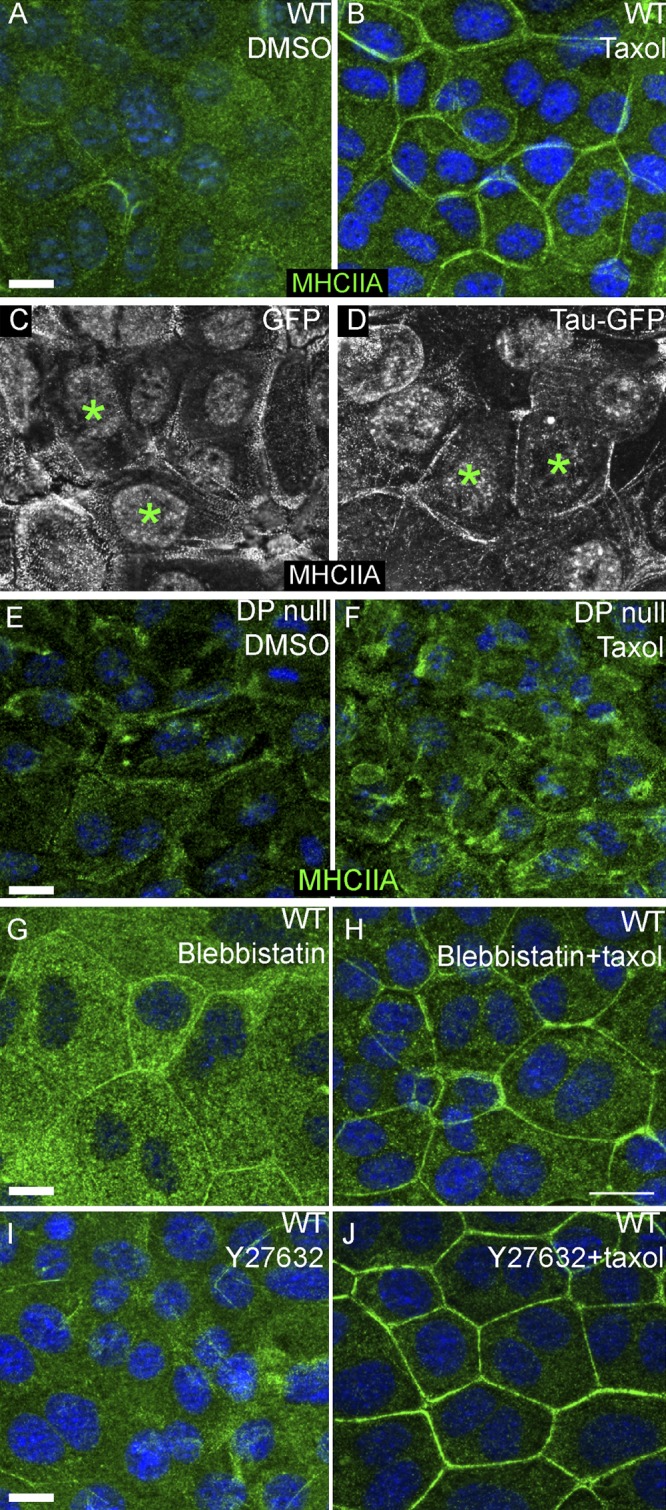
Myosin IIA accumulates at cell junctions upon microtubule stabilization. (A and B) Myosin IIA (MHCIIA, green in all images) localizes weakly at junctions and throughout the cytoplasm in control cells (A). In taxol-treated cells, myosin IIA junctional localization increases (B). (C and D) Myosin IIA accumulates at cell junctions of cells transfected with Tau-GFP (starred cells in D), but not in GFP-transfected cells (starred in C). (E and F) Myosin IIA does not accumulate at cell junctions after taxol treatment in desmoplakin-null cells. (G–J) Myosin IIA accumulation at cell junctions in response to taxol treatment is not affected by pharmacologic inhibition of myosin II or Rho kinase (blebbistatin or Y27632 treatment in H and J). Bars,10 µm.
Loss of myosin IIA/B in the epidermis leads to tight junction defects
In intact epidermis, myosin IIA was present at highest levels in the proliferative basal cells, but could be detected at low levels throughout the epidermis (Fig. S5 H). Myosin IIB was present in basal cells and at cell junctions in granular layer cells (Fig. S5 I). Myosin IIC was present at low levels throughout the epithelium (unpublished data). Our studies with cultured keratinocytes demonstrated that myosin II acted downstream of cortical microtubules to affect tight junctions. To genetically test this role in intact animals we ablated the genes for myosin IIA and IIB in the epidermis. Animals lacking myosin IIC in all tissues are viable and fertile and have no reported epidermal defects (Ma et al., 2010). Loss of myosin IIB in the epidermis did not result in a gross observable phenotype. Myosin IIA conditional null (cKO) mice exhibited an open-eye phenotype at birth, but pups were healthy, though they did develop additional phenotypes and showed growth retardation ∼1 wk later (to be described in detail elsewhere). In contrast, concurrent loss of both myosin IIA and B in the epidermis resulted in fully penetrant perinatal lethality. This phenotype is often the result of a barrier defect in the epidermis, resulting in dehydration. We first tested the outside-in barrier using a dye diffusion assay. Embryos at E18.5 were bathed in a solution containing X-gal. Control littermate mice had an intact barrier and did not turn blue (Fig. 6 A). Littermate double-myosin IIA/B cKO embryos showed loss of barrier activity over small focal areas—the digits, the open eyes, and the ears (Fig. 6 A). However, the epidermal barrier was intact over the rest of the embryos. This suggested a normal differentiation program and formation of functional cornified envelopes. Indeed, we saw no perturbations in the expression pattern of differentiation markers such as keratin 1/10 and filaggrin/loricrin (Fig. 6, B–E; and unpublished data). Despite this, the skin expressed the stress marker keratin 6 (Fig. 6, F and G).
Figure 6.

Loss of myosin IIA and B results in tight junction defects in the epidermis. (A) Barrier assay in E18.5 control littermate (left) and myosin IIA/B double cKO epidermis. Note the barrier defects on digits, eyes, and ears, and normal barrier function over the rest of the body. (B and C) Normal expression of differentiation markers: K5/14 (green) and K1 (red) in control (B) and myosin II dKO epidermis (C). (D and E) Normal expression of granular layer marker loricrin (green) in control (D) and myosin II dKO epidermis (E). β4-integrin (red) marks the basement membrane in these images. (F and G) Abnormal expression of the stress marker keratin 6 (K6, red) in myosin II dKO epidermis (G). Asterisks mark nonspecific cornified envelope staining in F and G. (H and I) Biotin diffusion assay in control (H) and myosin II dKO (I) epidermis. Biotin is detected with streptavidin (green) and tight junctions are marked with occludin (red). (J and K) Adherens junction components E-cadherin (green) and α-catenin (red) in control (J) and myosin II dKO epidermis (K). (L) Extracts from Myosin IIA/B dKO epidermis or littermate controls were immunoprecipitated with anti–β-catenin antibodies. Bound proteins were analyzed by Western blotting with vinculin and β-catenin. Bars, 10 µm.
Although the outside-in barrier is largely formed by the cornified envelopes and specialized lipids, the inside-out barrier is formed by tight junctions. We tested the function of tight junctions using the biotin diffusion assay. Littermate control epidermis exhibited occludin-puncta that acted as diffusion barriers (Fig. 6 H). In contrast, myosin IIA/B–null epidermis had a profound defect in tight junction function even though occludin was still present in observable puncta (Fig. 6 I). Therefore, myosin IIA/B are not required for the localization of tight junction proteins, but are required for their proper function.
Analysis of the adherens junction components α-catenin and E-cadherin revealed a largely normal localization in much of the backskin (Fig. 6, J and K), though focal areas with increased cytoplasmic pools were noted. To determine whether changes in adherens junctions that are not visible by immunofluorescence may underlie these tight junction defects, we immunoprecipitated β-catenin from myosin IIA/B dKO and littermate control epidermal extracts. Vinculin association with β-catenin was decreased in the dKO as compared with the control, mirroring the effect of loss of microtubules in the epidermis (Fig. 6 L). These data are consistent with our in vitro findings that myosin IIs are required for full adherens junction and tight junction activity in the epidermis.
Discussion
We propose that microtubule reorganization occurs in a two-step process in the epidermis. Upon differentiation, a subset of centrosomal proteins is selectively recruited to the desmosomes (Lechler and Fuchs, 2007; Sumigray et al., 2011). These centrosomal proteins work together with microtubule stabilizers to promote the formation of arrays of microtubules around the desmosomes. Although the molecular details of this stabilization remain to be elucidated, we note that MAP2, MAP4, and Tau are each sufficient to induce microtubule stabilization and cortical recruitment in keratinocytes, and all are up-regulated during terminal differentiation (Fabre-Jonca et al., 1999; Patel et al., 2006; Raymond et al., 2008; Mattiuzzo et al., 2011). It is therefore likely that a number of factors act cooperatively to induce microtubule stabilization in vivo. It is interesting to speculate that this stabilization may also be required to protect microtubules in the exposed skin from cold-induced depolymerization.
Our data demonstrate that cortical microtubules increase the mechanical strength of cell sheets by strengthening adherens junctions. A number of lines of evidence support this conclusion. We were first led to these findings by the observation that the cortical localization of β-catenin increased in the presence of cortical microtubules and that β-catenin was associated with increased levels of E-cadherin. In addition, we saw increased association of β-catenin with the actin-binding protein vinculin. Although the second result is consistent with previous reports on engaged adherens junctions, the changes in cortical β-catenin were unexpected. The molecular nature of this increased cortical association and whether it is a general result of tension on adherens junctions will require additional study. Further supporting a change in adherens junction architecture in the presence of cortical microtubules was our finding that a tension-sensitive epitope on α-catenin became exposed and the dynamics of α-catenin were reduced—all consistent with a stabilization of junctions by increased association with the underlying actin cytoskeleton. Finally, both genetic and pharmacologic perturbation of adherens junctions prevented cortical microtubules from increasing cell sheet integrity (though these treatments had no effect on the formation of cortical microtubules).
Previous work has indicated both positive and negative roles for microtubules in concentrating E-cadherin at cell–cell contacts. A study using cultured human keratinocytes demonstrated that microtubule disruption in low calcium conditions was sufficient to induce E-cadherin recruitment to the cell surface (Kee and Steinert, 2001). The mechanism underlying this is unclear and, as it appears to be a calcium-independent phenomena, the physiological relevance is unknown. In MCF-7 and Ptk2 cells, disruption of microtubules prevented robust cortical E-cadherin accumulation after calcium addition (Stehbens et al., 2006; Ligon and Holzbaur, 2007). This appears to be distinct from the effects reported here, as it required dynamic microtubules that extended radially into adherens junctions. These studies were done on short time scales after calcium addition, and the effect on turnover rather than formation has not been reported. In keratinocytes, we find that nondynamic stabilized microtubules are required for the strengthening of adherens junctions, though they do not affect cortical levels of E-cadherin. In addition, in keratinocytes we find that microtubule disruption by nocodazole does not significantly alter cell sheet integrity after cells have established their junctions. Therefore, although there are important roles for radial microtubules in establishing junctions, this is mechanistically distinct from the role of cortical microtubules in strengthening adherens junctions through an alternate mechanism of myosin II–induced tension.
Although adherens junctions are known mechanosensors and transducers, how these functions are used for normal physiology in an intact epithelium are not known. Our data demonstrate that differentiating epidermal cells undergo tension-induced adherens junction strengthening to increase the mechanical strength and barrier function of the tissue. This occurred in a myosin-dependent manner, consistent with the increased levels of myosin IIA at cell junctions and with contractility inducing more robust adherens junctions. Although microtubules have been proposed to both transport and organize myosin IIA in skeletal muscle (Pizon et al., 2005), the mechanism underlying microtubule-dependent recruitment of myosin II in epidermis is unknown. We do not see a strong colocalization between these two, so direct recruitment is unlikely.
Loss of myosin IIA or pharmacological inhibition of myosin IIs prevented the changes in adherens junctions and tight junctions normally induced by cortical microtubules. In intact epidermis, loss of both myosin IIA and IIB resulted in clear defects in tight junction activity. Although more work is required to test this possibility, we suggest that engaged adherens junctions in the granular cells are more similar to zonula adherens, whereas junctions in other cell layers are equivalent to lateral membrane adherens junctions in simple epithelia.
In contrast to the reported roles of microtubules in adherens junction assembly and function, there is little evidence in the literature that microtubules control tight junctions. Here, we have shown that cortical microtubules increase tight junction activity in cultured keratinocytes as measured by transepithelial resistance. Consistent with this, disruption of microtubules in the intact epidermis resulted in increased permeability of tight junctions. Although it is possible that microtubules may also play a more direct role in tight junction activity, our data suggest that some of the defects are secondary to changes in adherens junctions. Thus, although it is clear that adherens junctions are required for tight junction formation, our data suggest that the tension status of the adherens junction also controls the absolute level of tight junction activity.
The role of myosin II in adherens junctions and tight junctions has been explored in detail in cultured cells, but there is little in vivo data in mammals. Clear roles for both myosin IIA and IIB in adherens junction assembly in MCF-7 cells has been reported (Smutny et al., 2010). Loss of these proteins resulted in defects in zonula adherens structure. In intact epidermis, we detected no defects in skin architecture in single myosin IIA or IIB mutants at birth. This is a clear demonstration that these myosin IIs do not have essential independent functions. However, epidermal cells do not have zonula adherens, but rather have adherens junctions covering all cell–cell contacts. Loss of both myosin IIA and IIB resulted in perinatal lethality, but defects in the localization of adherens junction components were minimal. Importantly, despite the normal localization of adherens junction proteins, it is likely that adhesion is weakened, but this is difficult to detect because of the presence of robust desmosomes. Consistent with this idea, we detected occasional cell–cell separations in the double myosin IIA/B–null epidermis, likely induced by the mechanical trauma of sectioning the tissue.
Myosin IIA/B knockout epidermis exhibited severe defects in tight junction function. Much of the literature on this subject comes from cultured cells and points to a negative role for myosin IIs in tight junction barrier activity. However, much of this is indirect, as either MLCK levels/activity or Rho/Rho-kinase levels/activity have been modulated (Walsh et al., 2001; Hopkins et al., 2003; Shen et al., 2006; Yu et al., 2010). In a colonic epithelial cell line, knockdown of myosin IIA led to defects in tight junction activity, but these were associated with gross adherens junction defects and cell shape changes (Ivanov et al., 2007). These conflicting results may be due to cell type differences (desmosomes in epidermal cells may stabilize junctions against myosin II pulling forces), differences between in vivo and in vitro conditions and/or experimental methods of increasing or decreasing myosin II activity. Additional work will be required to define the roles for different type II myosins in simple epithelia in vivo. Our findings in the epidermis suggest that in vivo myosin II functions may be tuned to the tissue to meet its physiological needs.
Materials and methods
Cell culture
All keratinocyte cell lines were grown at 37°C, 7.5% CO2 in E low Ca2+ media. Once confluent, they were induced to differentiate by adding calcium to 1.2 mM. In microtubule organization assays, taxol or DMSO was added 24 h after induction of differentiation for 1 h at 10 µM. The E-cadherin–blocking antibody DECMA-1 (Abcam) was incubated with cells beginning 1 h before taxol addition, at 3 µg/ml of media. Blebbistatin (Sigma-Aldrich) was used at 10 µM.
Immunofluorescence staining
Cells were fixed in −20°C methanol for 2 min. For tissue, 8-µm sections were fixed in 4% paraformaldehyde for 8 min. Antibodies used were rabbit anti-Tau (Sigma-Aldrich), rabbit anti-occludin (Abcam), mouse anti–β-tubulin (Sigma-Aldrich), mouse anti–β-catenin (Sigma-Aldrich), rat anti-α18 (α-catenin; a gift from A. Nagafuchi, Kumamoto University, Kumamoto, Japan), rat anti–E-cadherin (a gift from C. Jamora, University of California, San Diego, La Jolla, CA), rabbit anti–α-catenin (Sigma-Aldrich), and rabbit anti-vinculin (Sigma-Aldrich). Secondary antibodies were Alexa 488 (Invitrogen) and Rhodamine Red X (Jackson ImmunoResearch Laboratories) conjugated. After staining, samples were mounted in a solution of 90% glycerol in PBS with 2.5 mg/ml p-Phenylenediamine (Sigma-Aldrich). Images were collected at room temperature using a microscope (AxioImager Z1; Carl Zeiss) with Apotome attachment, 63× 1.4 NA Plan Apochromat objective or 40× 1.3 NA EC Plan NEOFLUAR objective, immersion 518F oil, AxioCam MRm camera, and Axiovision software (Carl Zeiss). Photoshop (Adobe) was used for post-acquisition processing of brightness and contrast. Fluorescence intensity was measured using the Profile tool in the Axiovision software.
FRAP analysis
Mouse keratinocytes were grown on 35-mm glass-bottom culture dishes (MatTek Corporation). Cells were transfected with each GFP construct using TransIT-LT1 transfection reagent (Mirus) and were induced to differentiate 24 h after transfection. 24 h after calcium was added to the media, the cells were imaged at 37°C on a microscope (LSM 710; Carl Zeiss) with a 63× oil immersion objective, NA 1.4. FRAP experiments were performed using the Regions, Bleaching, and Time Series modules of Zen software (Carl Zeiss). Percent recovery was calculated by normalizing fluorescence intensity to background intensity, then normalizing intensity at each time point to initial intensity. Mobile fraction was determined as Mf = Imax − Io/1 − Io, where Imax was the maximum fluorescence intensity reached after photobleaching and Io was the initial fluorescence intensity immediately after photobleaching (Shen et al., 2008).
Transmission electron microscopy
Processing cell monolayers for ultrastructure analysis was adapted from previous studies (Reipert et al., 2008). Keratinocytes were grown on Falcon Easy Grip 35-mm culture dishes (BD). Once confluent, calcium was added to the media to 1.2 mM, and cells were fixed 24 h after calcium addition. After washing with PBS, 4% EM grade glutaraldehyde was added to the cells. They were rapidly fixed by microwave using a Pelco Biowave Pro microwave (Ted Pella, Inc.). The microwave frequency was 2.45 GHz and the power used was 108.5 W. Pulsed fixation was used with a setting of 1 min on, 1 min off, 1 min on. After fixation, the cells were washed twice in 0.1 M sodium cacodylate buffer for 5 min each. Next, 1% osmium tetroxide was added to the cells for 15 min; the cells were washed three times with 0.11 M veronal acetate buffer for 3 min each. En bloc stain was added for 15 min, and the cells were washed twice in 0.11 M veronal acetate buffer for 3 min each. Cell monolayers were dehydrated in a series of 70%, 95%, and 100% ethanol, 5 min per step. Cell monolayers were incubated in 50% Spurr resin in 100% ethanol for 30 min, changed once and incubated for an additional 15 min. Finally, 100% Spurr resin was added to the cells and incubated for 30 min, the resin was changed and incubated for an additional 30 min. Molds were filled with Spurr resin and placed on top of the cell monolayer. Once polymerized, the molds were peeled from the resin, removing the cell monolayer with it. After sectioning, samples were imaged with a transmission electron microscope (model CM12; Philips) run at 80 kV with a camera (model XR60; Advanced Microscopy Techniques). 2Vu software (Advanced Microscopy Techniques) was used for image acquisition.
Cell sheet integrity assays
Mouse keratinocytes were grown in 6-well dishes to confluency, then calcium was added to 1.2 mM. 24 h after calcium addition, cells were treated with drug for 1 h, then a 1:1 mixture of dispase II (Roche) in PBS was added to the cells for 1 h at 37°C. When the cells lifted off of the dish in a confluent sheet, they were subjected to mechanical disruption by pipetting up and down a set number of times. The number of sheet fragments was counted.
Transepithelial resistance
Cells were grown to confluence in 12-well Transwell dishes, pore size 0.4 µm (Corning), and induced to differentiate by addition of calcium to 1.2 mM. Transepithelial resistance measurements were taken using a MilliCell ERS-2 V-ohm-meter and probe (EMD Millipore). Readings were normalized to blank wells (wells with media but no cells) and area.
Immunoprecipitations
Keratinocyte lysates were prepared in 50 mM Hepes, pH 7.4, 100 mM NaCl, 1 mM MgCl2, 1% Triton X-100, and 1 mM DTT with protease inhibitor cocktail. After brief sonication, lysates were centrifuged for 5 min at 15,000 g. Soluble extracts were removed and added to protein G–Sepharose beads (EMD Millipore) prebound to rat anti–E-cadherin antibodies. After incubation for 1 h, beads were washed four times with lysis buffer and bound proteins were eluted in sample buffer and examined by Western blot analysis. For preparation of tissue lysates, full-thickness skin was immersed in dispase and either DMSO or 20 µM nocodazole for 2 h, epidermis was removed, minced with a razor blade, resuspended in the buffer above, sonicated, and centrifuged for 5 min at 15,000 g.
Biotin barrier assay
The biotin barrier assay was performed as described previously (Furuse et al., 2002). In brief, 50 µl of a 10-mg/ml solution of NHS-biotin (Sigma-Aldrich) was injected subcutaneously into the backskin of newborn mice in the presence or absence of 0.4 mM nocodazole. After 30 min, skin was removed and processed for cryosectioning and stained with occludin antibodies and Streptavidin-FITC (Invitrogen).
Epidermal barrier function assay
The biotin barrier assay was performed as described previously (Furuse et al., 2002). In brief, wild-type and experimental embryos at stage E18.5 were immersed in a solution consisting of 1.3 mM MgCl2, 100 mM NaPO4, 3 mM K3Fe(CN)6, 0.01% sodium deoxycholate, 0.2% NP-40, and 1 mg/ml X-gal (Hardman et al., 1998). After 6 h of treatment, embryos were washed with PBS and photographed.
E-cadherin biotinylation
Biotinylation was performed as described previously (Sumigray et al., 2011). In brief, a solution of sulfo-NHS-SS-biotin (Thermo Fisher Scientific) in PBS with 1 mM CaCl2 was added to control and taxol-treated cells. After 1 h at 4°C, cells were washed three times with PBS containing 20 mM Tris. Extracts were prepared in RIPA buffer and biotinylated proteins were isolated on avidin-agarose (Sigma-Aldrich). After four washes with RIPA buffer, bound proteins were analyzed by Western blot.
Online supplemental material
Fig. S1 shows Map4 localization in epidermis and microtubule organization by light and electron microscopy. In Fig. S2 we analyze the effect of taxol treatment on the localization of cell adhesion proteins. Fig. S3 reports the kinetics of junction localization of adherens junction components after a calcium switch in wild-type and myosin IIA–null cells. Fig. S4 documents the localization and turnover of junctional components after taxol treatment. In Fig. S5 the cortical localization requirements of myosin IIB are examined, as well as myosin light chain phosphorylation status and the localization of myosin IIA and B in the epidermis. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201206143/DC1.
Supplementary Material
Acknowledgments
We thank Akira Nagafuchi, Scott Soderling, Cagla Eroglu, Colin Jamora, Mark Rasenick, and Robert Adelstein for reagents. We also thank Julie Underwood for care of the mice, Phillip Christopher for assistance with electron microscopy, and Tim Oliver for assistance with FRAP. We are grateful to members of the Lechler laboratory for ideas and critical reading of the manuscript.
This work was supported by grants from the NIH/NIAMS (R01AR055926), the March of Dimes, and the Whitehead Foundation.
Footnotes
Abbreviation used in this paper:
- TER
- transepithelial resistance
References
- Cavey M., Lecuit T. 2009. Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harb. Perspect. Biol. 1:a002998 10.1101/cshperspect.a002998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehmelt L., Halpain S. 2005. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 6:204 10.1186/gb-2004-6-1-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgehyr N., Sillibourne J., Bornens M. 2005. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J. Cell Sci. 118:1565–1575 10.1242/jcs.02302 [DOI] [PubMed] [Google Scholar]
- Derry W.B., Wilson L., Jordan M.A. 1995. Substoichiometric binding of taxol suppresses microtubule dynamics. Biochemistry. 34:2203–2211 10.1021/bi00007a014 [DOI] [PubMed] [Google Scholar]
- Fabre-Jonca N., Viard I., French L.E., Masson D. 1999. Upregulation and redistribution of E-MAP-115 (epithelial microtubule-associated protein of 115 kDa) in terminally differentiating keratinocytes is coincident with the formation of intercellular contacts. J. Invest. Dermatol. 112:216–225 10.1046/j.1523-1747.1999.00500.x [DOI] [PubMed] [Google Scholar]
- Furuse M., Hata M., Furuse K., Yoshida Y., Haratake A., Sugitani Y., Noda T., Kubo A., Tsukita S. 2002. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J. Cell Biol. 156:1099–1111 10.1083/jcb.200110122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B., Stevenson B., Grimaldi A. 1988. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 107:1575–1587 10.1083/jcb.107.4.1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Yang Z., Song W., Chen Q., Wang F., Zhang Q., Zhu X. 2006. Nudel contributes to microtubule anchoring at the mother centriole and is involved in both dynein-dependent and -independent centrosomal protein assembly. Mol. Biol. Cell. 17:680–689 10.1091/mbc.E05-04-0360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman M.J., Sisi P., Banbury D.N., Byrne C. 1998. Patterned acquisition of skin barrier function during development. Development. 125:1541–1552 [DOI] [PubMed] [Google Scholar]
- Helfrich I., Schmitz A., Zigrino P., Michels C., Haase I., le Bivic A., Leitges M., Niessen C.M. 2007. Role of aPKC isoforms and their binding partners Par3 and Par6 in epidermal barrier formation. J. Invest. Dermatol. 127:782–791 10.1038/sj.jid.5700621 [DOI] [PubMed] [Google Scholar]
- Hopkins A.M., Walsh S.V., Verkade P., Boquet P., Nusrat A. 2003. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J. Cell Sci. 116:725–742 10.1242/jcs.00300 [DOI] [PubMed] [Google Scholar]
- Ivanov A.I., Bachar M., Babbin B.A., Adelstein R.S., Nusrat A., Parkos C.A. 2007. A unique role for nonmuscle myosin heavy chain IIA in regulation of epithelial apical junctions. PLoS ONE. 2:e658 10.1371/journal.pone.0000658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee S.H., Steinert P.M. 2001. Microtubule disruption in keratinocytes induces cell-cell adhesion through activation of endogenous E-cadherin. Mol. Biol. Cell. 12:1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Duc Q., Shi Q., Blonk I., Sonnenberg A., Wang N., Leckband D., de Rooij J. 2010. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 189:1107–1115 10.1083/jcb.201001149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechler T., Fuchs E. 2007. Desmoplakin: an unexpected regulator of microtubule organization in the epidermis. J. Cell Biol. 176:147–154 10.1083/jcb.200609109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon L.A., Holzbaur E.L. 2007. Microtubules tethered at epithelial cell junctions by dynein facilitate efficient junction assembly. Traffic. 8:808–819 10.1111/j.1600-0854.2007.00574.x [DOI] [PubMed] [Google Scholar]
- Liu Z., Tan J.L., Cohen D.M., Yang M.T., Sniadecki N.J., Ruiz S.A., Nelson C.M., Chen C.S. 2010. Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA. 107:9944–9949 10.1073/pnas.0914547107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Jana S.S., Conti M.A., Kawamoto S., Claycomb W.C., Adelstein R.S. 2010. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol. Biol. Cell. 21:3952–3962 10.1091/mbc.E10-04-0293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiuzzo N.R., Toulza E., Jonca N., Serre G., Guerrin M. 2011. A large-scale multi-technique approach identifies forty-nine new players of keratinocyte terminal differentiation in human epidermis. Exp. Dermatol. 20:113–118 10.1111/j.1600-0625.2010.01188.x [DOI] [PubMed] [Google Scholar]
- Müsch A. 2004. Microtubule organization and function in epithelial cells. Traffic. 5:1–9 10.1111/j.1600-0854.2003.00149.x [DOI] [PubMed] [Google Scholar]
- Orr G.A., Verdier-Pinard P., McDaid H., Horwitz S.B. 2003. Mechanisms of Taxol resistance related to microtubules. Oncogene. 22:7280–7295 10.1038/sj.onc.1206934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradez A., Wright A., Ehrhardt D.W. 2006. Microtubule cortical array organization and plant cell morphogenesis. Curr. Opin. Plant Biol. 9:571–578 10.1016/j.pbi.2006.09.005 [DOI] [PubMed] [Google Scholar]
- Patel S., Xi Z.F., Seo E.Y., McGaughey D., Segre J.A. 2006. Klf4 and corticosteroids activate an overlapping set of transcriptional targets to accelerate in utero epidermal barrier acquisition. Proc. Natl. Acad. Sci. USA. 103:18668–18673 10.1073/pnas.0608658103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M., Davis M.A., Wong E., Pasolli H.A., Reynolds A.B., Fuchs E. 2006. p120-catenin mediates inflammatory responses in the skin. Cell. 124:631–644 10.1016/j.cell.2005.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizon V., Gerbal F., Diaz C.C., Karsenti E. 2005. Microtubule-dependent transport and organization of sarcomeric myosin during skeletal muscle differentiation. EMBO J. 24:3781–3792 10.1038/sj.emboj.7600842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond A.A., Gonzalez de Peredo A., Stella A., Ishida-Yamamoto A., Bouyssie D., Serre G., Monsarrat B., Simon M. 2008. Lamellar bodies of human epidermis: proteomics characterization by high throughput mass spectrometry and possible involvement of CLIP-170 in their trafficking/secretion. Mol. Cell. Proteomics. 7:2151–2175 10.1074/mcp.M700334-MCP200 [DOI] [PubMed] [Google Scholar]
- Reipert S., Kotisch H., Wysoudil B., Wiche G. 2008. Rapid microwave fixation of cell monolayers preserves microtubule-associated cell structures. J. Histochem. Cytochem. 56:697–709 10.1369/jhc.7A7370.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Black E.D., Witkowski E.D., Lencer W.I., Guerriero V., Schneeberger E.E., Turner J.R. 2006. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J. Cell Sci. 119:2095–2106 10.1242/jcs.02915 [DOI] [PubMed] [Google Scholar]
- Shen L., Weber C.R., Turner J.R. 2008. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J. Cell Biol. 181:683–695 10.1083/jcb.200711165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smutny M., Cox H.L., Leerberg J.M., Kovacs E.M., Conti M.A., Ferguson C., Hamilton N.A., Parton R.G., Adelstein R.S., Yap A.S. 2010. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat. Cell Biol. 12:696–702 10.1038/ncb2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehbens S.J., Paterson A.D., Crampton M.S., Shewan A.M., Ferguson C., Akhmanova A., Parton R.G., Yap A.S. 2006. Dynamic microtubules regulate the local concentration of E-cadherin at cell-cell contacts. J. Cell Sci. 119:1801–1811 10.1242/jcs.02903 [DOI] [PubMed] [Google Scholar]
- Straight A.F., Cheung A., Limouze J., Chen I., Westwood N.J., Sellers J.R., Mitchison T.J. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 299:1743–1747 10.1126/science.1081412 [DOI] [PubMed] [Google Scholar]
- Sumigray K.D., Chen H., Lechler T. 2011. Lis1 is essential for cortical microtubule organization and desmosome stability in the epidermis. J. Cell Biol. 194:631–642 10.1083/jcb.201104009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K., Ishiuchi T., Takeichi M. 2011. Mechanosensitive EPLIN-dependent remodeling of adherens junctions regulates epithelial reshaping. J. Cell Biol. 194:643–656 10.1083/jcb.201104124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinkle C.L., Pasolli H.A., Stokes N., Fuchs E. 2008. New insights into cadherin function in epidermal sheet formation and maintenance of tissue integrity. Proc. Natl. Acad. Sci. USA. 105:15405–15410 10.1073/pnas.0807374105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunggal J.A., Helfrich I., Schmitz A., Schwarz H., Günzel D., Fromm M., Kemler R., Krieg T., Niessen C.M. 2005. E-cadherin is essential for in vivo epidermal barrier function by regulating tight junctions. EMBO J. 24:1146–1156 10.1038/sj.emboj.7600605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh S.V., Hopkins A.M., Chen J., Narumiya S., Parkos C.A., Nusrat A. 2001. Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology. 121:566–579 10.1053/gast.2001.27060 [DOI] [PubMed] [Google Scholar]
- Yonemura S., Wada Y., Watanabe T., Nagafuchi A., Shibata M. 2010. alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12:533–542 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
- Yu D., Marchiando A.M., Weber C.R., Raleigh D.R., Wang Y., Shen L., Turner J.R. 2010. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc. Natl. Acad. Sci. USA. 107:8237–8241 10.1073/pnas.0908869107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.