

Abstract
Polyamine oxidases are peroxisomal flavoproteins that catalyze the oxidation of an endo carbon nitrogen bond of N1-acetylspermine in the catabolism of polyamines. While no structure has been reported for a mammalian polyamine oxidase, sequence alignments of polyamine oxidizing flavoproteins identify a conserved histidine residue. Based on the structure of a yeast polyamine oxidase, S. cerevisiae Fms1, this residue has been proposed to hydrogen bond to the reactive nitrogen in the polyamine substrate. The corresponding histidine in mouse polyamine oxidase, His64, has been mutated to glutamine, asparagine, and alanine to determine if this residue plays a similar role in the mammalian enzymes. The kinetics of the mutant enzymes were examined with N1-acetylspermine and the slow substrates spermine and N, N′-dibenzyl-1,4-diaminobutane. On average the mutations result in a decrease of ~15-fold in the rate constant for amine oxidation. Rapid -reaction kinetic analyses established that amine oxidation is rate-limiting with spermine as substrate for the wild -type and mutant enzymes and for the H64N enzyme with N1-acetylspermine as substrate. The kcat/KO2 value was unaffected by the mutations with N1-acetylspermine as substrate, but decreased ~55-fold with the two slower substrates. The results are consistent with this residue assisting in properly positioning the amine substrate for oxidation.
The polyamines spermine, spermidine, and putrescine are natural aliphatic polycations ubiquitous in living organisms and essential for cell growth and differentiation [1]. As a result polyamine metabolism has been a frequent target for the development of antineoplastic agents [2–4]. Catabolism of spermine and spermidine requires the sequential action of two enzymes [5]. Acetylation of spermine by spermidine/spermine N1-acetylspermine acetyltransferase forms N1-acetylspermine [6]. N1-acetylspermine is then converted to spermidine by the peroxisomal flavoenzyme polyamine oxidase (PAO)1 as shown in Scheme 1 [7]. These two enzymes also catalyze the acetylation of spermidine to N1-acetylspermidine and its subsequent oxidation to putrescine. While spermine is a slow substrate of PAO [7, 8], it can be directly oxidized to spermidine in the cytosol by the flavoprotein spermine oxidase (SMO) [9–11], which shows low activity with N1-acetylspermine.
Scheme 1.
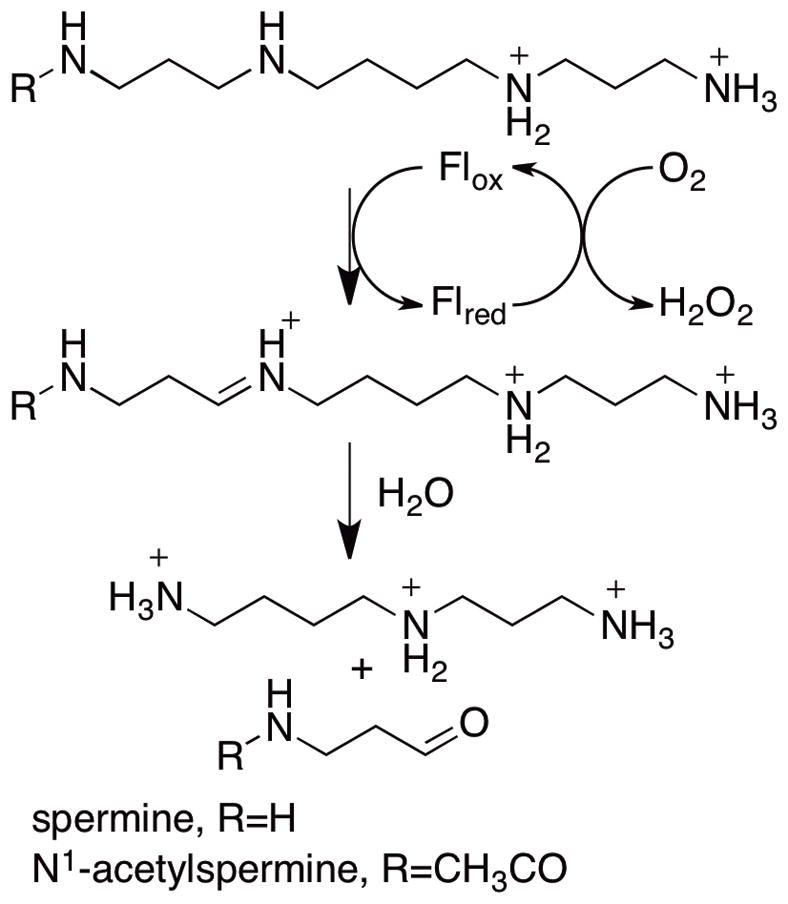
Mammalian PAOs and SMOs cleave the exo carbon-hydrogen bond of polyamines, producing spermidine and N-acetyl-3-aminopropanaldehyde from N1-acetylspermine or putrescine and N-acetyl-3-aminopronaldehyde from N1-acetylspermidine (Scheme 1). In contrast, plant PAOs oxidize the endo carbon hydrogen bond of spermine to produce propane-1,3-diamine and N-(3-aminopropyl)-4-aminobutyraldehyde [12]. The structural bases for the different substrate specificities of PAOs and SMOs and for the different sites of substrate oxidation for plant and animal enzymes are not known. There is as yet no crystal structure of a mammalian PAO or SMO, but there are structures of the S. cerevisiae PAO Fms1 [13] and of maize PAO [14]. While the sequences of mammalian PAOs are only about 20% identical to those of maize PAO and Fms1, a sequence alignment of mammalian PAOs, SMOs and Fms1 shows a conserved sequence around His67 of Fms1 and His64 of mouse PAO (Figure 1). The crystal structure of Fms1 (Figure 2) shows that this histidine forms a part of a catalytic site motif in which the histidine forms hydrogen bonds with the flavin, the substrate, and an asparagine residue (Asn195). This asparagine residue also interacts with an aspartate residue (Asp94). Neither the asparagine nor the aspartate are conserved in PAOs. Mutagenesis of His67 in Fms1 decreases the first-order rate constant for amine oxidation by 2–3 orders of magnitude but does not affect the protein structure [15], consistent with this residue playing a critical role in positioning the polyamine substrate in the active site. The work presented here describes an analysis of the effect of mutating His64 of mouse PAO to probe the role that this histidine residue plays in mammalian PAOs.
Figure 1.

Sequence alignment of polyamine-oxidizing flavoenzymes from different sources showing the sequence around His64 (in bold) of mouse PAO.
Figure 2.

The active site of yeast Fms1 with spermine bound. This figure was composed from the PDB code 1XPQ, subunit B, and it was generated using the program Chimera [30]. Proposed hydrogen bonds between active site residues (distance ≤ 3.0 Å) [13] are shown as dashed lines.
EXPERIMENTAL PROCEDURES
Materials
Spermine tetrahydrochloride, acrylonitrile, 1,4-diaminobutane, benzaldehyde, phenylmethylsufonyl fluoride, 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) and 2-(cyclohexylamino)ethanesulfonic acid (CHES) were purchased from Sigma-Aldrich (St. Louis, MO). N1-Acetylspermine trihydrochloride was purchased from Fluka, (Switzerland). D7-Benzaldehyde, NaBD4, and D3-acrylonitrile were purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA). Isopropyl-thio-2-D-galactopyranoside was purchased from Research Products International Corp., (Prospect, IL). Kanamycin monosulfate was from Fisher Bioreagents (Fair Lawn, NJ). The nickel-nitrilotriacetic acid column was obtained from Invitrogen (Carlsbad, CA). N,N′-Dibenzyl-1,4-diaminobutane dihydrochloride and N,N′-di(D7-benzyl)-1,4-diaminobutane dihydrochloride were synthesized as described previously [16]. 2,3,3,10,10,11-D6-Spermine tetrahydrochloride was synthesized by an adaptation of the method described by Edwards et al. [17]. D3-Acrylonitrile (1.0 g, 17.8 mmol) was added to a stirring solution of 1,4-diaminobutane (0.63 g, 7.12 mmol) in 10 mL of diethyl ether. After reflux overnight, the solvent was removed under pressure. The resulting oil was loaded onto a silica column using a mobile phase system of chloroform:methanol (85:15). Elution of the desired 1,4-bis[(cyanoethyl)amino]butane was monitored by thin layer chromatography. Fractions containing the product were combined, and the volume was reduced under vacuum to yield ~5 mL of a pale yellow oil. PtO2 (200 mg) was added to a solution of the 1,4-bis[(cyanoethyl)amino]butane (0.7 g, 3.5 mmol) in 10 mL of 12.1 M HCl in the reaction flask of a Parr apparatus and was reduced using D2 at 40 – 45 psi for 48 hours. The reaction mixture was filtered to remove the PtO 2. The solution was washed three times with dichloromethane and the aqueous phase was reduced under vacuum to ~5 mL. Isopropanol was added dropwise to the remaining solution until a white precipitate appeared. The precipitate was collected by filtration, washed with diethyl ether and recrystallized from water/ethanol. D6-Spermine tetrahydrochloride: yield 0.4 g (57%; D 6,97%); 1H NMR (D2O) δ 3.1 (8H, m); δ 1.95 (2H, m) and δ 1.73 (4H, s).
Expression and Purification of Polyamine Oxidase
The H64A, H64N and H64Q mutations were introduced into the pET28b-based expression vector for mouse PAO [18] using the QuikChange site-directed mutagenesis kit (Stratagene) and the mutagenic primers 5′-CATTGGATCGCTGGTCCAGCCAGGACAACCCAG-3′, 5′-GGGCGCGCATTGGATCCAGGGTCCAAGCC-3′ and 5′-GCGCGCATTGGATCAATGGTCCAAGCCAG-3′, for the H64A, H64Q and H64N mutations (in bold), respectively. The DNA sequence of the entire gene was determined to ensure that no unwanted mutations occurred. All enzymes were purified using the protocol developed for the wild -type enzyme [18] and stored with 10% glycerol at −80 °C. Enzyme concentrations were determined using an ε458 value of 10,400 M−1 cm−1.
Assays
Steady-state kinetic assays were performed by monitoring the rate of oxygen consumption with a computer-interfaced YSI Model 5300A biological oxygen electrode at 30 °C as described previously [18]. Assays were initiated by the addition of enzyme. All buffers contained 10% glycerol; the specific buffers used are indicated in the legends for figures and tables. To vary the concentration of oxygen, oxygen and argon were combined in different ratios with a MaxBlend low-flow air/oxygen blender (Maxtec Inc. Salt Lake City, UT) and bubbled into the cell of the oxygen electrode containing the assay buffer.
Rapid-reaction kinetic experiments were conducted at 30 °C on an Applied Photophysics SX-20MV stopped-flow spectrophotometer. The instrument was incubated with anaerobic buffer containing 5 mM glucose and 36 nM glucose oxidase overnight before the experiment. For enzyme solutions, anaerobic conditions were established by applying cycles of vacuum and argon, while substrate solutions were bubbled with argon for 10 minutes. Glucose oxidase and glucose were added to all anaerobic solutions at final concentrations of 36 nM and 5 mM, respectively, before they were loaded onto the stopped-flow spectrophotometer.
Data Analysis
Kinetic data were analyzed using the programs KaleidaGraph (Synergy Software, Reading, PA) and IgorPro (WaveMetrics, Inc., Lake Oswego, OR). Steady-state kinetic parameters were determined by fitting the data to the Michaelis-Menten equation. Deuterium kinetic isotope effects were determined by fitting the data to equations 1 and 2. Equation 1 applies for equal isotope effects on the kcat and kcat/KM values, while equation 2 applies when the isotope effects on the kcat and kcat/KM values are different [19]. Fi is the atom fraction of deuterium label in the substrate, Evk and Ev are the isotope effects on the kcat/KM and kcat values, respectively, and S is the substrate. Rapid reaction kinetic traces were fit as single exponential decays (equation 3); here, k is the observed first-order rate constant for the absorbance change, At is the absorbance at time t, ΔA is the absorbance change, and A∞ is the final absorbance.
| (1) |
| (2) |
| (3) |
RESULTS
Steady-State Kinetics of His64 Mutant Proteins
To probe the role of the active site residue His64 in mouse PAO, this residue was mutated to glutamine, asparagine, and alanine. The steady-state kinetic parameters for the wild-type and mutant enzymes were determined with the best substrate, N1-acetylspermine, and with two slow substrates, spermine and N,N′-dibenzyl-1,4-diaminobutane. In each case the assays were carried out at the pH optimum for that substrate [7, 8, 16]. The results are shown in Table 1.
Table 1.
Kinetic Parameters for Wild-Type and Mutant Polyamine Oxidases
| Kinetic Parameter | Wild-type PAO | H64A | H64Q | H64N |
|---|---|---|---|---|
| N1-acetylsperminea
| ||||
| kcat (s−1)b | 49 ± 12 | 3.1 ± 0.1 | 4.5 ± 0.2 | 4.1 ± 0.1 |
| kcat/Kamine (mM−1s−1)c | 230 ± 78 | 62 ± 14 | 84 ± 9 | 17 ± 1 |
| Kamine (mM) | 0.21 ± 0.09 | 0.05 ± 0.01 | 0.053 ± 0.006 | 0.24 ± 0.02 |
| kcat/KO2 (mM−1s−1)b | 39 ± 6 | 39 ± 7 | 53 ± 10 | 39 ± 4 |
| KO2 (mM)b | 1.3 ± 0.5 | 0.08 ± 0.02 | 0.09 ± 0.02 | 0.11 ± 0.01 |
| kred (s−1)d | 436 ± 18 | - | 39.0 ± 0.2 | 3.18 ± 0.04 |
| Kd (μM)d | 453 ± 69 | - | 125 ± 2 | 99 ± 6 |
|
| ||||
| spermine
| ||||
| kcat (s−1)e | 4.7 ± 0.2 | 0.30 ± 0.03 | 0.34 ± 0.04 | 0.12 ± 0.02 |
| kcat/Kamine (mM−1s−1)c | 29 ± 1 | 1.4 ± 0.3 | 1.6 ± 0.3 | 3 ± 1 |
| Kamine (mM) | 0.16 ± 0.01 | 0.21 ± 0.05 | 0.21 ± 0.05 | 0.04 ± 0.01 |
| kcat/KO2 (mM−1s−1)e | 66 ± 13 | 1.0 ± 0.2 | 0.7 ± 0.1 | 0.4 ± 0.1 |
| KO2 (mM)e | 0.07 ± 0.02 | 0.3 ± 0.1 | 0.5 ± 0.1 | 0.3 ± 0.1 |
|
| ||||
| N,N′-dibenzyl-1,4-diaminobutanef
| ||||
| kcat (s−1)g | 0.90 ± 0.03 | 0.16 ± 0.01 | 0.20 ± 0.01 | 0.22 ± 0.01 |
| kcat/Kamine (mM−1s−1)g | 33 ± 2 | 0.6 ± 0.1 | 1.6 ± 0.6 | 0.6 ± 0.1 |
| Kamine (mM) | 0.027 ± 0.01 | 0.27 ± 0.05 | 0.13 ± 0.05 | 0.37 ± 0.06 |
| kcat/KO2 (mM−1s−1)g | 5.5 ± 0.6 | 1.0 ± 0.2 | 1.4 ± 0.1 | 1.1 ± 0.1 |
| KO2 (mM)g | 0.16 ± 0.02 | 0.15 ± 0.03 | 0.15 ± 0.02 | 0.20 ± 0.03 |
Conditions: 200 mM CAPS, pH 10.0, 30°C.
Determined by varying the concentration of oxygen at 10 mM N1-acetylspermine.
Determined by varying the concentration of the amine at 250 μM oxygen.
200 mM CAPS, pH 9.5.
Determined by varying the concentration of oxygen at 30 mM spermine.
Conditions: 200 mM Tris-HCl, pH 8.6, 30°C.
Determined by varying the concentration of oxygen at 20 mM N,N′-dibenzyl-1,4-diaminobutane.
With N1-acetylspermine as the substrate, the mutations result in similar decreases in the kcat value of ~15-fold. The kcat/Kamine value decreases only 3–4 fold for the H64A and H64Q enzymes and somewhat more, ~13-fold, in the case of the H64N enzyme. The mutations have no effect on the kcat/KO2 value when N1-acetylspermine is the polyamine substrate.
The effects of the mutations are larger with the slower substrate spermine. Again the kcat value decreases ~15-fold with the H64A and H64Q mutants, and about three times more with the H64N enzyme. With all three mutants the kcat/Kamine values decrease to a comparable extent, with an average reduction of 15-fold. The kcat/KO2 value decreases 2 orders of magnitude with all three mutant enzymes when spermine is the polyamine substrate.
The effects of the mutations on the steady-state kinetic parameters with the slowest substrate, N,N′-dibenzyl-14-diaminobutane, are intermediate between the effects with the two faster substrates. The kcat/Kamine value decreases ~30-fold with all three mutant enzymes, while the kcat/KO2 and kcat values only decrease ~5-fold on average. Overall, the kinetic parameters for the mutant enzymes are quite similar with spermine and N,N′dibenzyl-1,4-diaminobutane as substrates, even though the wild - type enzyme has significantly greater kcat and kcat/KO2 values with the former.
Identification of the rate-limiting step
Previous analyses have established that product release is rate-limiting for turnover when N1-acetylspermine is the substrate for wild-type PAO [8, 20], while amine oxidation is rate-limiting with N,N′-dibenzyl-1,4-diaminobutane as substrate [16]. To determine whether amine oxidation is also the rate-limiting step for the mutant enzymes with N1-acetylspermine, the kinetics of flavin reduction were examined with the H64Q and H64N enzymes. With both, flavin reduction occurred as a single exponential step. The results for the mutant enzymes are shown in Table 1. Both enzymes exhibit a significant decrease in the value of kred, the rate constant for flavin reduction at saturating concentrations of the amine. In the case of the H64N enzyme, the value decreases ~140-fold so that it is equivalent to the kcat value. With the H64Q enzyme, kred is still about 10-fold greater than kcat, suggesting that product release remains rate-limiting with this enzyme.
The rate-limiting step with spermine has not been determined. Two approaches were taken to determining whether chemistry or product release is rate limiting with this substrate. In the first, stopped-flow experiments were conducted with the wild-type enzyme with spermine as substrate to determine the kred value, the rate constant for flavin reduction at saturating substrate concentrations. The analysis was carried out at pH 9.0 because the enzyme was not stable to the prolonged incubation at pH 10 required for the analysis. The absorbance decrease at 458 nm upon mixing enzyme and spermine could be fit to a single exponential decay (Figure 2A). The observed rate first-order rate constants for reduction at different spermine concentrations were fitted to the Michaelis-Menten equation (Figure 2B) to obtain a kred value for the wild-type enzyme of 0.71 ± 0.03 s−1 and a Kd value of 24 ± 6 μM at pH 9.0. This kred value is comparable to the kcat value of 1.2 ± 0.1 s−1 at pH 9.0. Second, the deuterium kinetic isotope effects on the kcat and kcat/Kamine values were determined with spermine. The initial rate data for the deuterated and non-deuterated substrates fit well to eq 1, which applies for equal isotope effects on kcat and kcat/Kamine, to yield an isotope effect of 11 _ 2. Fitting the data to the more complex eq 2 gave a comparable χ2 value and larger errors (Dkcat =13 ± 3 and D(kcat/Kamine) = 5 ± 4). Equal isotope effects on kcat and kcat/Kamine is consistent with amine oxidation being rate-limiting, since the rate constant for CH bond cleavage is the only rate constant in both the kcat and kcat/Kamine values for this enzyme [18]. Thus, both the rapid-reaction analyses and the deuterium kinetic isotope effects establish amine oxidation as rate-limiting when spermine is the substrate for mouse PAO, just as it is with N, N′-dibenzyl-1,4-diaminobutane as substrate.
DISCUSSION
Oxidation of amine substrates by flavoprotein amine oxidases such as PAO is generally accepted to occur by direct hydride transfer from the neutral amine substrate to the flavin [21–24], and studies of mouse PAO to date are fully consistent with such a mechanism [8, 16]. The amine must bind to the enzyme with the nitrogen in the reactive carbon-nitrogen bond in the neutral form [8, 25], so that there is no need for an active site base to deprotonate the amine. Instead, the role of active site residues is to properly position the amine substrate relative to the flavin [26]. The structure of the yeast polyamine oxidase Fms1 suggests that the role of the conserved histidine in polyamine oxidases is to hydrogen bond to the reacting nitrogen in the substrate, and the effects of site-directed mutagenesis of Fms1 support this conclusion [15]. While the identities of the amino acid sequences of Fms1 and mammalian PAOs are low, the relatively high sequence conservation around His67 of Fms1 and His64 of PAO suggest that these residues play similar roles in the two enzymes. The effects of mutating His64 in PAO described here are fully consistent with a model in which this residue forms hydrogen bonds with the reacting nitrogen in the polyamine substrate in order to properly position the amine for oxidation.
The steady-state kintic mechanism for Fms1 is given in Scheme 2. The effects of the different mutations on the kinetic parameters of PAO vary among the three substrates examined here. N1-Acetylspermine is the best substrate for the enzyme, while spermine and N,N′-dibenzyl-1,4-diaminobutane are both slow substrates [7, 8, 16]. With N1-acetylspermine as substrate, product release is rate-limiting for the wild-type enzyme, so that changes in kcat need not reflect changes in rate constants for chemical steps. In addition, this substrate has a significant forward commitment to catalysis [8], so that the kcat/Kamine value with this substrate reflects both binding and chemistry. Thus, while the kcat/Kamine value only decreases about 3-fold for the H64A and H64Q enzyme, the rate constant for the chemical step, kred, decreases over an order of magnitude with the latter enzyme. Mutating His64 to asparagine decreases kred another order of magnitude, so that chemistry becomes rate-limiting for this enzyme. The H67N mutation in Fms1 similarly had the largest effect on the value of kred when the same three mutations were introduced into that enzyme [15]. Mutagenesis of Fms1 His67 to glutamine or alanine also decreases the rate constant for product release when N1-acetylspermine is the substrate. This suggests that the oxidized amine product actually binds more tightly when this histidine is mutated.
Scheme 2.
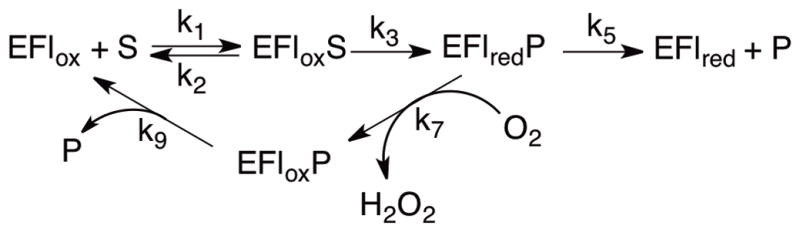
When spermine or N,N′-dibenzyl-1,4-diaminobutane is the substrate for PAO, amine oxidation rather than product release becomes rate-limiting. The kinetic parameters for the mutant enzymes are remarkably similar with the two substrates, although the kcat value for the wild-type enzyme is larger with spermine. With both substrates the kcat/Kamine value decreases ~15-fold for all three mutants. The rate constant for amine oxidation, equal to the kcat value, similarly decreases 15-fold with spermine as substrate. The rate constant for the chemical step is less affected with the slower N,N′-dibenzyl-1,4-diaminobutane as substrate; in this case there is instead a decrease in the substrate affinity as measured by the Kamine value.
While the reactivity of the reduced enzyme with oxygen, given by the kcat/KO2, is unaffected by the mutations when N1-acetylspermine is the substrate, this kinetic parameter decreases ~5-fold with N,N′-dibenzyl-1,4-diaminobutane as substrate and ~100-fold with spermine. The values for the mutant enzymes are the same within error with the two slow substrates, ~1000 M−1s−1. The reaction of reduced flavoproteins amine oxidases with oxygen is typically 10–20 faster when the oxidized amine is still bound compared to the reaction of the free reduced enzyme [27–29], and the kcat/KO2 values for the mutant enzymes in Table 1 are similar to reported values for the reactions of reduced amine oxidases when the produce is not bound. The oxygen reactivity of the PAO mutant enzymes with the two slow amine substrates is consistent with dissociation of the oxidized amine from the reduced enzyme occurring before the reduced flavin reacts with oxygen. In contrast, when N1-acetylspermine is substrate, the oxidized amine dissociates more slowly from the reduced enzyme, so that it is still bound when the reaction with oxygen occurs and the rate constant for the reaction is not affected.
Taken all together, the present data suggest that the interaction of His464 of mouse PAO with the polyamine substrate is worth about 1.6 kcal/mol for amine oxidation and is important for positioning the amine substrate for oxidation. Mutagenesis of the homologous histidine in Fms1 decreases the rate constant for amine oxidation about 10-fold more than mutation of PAO His64 [15]. The kred values for the yeast enzyme with both spermine and N1-acetylspermine are both over an order of magnitude greater than the values for wild-type PAO with the same substrates. Thus, the smaller effect of the mutation in PAO may be in part due to the mammalian enzyme being less optimized for catalysis of the reaction.
Figure 3.
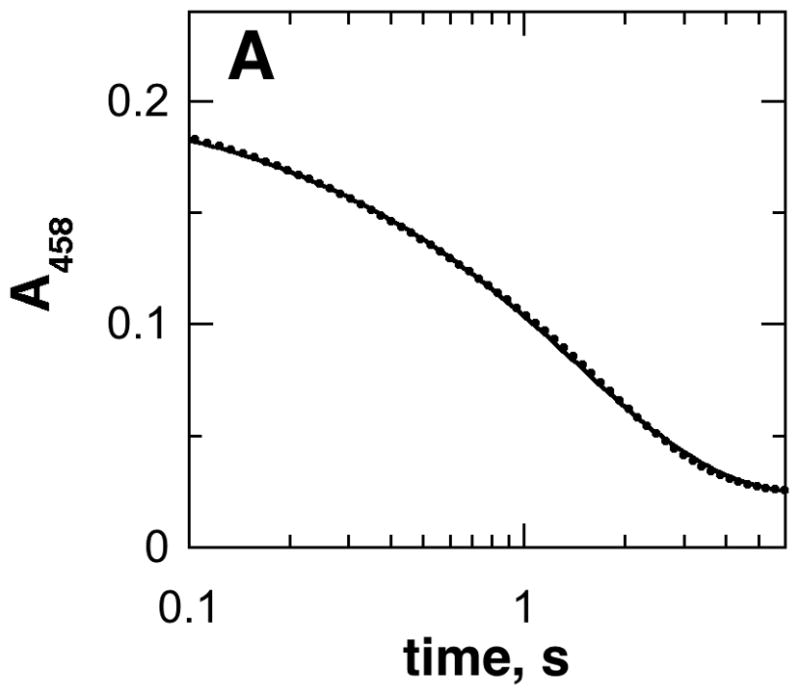
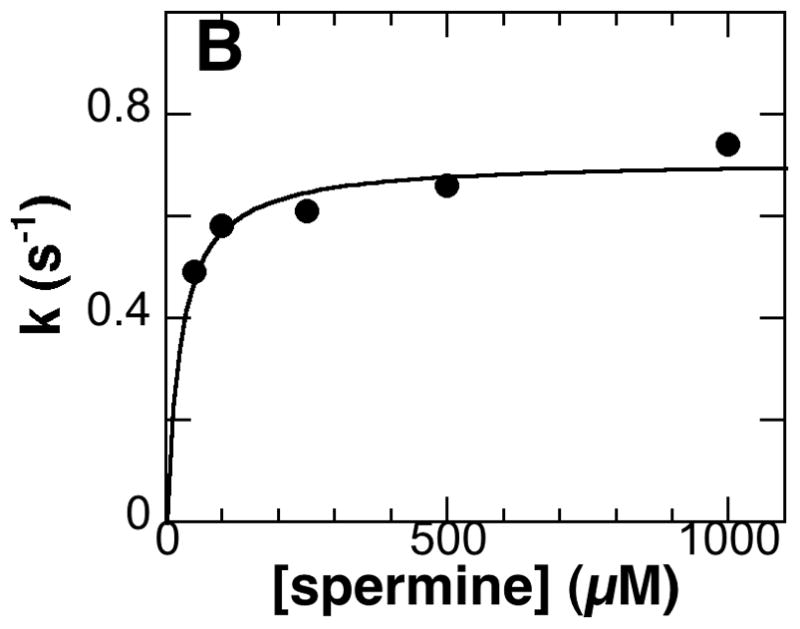
A, Spectral changes at 458 nm during reduction of PAO by spermine in 200 mM CHES, pH 9.0, 30 °C. (Only 20% of the points are shown for clarity.) The line is from a fit to eq 5. B, Dependence of the flavin reduction constant (kred) on the spermine concentration for the wild-type enzyme. The line is from a fit of the data to the Michaelis-Menten equation.
Research Highlights.
His64 of mouse polyamine oxidase is conserved in polyamine-oxidizing enzymes.
His64 of mouse polyamine oxidase was mutated to asparagine, glutamine, and alanine.
The mutations decrease the rate constant for amine oxidation with fast and slow substrates.
The mutations only affect the rate constant for flavin oxidation with the slow substrates.
The results are consistent with His64 forming a hydrogen bond with the polyamine.
Acknowledgments
This work was supported in part by NIH grant R01 GM058698.
Footnotes
Abbreviations: PAO, polyamine oxidase; SMO, spermine oxidase; CAPS, 3-(cyclohexylamino)-1-propanesulfonic acid; CHES, 2-(cyclohexylamino)ethanesulfonic acid.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pegg AE. IUBMB Life. 2009;61:880–894. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tavladoraki P, Cona A, Federico R, Tempera G, Viceconte N, Saccoccio S, Battaglia V, Toninello A, Agostinelli E. Amino Acids. 2012;42:411–426. doi: 10.1007/s00726-011-1012-1. [DOI] [PubMed] [Google Scholar]
- 3.Casero RA, Jr, Marton LJ. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 4.Bachrach U. Amino Acids. 2004;26:307–309. doi: 10.1007/s00726-004-0076-6. [DOI] [PubMed] [Google Scholar]
- 5.Seiler N. Amino Acids. 2004;26:217–233. doi: 10.1007/s00726-004-0070-z. [DOI] [PubMed] [Google Scholar]
- 6.Pegg AE. Am J Physiol Endocrinol Metab. 2008;294:E995–1010. doi: 10.1152/ajpendo.90217.2008. [DOI] [PubMed] [Google Scholar]
- 7.Wu T, Yankovskaya V, McIntire WS. J Biol Chem. 2003;278:20514–20525. doi: 10.1074/jbc.M302149200. [DOI] [PubMed] [Google Scholar]
- 8.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Biochemistry. 2009;48:1508–1516. doi: 10.1021/bi802227m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cervelli M, Polticelli F, Federico R, Mariottini P. J Biol Chem. 2003;278:5271–5276. doi: 10.1074/jbc.M207888200. [DOI] [PubMed] [Google Scholar]
- 10.Vujcic S, Diegelman P, Bacchi CJ, Kramer DL, Porter CW. Biochem J. 2002;367:665–675. doi: 10.1042/BJ20020720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster PM, Casero RA., Jr Biochem Biophys Res Commun. 2003;304:605–611. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- 12.Sebela M, Radova A, Angelini R, Tavladoraki P, Frebort I, Pec P. Plant Sci. 2001;160:197–207. doi: 10.1016/s0168-9452(00)00380-0. [DOI] [PubMed] [Google Scholar]
- 13.Huang Q, Liu Q, Hao Q. J Mol Biol. 2005;348:951–959. doi: 10.1016/j.jmb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. Structure. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- 15.Adachi MS, Taylor AB, Hart PJ, Fitzpatrick PF. Biochemistry. 2012;51:4888–4897. doi: 10.1021/bi300517s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Biochemistry. 2009;48:12305–12313. doi: 10.1021/bi901694s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edwards ML, Stemerick DM, Bitonti AJ, Dumont JA, McCann PP, Bey P, Sjoerdsma A. J Med Chem. 1991;34:569–574. doi: 10.1021/jm00106a015. [DOI] [PubMed] [Google Scholar]
- 18.Royo M, Fitzpatrick PF. Biochemistry. 2005;44:7079–7084. doi: 10.1021/bi050347k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook PF, Cleland WW. Biochemistry. 1981;20:1797–1805. doi: 10.1021/bi00510a014. [DOI] [PubMed] [Google Scholar]
- 20.Henderson Pozzi M, Fitzpatrick PF. Arch Biochem Biophys. 2010;498:83–88. doi: 10.1016/j.abb.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzpatrick PF. Arch Biochem Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan H, Xin Y, Hamelberg D, Gadda G. J Am Chem Soc. 2011 doi: 10.1021/ja2082729. [DOI] [PubMed] [Google Scholar]
- 24.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. J Am Chem Soc. 2000;122:12896–12897. [Google Scholar]
- 25.Dunn RV, Marshall KR, Munro AW, Scrutton NS. FEBS Journal. 2008;275:3850–3858. doi: 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]
- 26.Fraaije MW, Mattevi A. TIBS. 2000;25:126–132. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- 27.Porter DJT, Voet JG, Bright HJ. J Biol Chem. 1977;252:4464–4473. [PubMed] [Google Scholar]
- 28.Adachi MS, Juarez PR, Fitzpatrick PF. Biochemistry. 2010;49:386–392. doi: 10.1021/bi9017945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gadda G. Biochemistry. 2012;51:2662–2669. doi: 10.1021/bi300227d. [DOI] [PubMed] [Google Scholar]
- 30.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]