

Abstract
The heat shock protein (HSP) 70 family has been implicated in the pathology of Alzheimer’s disease (AD). In this study, we examined common genetic variations in the 80 genes encoding HSP70 and its co-chaperones. We conducted a study in a series of 462 patients and 5238 unaffected participants derived from the Rotterdam Study, a population-based study including 7983 persons aged 55 years and older. We genotyped a total of 12,053 Single Nucleotide Polymorphisms (SNPs) using the HumanHap550K Genotyping BeadChip from Illumina. Replication was performed in two independent cohort studies, the Framingham Heart study (FHS; N=806) and Cardiovascular Health Study (CHS; N=2150). When adjusting for multiple testing, we found a small but consistent, though not significant effect of rs12118313 located 32kb from PFDN2, with an OR of 1.19 (p-value from meta-analysis =0.003). However this SNP was in the intron of another gene, suggesting it is unlikely this SNP reflects the effect of PFDN2. In a formal pathway analysis we found nominally significant evidence for an association of BAG, DNAJA and prefoldin with AD. These findings corroborate with those of a study of 2032 AD patients and 5328 controls, in which several members of the prefoldin family showed evidence for association to AD. Our study did not reveal evidence for a genetic variant if the HSP70 family with a major effect on AD. However, our findings of the single SNP analysis and pathway analysis suggest that multiple genetic variants in prefoldin are associated with AD.
Keywords: Heat-Shock Proteins, Alzheimer Disease, prefoldin, Genetic Association Studies
Introduction
Heat shock proteins (HSPs) have been implicated in the pathophysiology of Alzheimer’s disease (AD) and its major pathological characteristics, neurofibrillary tangles (NFTs) and β-amyloid (Aβ) [1-3]. AD is an example of a protein-folding disorder, with Aβ plaques as main misfolded protein [1, 3]. HSPs are the major chaperones mediating proper (re)folding of proteins [4].
There is increasing evidence for the involvement of the heat shock protein 70 (HSP70) family in AD. It has been shown that Aβ plaques co-localize with HSP70 [1, 5-6]. Further, HSP70 participates in the neuroprotective response to Aβ plaques [7]. HSP70 has also been found to promote tau solubility and tau binding to microtubules, thereby suppressing formation of NFTs [8].
HSP70 has numerous co-chaperones that support its function. The largest group of co-chaperones comprises the HSP40 family, also called J domain protein family. HSP40 can be subdivided into 3 subclasses (A, B and C) and each member has both common and unique functions in the cell [9]. A role of HSP40 proteins in neurodegenerative disorders, like AD, has been suggested in several studies [10]. Other important groups of co-chaperones include the BCL2-associated anthanogene (BAG) proteins, Hip, Hop, CHIP and prefoldin [11-12]. At autopsy, the prefoldin protein complex was found to be up-regulated in brains of patients with AD [13].
We have tested 79 genes encoding all members of these HSP families and assessed their association with AD. First we have conducted a single SNP analysis. Second we performed a formal pathway analysis in which we test for evidence of multiple SNPs in various genes in a single pathway which contribute jointly to the evidence of association. To our knowledge, this is the first large-scale genomic study of the association of HSP70 and its co-chaperones with AD to be performed.
Methods
Discovery Study
Our discovery cohort was the Rotterdam Study (RS1). RS1 is a population-based cohort study that investigates the occurrence and determinants of diseases in the elderly [14]. Baseline examinations, including a detailed questionnaire, physical examination and blood collection, were conducted between 1990 and 1993. The Medical Ethics Committee at Erasmus Medical Center approved the study protocol.
Dementia is one of the focus disorders of the Rotterdam Study. The diagnosis of dementia was made following a stringent three-step protocol [15]. Briefly, all subjects were screened at follow-up visits (1997-1999, 1999-2000 and 2000-2003) using two tests of cognition: the Mini-Mental State Examination (MMSE) [16] and Geriatric Mental State Schedule (GMS) [17]. Participants that were screen-positive (MMSE score < 26 or GMS organic level > 0) underwent the Cambridge examination for mental disorders of the elderly (CAMDEX). When additional neuropsychological testing was required for diagnosis, a neuropsychologist examined subjects who were suspected of having dementia. In addition, the total cohort was continuously monitored for incident dementia through a computerized link between the study databases and digitalized medical records from general practitioners and the Regional Institute for Outpatient Mental Health Care until January 1, 2005. The diagnosis of dementia and subtypes of dementia was made in accordance with internationally accepted criteria for dementia (DSM-III-R) [18] and Alzheimer disease (NINDS-ADRDA) [19] by a panel consisting of a neurologist, a neuropsychologist and a research physician. We used only incident patients in the discovery study.
Genomic DNA was extracted from whole blood samples using standard methods [20]. Genome-wide SNP genotyping was performed using Infinium II assay on the HumanHap550 Genotyping BeadChips (Illumina Inc, San Diego, USA). Approximately 2 million SNPs were imputed using release 22 HapMap CEU population as reference. The imputations were performed using MACH software10 (http://www.sph.umich.edu/csg/abecasis/MACH/). The quality of imputations were checked by contrasting imputed and actual genotypes at 78,844 SNPs not present on Illumina 550K for 437 individuals for whom these SNPs were directly typed using Affymetrix 500K. Using the “best guess” genotype for imputed SNPs the concordance rate was 99% for SNPs with R2 (ratio of the variance of imputed genotypes to the binomial variance) quality measure greater than 0.9; concordance was still over 90% (94%) when R2 was between 0.5 and 0.9.
The genome wide association study has been analyzed and is submitted elsewhere [21]. For the study of HSP proteins presented here, a total of 12,053 SNPs in 79 genes were initially selected for the association test on the basis of the following criteria: (1) position within the genes of interest with a margin of 100kb on each side of the genes according to NCBI build 36.3, (2) P-value for Hardy-Weinberg equilibrium test ≥ 0.0001, and (3) call rate ≥95%. For further analysis and selection of SNPs selected for evaluation of the consistency of findings a R2 higher than 0.8 and a minor allele frequency (MAF) higher than 0.05 was used as selection criterion for a SNP.
Single SNP analyses
First we analyzed the individual SNPs using ProbABEL [22]. We used allele-based logistic regression to test the association between a single SNP and AD. Odds Ratios (ORs) for each SNP were derived adjusting for age and sex. To calculate empirical significance for SNPs, permutations were performed per region of interest (ROI) [23-24]. Briefly, the empirical distribution of the region-wide maximum of the test statistic under the null was obtained in 10,000 replications. To estimate empirical significance each observed test statistic was compared with null statistics obtained empirically and the p-value was estimated as the proportion of replicas generating the test statistics greater than or equal to the observed statistic. The permutation analysis keeps the original genotypes for each individual, but randomly allocates the phenotypes for each consecutive permutation. Therefore the linkage disequilibrium (LD) structure of genes is not broken up. We did not perform any additional correction for the number of ROIs after permutations as we used the permutation analysis to significantly reduce the number of SNPs to be sent for validation of the effect of the SNP. For each suggestively associated ROI (ppermuted<0.10) we next selected the ‘independently associated’ SNPs in a backward stepwise logistic regression until only nominally significant SNPs remained in the model. These independently associated SNPs were then followed up in two independent cohorts. As effect estimates are expected to be small, a positive replication based on the p-value and odds ratio (OR) observed in individual replication studies, might be difficult to achieve due to power issues. Therefore we evaluated whether a SNP was showing an OR in the same direction in all three cohorts, i.e., whether findings were consistent over the various cohorts. Similar to genome wide association studies, we performed a joint meta-analysis of the discovery and the two independent cohorts and tested whether the joint p-value was significant using a Bonferoni correction for the number of SNPs validated in independent cohorts.
We used two studies that are part of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium to evaluate whether findings were consistent. Data on AD were available in the Cardiovascular Health Study (CHS) and the Framingham Heart Study (FHS) [25]. Again only incident patients were studied. The CHS is a population-based cohort study of risk factors for CHD and stroke in adults ≥65 years conducted across four field centers. The original predominantly European ancestry cohort of 5201 persons was recruited in 1989- 1990 from random samples of the Medicare eligibility lists and an additional 687 African-Americans were enrolled subsequently for a total sample of 5888 [26]. For compatibility with the discovery samples, however, these analyses were limited to the participants of self-described European ancestry. Blood was drawn for genotyping at the baseline examination. Persons were examined annually from enrollment to 1999, and the examination included a 30 minute screening cognitive battery [27]. In 1992- 94 and again, in 1997-99, participants were invited to undergo brain MRI and detailed cognitive and neurological assessment as part of the CHS Cognition Study [27]. Persons with prevalent dementia were identified, and all others were followed until 1999 for the development of incident dementia and AD. Since then, CHS participants at the Maryland and Pennsylvania centers have remained under ongoing dementia surveillance [28]. The sample for this study included all incident cases between 1992 and December 2006.
FHS is a single-site study that comprises three generations of participants including the Original cohort followed since 1948 (n=5209), [29] and their offspring and spouses of the offspring (n=5124) followed since 1971 [30]. The original cohort has been evaluated biennially since 1948, was screened for prevalent dementia in 1974-76 and has been under surveillance for incident dementia since then [31-32]. The offspring were examined once every 4 years, were screened for prevalent dementia in 1979-1983 and have since been under surveillance for incident dementia [33-34]. In order to be consistent with the sampling frame for the CHS sample, we excluded FHS subjects with a baseline age <65yrs at the time of DNA draw which was in the 1990s. To minimize survival biases persons who developed dementia prior to the date of DNA draw were treated as prevalent cases and subsequent events occurring prior to December 2006 were included in the incident analyses.
Pathway analysis
As we targeted distinct families within a larger pathway we next conducted a pathway analysis, using the SNP Ratio Test (SRT) tool, which allows for the testing of a specific, ‘user defined’ hypothesis [35]. Users can define their own pathways to test. We tested each HSP family, namely HSP40, its three subclasses, BAG, CHIP-like, prefoldin and HSP70 separately. We created 1000 simulation studies to test whether our observed number of ‘significant’ SNPs superseded what could be expected by chance. According to the principle of permutation analysis, all original genotypes remain and the phenotypes are randomly allocated with each simulation before rerunning the association analysis. This deals with any potential bias due to LD, as the same LD patterns are present in the original analysis and in each simulation analysis. All SNPs previously selected for the individual SNP analysis (SNPs within a 100kb window for the flanking region and a cut-off for the R2 0f 0.8 and MAF of 0.05) in RS1 were entered in SRT. SRT deals with differences in gene size and number of SNPs per gene, by completely discarding the concept ‘gene’. SRT simply counts the number of significant SNPs in a defined pathway and divides this by the total number of SNPs in that pathway to create a ratio. Significance of the original SNPs is determined by one of three cut-off p-values, namely 0.05, 0.01 or 0.001. In each simulation analysis SRT deals with LD and sizes of genes similar to the original analysis. The p-value is determined by dividing the number of simulations that have a more extreme ratio by the total number of simulations performed. The analysis was adjusted for age and sex. The findings of the SRT analyses were compared to a genome-wide pathway analyses published earlier by Hong et al [36].
Results
General characteristics of the discovery population as well as the two samples used to check the ORs for consistency are shown in Table 1. As expected in all cohorts the cases are significantly older than controls and there are significantly more women and APOE*4 carriers among them. A summary of all SNPs tested in HSP70 and its co-chaperones in RS1 can be found in the Supplementary Table 1. Of note is that 3 HSP70 genes located on chromosome 6 have overlapping regions and are taken as 1 Region of Interest (ROI) with 148 SNPs. Also of note is that the HSPs are currently undergoing a change in nomenclature [37]. In the table are the official gene names and the gene names according to the new nomenclature. Here we will use the names of the new nomenclature.
Table 1.
General Characteristics for the discovery and cohorts used to test the consistency of effects
| RS1 |
CHS |
FHS |
||||
|---|---|---|---|---|---|---|
| Cases | Controls | Cases | Controls | Cases | Controls | |
| N | 462 | 5238 | 366 | 1784 | 76 | 730 |
| Age (mean+se) | 76 (7) | 68 (9) | 80 (6) | 75 (5) | 87 (6) | 76 (7) |
| Female (%) | 74 | 57 | 53 | 62 | 81 | 57 |
| APOE*4 carrier (%) | 45 | 27 | 38 | 24 | 38 | 20 |
RS1: Rotterdam Study; CHS: Cardiovascular Health Study
FHS: Framingham Heart Study
Figure 1 shows the observed versus the expected chi2 of the 12,053 SNPs tested in RS1. The plot shows some excess of low p-values for the tested SNPs. Supplementary Figure 1a-1c shows an overview of the p-values in all genes tested in RS1. After adjusting for multiple testing by permutation analysis, 8 genes showed suggestive evidence (p-permuted<0.10) for association to AD in RS1 (Table 2). These genes encode four members of the HSP40 family (DNAJA4, DNAJC14, DNAJC17 and DNAJC28), one Prefoldin subunit (PFDN2), one BAG gene (BAG2) and two HSP70 genes (HSPA5 and HSPA12B). The following SNPs were selected for validation of their effects in two independent studies: rs8027394 in DNAJA4, rs1463592 in DNAJC14, rs7915 in DNAJC17, rs7280365 in DNAJC28, rs12118313 in PFDN2, rs7760349 and rs13213618 in BAG2, rs13294021 in HSPA5 and rs3899452 in HSPA12B. Two SNPs in BAG2 were selected as both showed independent evidence for association to AD in RS1 when including all SNPs in the ROI in the model. In the validation phase 2 SNPs were consistent in direction of ORs across populations (Table 2), with a nominal p-value of the meta-analysis at or close to a Bonferoni adjusted p-value for significance (0.05/9=0.0056). One SNP (rs1463592) did not surpass the adjustment for multiple testing (DNAJC14; p=0.007). Figure 2a shows the Forrest plot of the meta-analysis of PFDN2 rs12118313. The summary OR for the SNP rs12118313 is 1.19 (95% CI 1.06-1.33; p=0.003) for the C allele. Though this SNP is not replicated when considering the p-value corrected for testing multiple SNPs < 0.05, findings are consistent across cohorts in terms of effect size and direction. In figure 2b a regional plot of the SNPs tested in PFDN2 from the original RS1 cohort can be found. The figure shows that the p-value was most significant in RS1. Although there are multiple marginally associated SNPs in and flanking the PFDN2 gene (see supplementary table 2), rs12118313 in located in the intron of another gene (ARHGAP30).
Figure 1.
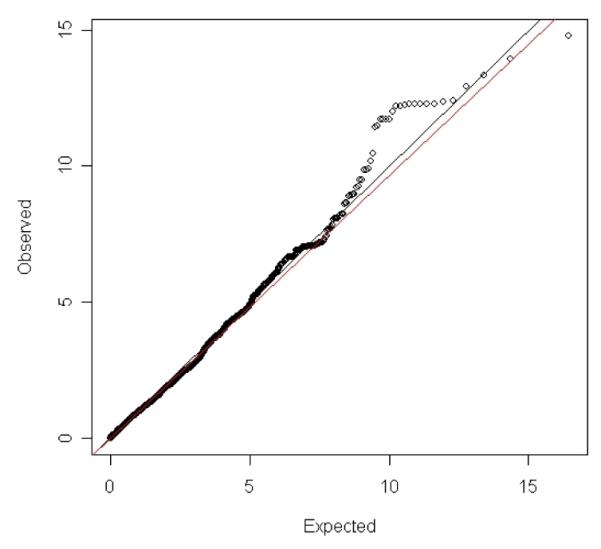
Observed versus Expected p-value plot for the 12053 SNPs tested in the Rotterdam study (chi2 are given at the x-axis and y-axis)
Table 2.
Consistency of the effects in the CHS and FHS cohorts of the SNPs significantly associated with AD in the Rotterdam study
| RS1 |
CHS |
FHS |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | SNP | Chr | Position | Coded allele |
Freq controls |
OR | 95% CI | P- value |
Freq controls |
OR | 95% CI | P- value |
Freq controls |
OR | 95% CI | P- value |
| HSPA12B | rs3899452 | 20 | 3775265 | C | 0.13 | 1.49 | 1.22-1.85 | 0.0002 | 0.14 | 0.90 | 0.70-1.16 | 0.413 | 0.14 | 0.63 | 0.30-1.31 | 0.216 |
| HSPA5 | rs13294021 | 9 | 127108209 | G | 0.51 | 1.21 | 1.05-1.39 | 0.008 | 0.53 | 0.93 | 0.80-1.07 | 0.297 | 0.52 | 0.84 | 0.62-1.15 | 0.278 |
| PFDN2 | rs12118313 | 1 | 159304603 | C | 0.22 | 1.37 | 1.16-1.61 | 0.0001 | 0.22 | 1.01 | 0.84-1.22 | 0.891 | 0.21 | 1.06 | 0.70-1.61 | 0.770 |
| BAG2_1 | rs7760349 | 6 | 57220087 | A | 0.45 | 1.28 | 1.11-1.47 | 0.0007 | 0.45 | 0.97 | 0.83-1.12 | 0.658 | 0.44 | 1.09 | 0.78-1.53 | 0.602 |
| BAG2_2 | rs13213618 | 6 | 57229922 | G | 0.17 | 1.37 | 1.15-1.64 | 0.0005 | 0.16 | 1.07 | 0.87-1.33 | 0.520 | 0.18 | 0.76 | 0.48-1.22 | 0.261 |
| DNAJC14 | rs1463592 | 12 | 54543559 | A | 0.22 | 1.28 | 1.09-1.52 | 0.003 | 0.18 | 1.06 | 0.87-1.29 | 0.556 | 0.22 | 1.27 | 0.88-1.84 | 0.208 |
| DNAJC17 | rs7915 | 15 | 38893777 | C | 0.37 | 1.24 | 1.08-1.43 | 0.003 | 0.35 | 0.83 | 0.71-0.96 | 0.013 | 0.37 | 0.81 | 0.56-1.15 | 0.230 |
| DNAJA4 | rs8027394 | 15 | 76404968 | C | 0.70 | 1.33 | 1.14-1.57 | 0.0005 | 0.67 | 0.99 | 0.85-1.16 | 0.892 | 0.66 | 1.02 | 0.73-1.41 | 0.921 |
| DNAJC28 | rs7280365 | 21 | 33874453 | G | 0.62 | 1.28 | 1.10-1.49 | 0.001 | 0.64 | 1.01 | 0.86-1.18 | 0.953 | 0.61 | 0.95 | 0.67-1.37 | 0.800 |
RS1: Rotterdam study; CHS: Cardiovascular Health Study; FHS: Framingham Heart Study; Chr: chromosome; Freq: frequency; OR: Odds Ratio; 95% CI: 95% Confidence Interval; Bold: The results for this gene are consistent across populations, with all ORs in the same direction.
Figure 2.

The meta-analysis of the PFDN2 rs12118313 to AD in the three population-based studies
In order to evaluate whether there is a joint effect of multiple SNPs in the PFDN2 region and elsewhere we next conducted a formal pathway analysis. Table 3 presents the results for the SRT pathway analysis on the different HSP families in RS1. We chose three cut-off p-values for selection of the SNPs in the discovery set (0.05, 0.01, 0.001) as was suggested in the original paper on the SRT approach. For the prefoldin HSP family we found an effect at the higher p-value cut-off (0.01), this effect is explained by 29 SNPs including 3 SNPs in ARGHAP30. When excluding the SNPs in ARGHAP30 from the analysis, the p-value for the pathway was 0.03, suggesting that there are many prefoldin SNPs with smaller effects implicated. We further find nominally significant evidence for a role of the DNAJA and BAG families of HSPs when applying the smallest p-value cut-off (0.001). For the replication we used a data mining approach of a genome-wide pathway analysis of 2032 patients with AD and 5328 controls [36]. The major pathway identified in this paper was related to intracellular transmembrane protein transport. We used supplementary data to validate our SRT analyses. The PFDN gene family emerges with different genes (PFDN1, PFDN2, PFDN6) with p-values varying from 3.85*10−2 (PFDN6) to 6.80*10−8 (PFND1). A total of 42 SNPs in the gene family were associated with marginal p-values <0.05. There was little support for the BAG family (best p-value 0.02 for BAG2) nor for the DNAJA family (DNAJA4: p=6.11*10−6).
Table 3.
SNP Ratio Test (SRT) pathway analysis results
| HSP family | Total # SNPs | P=0.05 | P=0.01 | P=0.001 |
|---|---|---|---|---|
| HSP40 | 6352 | 0.72 (279) | 0.68 (50) | 0.31 (7) |
| DNAJA | 542 | 0.62 (13) | 0.19 (9) | 0.02 (7) |
| DNAJB | 1390 | 0.49 (70) | 0.96 (1) | 1.00 (0) |
| DNAJC | 4420 | 0.67 (196) | 0.56 (40) | 1.00 (0) |
| BAG | 517 | 0.68 (16) | 0.20 (10) | 0.02 (9) |
| CHIPlike | 292 | 1.00 (0) | 1.00 (0) | 1.00 (0) |
| Prefoldin | 912 | 0.10 (93) | 0.03 (29) | 0.11 (3) |
| HSP70 | 1880 | 0.78 (94) | 0.34 (29) | 0.37 (2) |
Pathway analysis results of the different HSP families at three different cut-offs for significance (P=0.05, P=0.01 and P=0.001). The p-values given in the table depict the chance that the number of SNPs found to be significant at the given p-value cut-off is more than expected by chance. In bold are those p-values passing a nominal threshold of significance. In brackets are the number of SNPs passing the threshold of significance depicted. DNAJA, DNAJB and DNAJC are sub-families of HSP40.
Discussion
In our study, we did not find significant evidence for a role of the individual SNPs in or directly flanking the HSP genes. However, in the pathway analysis we found nominally significant evidence for a joint effect of multiple SNPs in the DNAJA, BAG and prefoldin families. In this paper we did not adjust for multiple testing of the pathway analysis. Multiple testing adjustments on pathway analysis are difficult because many, if not all, pathways are not independent of one another. HSP70 cannot function properly without its co-chaperones and the different groups of co-chaperones compete for association to HSP70. Most other pathway tools therefore often clump all HSP families together as one huge pathway. As we wanted to test specifically which HSP family is the best candidate for association with AD we decided to divide them in separate, but not independent, pathways. It is therefore arguable whether or not to adjust for multiple testing in the pathway analysis. We have analyzed 8 pathways for association with AD. Although the HSP pathway was not the major pathway implicated in AD in an analysis of 2032 patients with AD and 5328 controls [36], there also is marginal evidence for the prefoldin family in this study. Specifically, PFDN1, PFDN2 (the gene we detected), PFDN6 surface also in this GWAs supporting our findings at the pathway level.
PFDN2 showed evidence for association with AD both in the single SNP analysis as in the pathway analysis as a part of the prefoldin HSP family. As the single SNP analysis showed an association with a SNP in another gene, the findings of the pathway analyses were most convincing, although the 3 SNPs located in ARGHAP30 have contributed to the p-value, these do not explain the association fully. When excluding the 3 SNPs the pathway was still significant. From a biological perspective, there is no evidence that these 3 SNPs are involved in the expression of PFDN2, however we cannot exclude this as this is based on eSNP analyses in lymphocytes. PFDN2 is located in a chromosomal region known to be associated with AD in linkage studies [38] and it is a subunit of the prefoldin complex. Prefoldin is an intermediary factor between HSP70 and the TCP-1 ring complex (TRiC). The TRiC complex is involved in about 10% of the protein folding in the cytosol. Prefoldin is necessary for the transport of unfolded proteins to this complex [11]. Prefoldin has also been shown to induce in vitro formation of soluble Aβ oligomers similar in size to those found in AD brains [39]. It is speculated that the function of Prefoldin is to prevent aggregation, causing more of the highly toxic soluble Aβ oligomers to be present in the brain [39]. Moreover, the PFDN2 protein has been found to be upregulated in the brains of patients with AD [13]. This makes the PFDN2 gene a plausible candidate gene for AD.
There is some evidence in our study and the study of Hong et al [36] for a role of DNAJA. The HSP family DNAJA is a subfamily of the larger HSP40 family. HSP40 has been shown to reduce aggregate formation in other neurodegenerative diseases [40] and recruits HSP70 to aggregates [41]. It is required for HSP70 to bind these aggregated proteins and process them [42]. Genetic variants in HSP40 genes may modify their ability to recruit and guide HSP70 to aggregates, at least partly explaining their association to AD at the protein level [5].
The last group of genes we find evidence for association for is the BAG domain protein family. A direct physical interaction with HSP70 was shown for all members of the BAG family [43]. The BAG proteins have therefore been proposed to serve as targeting factors for HSP70 [44]. However there is little evidence for association of the BAG family to AD in the paper of Hong et al [36].
Our study has a major advantage since the discovery cohort is embedded within a large, population based-study (RS1). Further, the studies used to evaluate the consistency of effects are also established epidemiological cohorts. We did not reach genome wide significance in the present study. A limitation in the interpretation of our data is that our findings were significant in the meta-analysis but did not reach significance in the individual cohorts except for in the RS1 cohort. The smaller number of cases in CHS and FHS in combination with the small effect in terms of OR could explain why the finding was not significant in those populations.
Until now, no large-scale studies have been performed on the role of HSP70 and its co-chaperone genes in patients with AD. In our genomic study in three population-based cohorts, we found evidence suggesting that genetic variants in the prefoldin family are associated with the risk of AD. Although larger case-series are needed to achieve genome wide significance for individual SNPs, combining these data with those of earlier functional studies makes it likely that this family of genes plays a role in AD.
Supplementary Material
Supplementary Figure 1: -log(p-values) of all SNPs tested. Each gene tested is a bar. 1a: HSP70 genes
1b: HSP40 genes, ordered on subclass A, B and C
1c: BAG genes, CHIP-like genes and Prefoldin genes.
Supplementary Table 1: overview of all genes tested in RS1 and the number of SNPs per gene
Supplementary Table 2:All SNPs in PFDN2
Acknowledgements
RS1
The generation and management of GWAS genotype data for the Rotterdam Study is supported by the Netherlands Organisation of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012). This study is funded by the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) project nr. 050-060- 810. We thank Pascal Arp, Mila Jhamai, Marijn Verkerk, Lizbeth Herrera and Marjolein Peters for their help in creating the GWAS database, and Karol Estrada and Maksim V. Struchalin for their support in creation and analysis of imputed data. The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists.
Additional funding was obtained from the Alzheimer Association IIRG-05-14359, Hersenstichting Nederland and Internationale Stichting Alzheimer Onderzoek (ISAO).
CHS
The research reported in this article was supported by contract numbers N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, grant numbers U01 HL080295 and R01 HL087652 from the National Heart, Lung, and Blood Institute, with additional contribution from the National Institute of Neurological Disorders and Stroke and grants AG15928 and AG20098, and AG05133 from the National Institute on Aging. DNA handling and genotyping was supported in part by National Center for Research Resources grant M01RR00425 to the Cedars-Sinai General Clinical Research Center Genotyping core, National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center, and the Cedars-Sinai Board of Governors’ Chair in Medical Genetics (JIR). A full list of principal CHS investigators and institutions can be found at http://www.chsnhlbi.org/pi.htm.
FHS
From the Framingham Heart Study of the National Heart Lung and Blood Institute of the National Institutes of Health and Boston University School of Medicine. This work was supported by the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contract No. N01-HC-25195) and its contract with Affymetrix, Inc for genotyping services (Contract No. N02-HL-6-4278). A portion of this research utilized the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center. Analyses reflect intellectual input and resource development from the Framingham Heart Study investigators participating in the SNP Health Association Resource (SHARe) project. This study was also supported by grants from the National Institute of Neurological Disorders and Stroke (NS17950) and the National Institute of Aging (AG08122, AG16495, AG033193, AG031287, P30AG013846, and AG025259).
References
- [1].Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- [2].Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- [3].Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- [4].Chen S, Brown IR. Neuronal expression of constitutive heat shock proteins: implications for neurodegenerative diseases. Cell Stress Chaperones. 2007;12:51–58. doi: 10.1379/CSC-236R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Magrane J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- [7].Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16:601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- [8].Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63:2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gibbs SJ, Braun JE. Emerging roles of J proteins in neurodegenerative disorders. Neurobiol Dis. 2008;32:196–199. doi: 10.1016/j.nbd.2008.07.016. [DOI] [PubMed] [Google Scholar]
- [11].Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- [12].Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer’s disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- [14].Hofman A, Breteler MM, van Duijn CM, Janssen HL, Krestin GP, Kuipers EJ, Stricker BH, Tiemeier H, Uitterlinden AG, Vingerling JR, Witteman JC. The Rotterdam Study: 2010 objectives and design update. Eur J Epidemiol. 2009;24:553–572. doi: 10.1007/s10654-009-9386-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ruitenberg A, Ott A, van Swieten JC, Hofman A, Breteler MM. Incidence of dementia: does gender make a difference? Neurobiol Aging. 2001;22:575–580. doi: 10.1016/s0197-4580(01)00231-7. [DOI] [PubMed] [Google Scholar]
- [16].Folstein MF, Folstein SE, McHugh PR. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- [17].Copeland JR, Kelleher MJ, Kellett JM, Gourlay AJ, Gurland BJ, Fleiss JL, Sharpe L. A semi-structured clinical interview for the assessment of diagnosis and mental state in the elderly: the Geriatric Mental State Schedule. I. Development and reliability. Psychol Med. 1976;6:439–449. doi: 10.1017/s0033291700015889. [DOI] [PubMed] [Google Scholar]
- [18].Association AP . Diagnostic and statistical manual of mental disorders. American Psychiatric In; 1987. p. 608. [Google Scholar]
- [19].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- [20].Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT, Jr., Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM, Consortium C, Consortium G, Consortium E. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aulchenko YS, Struchalin MV, van Duijn CM. ProbABEL package for genome-wide association analysis of imputed data. BMC Bioinformatics. 2010;11:134. doi: 10.1186/1471-2105-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fisher R. In: The Design of Experiments. Ltd. OB, editor. London: 1935. [Google Scholar]
- [25].Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, Uitterlinden AG, Harris TB, Witteman JC, Boerwinkle E, Consortium C. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- [27].Fitzpatrick AL, Kuller LH, Ives DG, Lopez OL, Jagust W, Breitner JC, Jones B, Lyketsos C, Dulberg C. Incidence and prevalence of dementia in the Cardiovascular Health Study. J Am Geriatr Soc. 2004;52:195–204. doi: 10.1111/j.1532-5415.2004.52058.x. [DOI] [PubMed] [Google Scholar]
- [28].Lopez OL, Kuller LH, Fitzpatrick A, Ives D, Becker JT, Beauchamp N. Evaluation of dementia in the cardiovascular health cognition study. Neuroepidemiology. 2003;22:1–12. doi: 10.1159/000067110. [DOI] [PubMed] [Google Scholar]
- [29].Dawber TR, Kannel WB. The Framingham study. An epidemiological approach to coronary heart disease. Circulation. 1966;34:553–555. doi: 10.1161/01.cir.34.4.553. [DOI] [PubMed] [Google Scholar]
- [30].Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;110:281–290. doi: 10.1093/oxfordjournals.aje.a112813. [DOI] [PubMed] [Google Scholar]
- [31].Bachman DL, Wolf PA, Linn RT, Knoefel JE, Cobb JL, Belanger AJ, White LR, D’Agostino RB. Incidence of dementia and probable Alzheimer’s disease in a general population: the Framingham Study. Neurology. 1993;43:515–519. doi: 10.1212/wnl.43.3_part_1.515. [DOI] [PubMed] [Google Scholar]
- [32].Beiser A, D’Agostino RB, Sr., Seshadri S, Sullivan LM, Wolf PA. Computing estimates of incidence, including lifetime risk: Alzheimer’s disease in the Framingham Study. The Practical Incidence Estimators (PIE) macro. Stat Med. 2000;19:1495–1522. doi: 10.1002/(sici)1097-0258(20000615/30)19:11/12<1495::aid-sim441>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- [33].Au R, Seshadri S, Wolf PA, Elias M, Elias P, Sullivan L, Beiser A, D’Agostino RB. New norms for a new generation: cognitive performance in the framingham offspring cohort. Exp Aging Res. 2004;30:333–358. doi: 10.1080/03610730490484380. [DOI] [PubMed] [Google Scholar]
- [34].DeCarli C, Massaro J, Harvey D, Hald J, Tullberg M, Au R, Beiser A, D’Agostino R, Wolf PA. Measures of brain morphology and infarction in the framingham heart study: establishing what is normal. Neurobiol Aging. 2005;26:491–510. doi: 10.1016/j.neurobiolaging.2004.05.004. [DOI] [PubMed] [Google Scholar]
- [35].O’Dushlaine C, Kenny E, Heron EA, Segurado R, Gill M, Morris DW, Corvin A. The SNP ratio test: pathway analysis of genome-wide association datasets. Bioinformatics. 2009;25:2762–2763. doi: 10.1093/bioinformatics/btp448. [DOI] [PubMed] [Google Scholar]
- [36].Hong MG, Alexeyenko A, Lambert JC, Amouyel P, Prince JA. Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. J Hum Genet. 2010;55:707–709. doi: 10.1038/jhg.2010.92. [DOI] [PubMed] [Google Scholar]
- [37].Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Butler AW, Ng MY, Hamshere ML, Forabosco P, Wroe R, Al-Chalabi A, Lewis CM, Powell JF. Meta-analysis of linkage studies for Alzheimer’s disease--a web resource. Neurobiol Aging. 2009;30:1037–1047. doi: 10.1016/j.neurobiolaging.2009.03.013. [DOI] [PubMed] [Google Scholar]
- [39].Sakono M, Zako T, Ueda H, Yohda M, Maeda M. Formation of highly toxic soluble amyloid beta oligomers by the molecular chaperone prefoldin. FEBS J. 2008;275:5982–5993. doi: 10.1111/j.1742-4658.2008.06727.x. [DOI] [PubMed] [Google Scholar]
- [40].Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- [41].Acebron SP, Fernandez-Saiz V, Taneva SG, Moro F, Muga A. DnaJ recruits DnaK to protein aggregates. J Biol Chem. 2008;283:1381–1390. doi: 10.1074/jbc.M706189200. [DOI] [PubMed] [Google Scholar]
- [42].Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Takayama S, Xie Z, Reed JC. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J Biol Chem. 1999;274:781–786. doi: 10.1074/jbc.274.2.781. [DOI] [PubMed] [Google Scholar]
- [44].Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins. Nat Cell Biol. 2001;3:E237–241. doi: 10.1038/ncb1001-e237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: -log(p-values) of all SNPs tested. Each gene tested is a bar. 1a: HSP70 genes
1b: HSP40 genes, ordered on subclass A, B and C
1c: BAG genes, CHIP-like genes and Prefoldin genes.
Supplementary Table 1: overview of all genes tested in RS1 and the number of SNPs per gene
Supplementary Table 2:All SNPs in PFDN2