

Abstract
Sequencing data analysis remains limiting and problematic, especially for low complexity repeat sequences and transposon elements due to inherent sequencing errors and short sequence read lengths. We have developed a program, ReviSeq, which uses a hybrid method comprised of iterative remapping and local assembly upon a bacterial sequence backbone. Application of this method to six Brucella suis field isolates compared to the newly revised Brucella suis 1330 reference genome identified on average 13, 15, 19 and 9 more variants per sample than STAMPY/SAMtools, BWA/SAMtools, iCORN and BWA/PINDEL pipelines, and excluded on average 4, 2, 3 and 19 variants per sample, respectively. In total, using this iterative approach, we identified on average 87 variants including SNVs, short INDELs and long INDELs per strain when compared to the reference. Our program outperforms other methods especially for long INDEL calling.
The program is available at http://reviseq.sourceforge.net.
Keywords: Brucella, sequence assembly, resequencing, variant calling, comparative genomics, iterative mapping
1. Introduction
The genome sequences of microbial field isolates often contain a substantial number of loci different from the published references due to the high rate of mutation in bacterial replication (ca. 1/300 per genome per replication) [1]. Fortunately variant calling in bacterial genomes is relatively straightforward compared to that for eukaryotic studies because bacterial genomes are haploid. Incorrect variant calling in bacterial genomes is often caused by structural variants or incorrect mapping due to sequence variants in diverse repeat sequences including tandem repeats and transposon elements. Sequencing errors rarely cause incorrect variant calling because they are easily identified by designing the study to have a high depth of raw sequence coverage (i.e. >20×). Variants occurring in repeat sequences can incorrectly fool mapping programs into assigning high quality scores to incorrectly mapped reads when the sequence reads from the repeat loci are significantly different from the reference sequence (e.g. length variation at two or more tandem repeat loci containing the same motif often causes incorrect mapping of sequence reads and high quality scores to the reads). This leads directly to invalid variant calls in repeat loci because the variation calling programs rely only on the mapping quality scores to filter out false positive variants from incorrectly mapped reads. Several programs have been developed to find structural variations such as insertions, deletions and copy number variation, but they also have a limitation in searching for long (i.e. > 8 bases) insertions or deletions when the number of incorrectly mapped sequences at a locus is high. An improved mapping post processing step is necessary to correct for this class of incorrect variant calls.
To address these issues in variant calling, we have developed ReviSeq which uses an iterative backbone remapping and local assembly method to generate and revise bacterial genome sequences from short sequence reads and a reference sequence. Previous iterative retrieval approaches used in several de novo assembly methods [2, 3] are limited in application to resequencing analysis because they do not assemble contigs into large structural sequences, especially in or near low complexity repetitive sequences. iCORN, which uses an iterative mapping approach to revise a genome sequence, was developed for resequencing, but it does not correct long INDELs because iCORN's approach uses simple iterative mapping and does not benefit from local re-assembly [4]. Here, we report an advanced iterative remapping and local assembly approach which generates the revised whole genome sequence structure at each iteration based upon a backbone sequence structure.
We demonstrated the effectiveness of this approach for identifying accurate sequence variants found within the bacterial mutants of Brucella field isolates. Brucella is a gram-negative pathogenic bacteria that causes zoonotic disease in domestic animals [5] and has been designated as a category B priority pathogen. Consuming milk products from or having direct contact with infected animals may result in transmission to humans via penetration of skin or mucosal membranes [6]. At the start of resequencing analysis project, it is important to choose a suitable reference genome sequence against which high probability variants can be identified. The variation identified is a foundation for many downstream analyses. Due to the pathogenicity of Brucella, the results of variation detection are the basis for developing assays that are critical to the detection and mitigation of Brucella, as both a potential bioterrorism threat and as an infectious agent.
The Brucella genome is composed of two circular chromosomes of 2.1 Mbp and 1.2 Mbp. The first fully sequenced organism in the genus Brucella was B. melitensis biovar 1 which was published in 2002 [7]. Currently, the complete genome sequences of 11 additional Brucella organisms are publically available and many other genomes from other strains in the genus Brucella are in the process of being sequenced. Here we used the genome sequence of Brucella suis 1330 as a reference for detection of variants in six field isolates of Brucella collected from several different hosts that exhibited highly similar characteristics to Brucella suis 1330 in the ‘gold standard’ antibody diagnostic tests and biochemical tests [8]. Currently, two different versions of Brucella suis 1330 genome sequences are available. The original sequence was published in 2002 [9] and has been used as a reference in several resequencing studies [10, 11], and the revised sequence of the ‘same’ sample was published recently [12].
The Brucella genome contains an 842 bp IS711 transposon element [13] that is unique to Brucella and exists at several different locations in the genome. The published Brucella suis 1330 reference genomes have seven copies of IS711. If two or more close proximity variants exist within a transposon element, then this can lead to incorrect mapping of sequencing reads and therefore wrong variant calling in these regions.
The published Brucella suis 1330 genome sequences also have 10 loci containing 8-mer tandem repeats (≥ 3 motif copies) which are highly variable in its field isolate genomes. When the lengths of tandem repeats at these loci are dramatically different from the reference, the reads containing these elements can produce invalid alignments or be mapped to incorrect loci, leading again to incorrect variant calls.
2. Results
2.1. Overview of the variant calling results
Through this iterative remapping and local assembly approach, we identified an average of 87 variants in genomes of field isolate samples with respect to the reference sequence (Supplemental Table 1). Conversely, traditional mapping approaches called approximately 70 variants from genome sequences of each field isolate with respect to the revised Brucella suis 1330 genome [12]. The largest differences observed between the reference genome and field isolates were observed within two long additional sequences (69 bp and 78 bp) in genomes of all isolates (Fig. 1), which were not detected by traditional mapping methods. Interestingly, since the sequences exist not only in genome sequences of all six field isolates but also in seven other sequenced Brucella species - Brucella abortus S19, Brucella abortus biovar 1, Brucella canis ATCC 23365, Brucella melitensis 16M, Brucella microti CCM 4915, Brucella ovis ATCC 25840 and Brucella suis ATCC 23445 - the sequences are likely to be deletions in the specific sample used to generate the Brucella suis 1330 reference sequences rather than new insertions in the field isolates or closely related species.
Figure 1. Two long deletion sites in the genome sequence of the Brucella suis 1330 ‘reference’ sample.
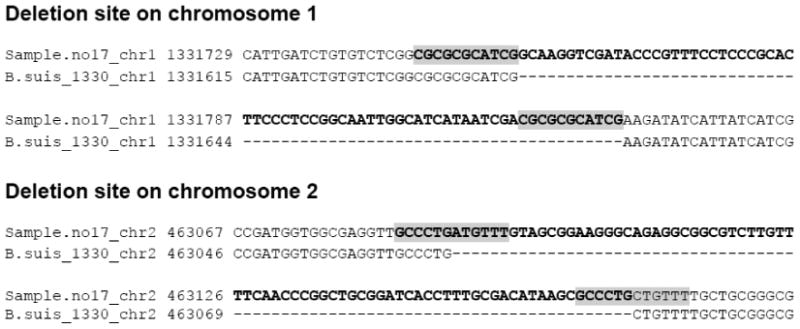
These loci were not detected by traditional mapping methods. The similarities of the highlighted sequences were possible sources of deletion events and may have contributed to misalignments.
2.2. Comparison with other resequencing analysis pipelines
To address the limitation of the traditional resequencing methods in correcting mismapping/misalignment of sequence reads, variant calling results of ReviSeq were compared with that from the traditional resequencing analysis method which uses a simple mapping and variant calling pipeline. The most well known pipelines are combinations of BWA/SAMtools and STAMPY/SAMtools. Both mapping programs, BWA (ver. 0.5.9) [14] and STAMPY (ver. 1.0.13) [15], were executed with default options, and SAMtools [16] (mpileup) was used to generate pileup files. Since the purpose of this comparison was to evaluate the limitation of the methods in variant calling for haploid genomes due to incorrect read alignments, we used a simple filtering approach to call variants instead of using variant calling programs such as ‘bctools’ of SAMtools or SNVer [17]. The variants were called from the pileup files only if they were supported by at least 40% of reads covering each locus which were themselves covered by at least 10 reads. For an insertion, we applied 40% × (read length-insertion length)/read length, considering the probability of a read completely covering the inserted sequence. Another program, iCORN (ver. 0.97), which uses an iterative mapping method, was also compared. To assess the reliability of variant calling of each pipeline, we generated a consensus sequence for the pipeline by replacing the reference bases with the variants identified by the pipeline using sample 13. Then we remapped sequence reads of sample 13 to the consensus sequence using BWA and counted the number of problematic reads including unmapped reads, reads with mismatches, clipped (partially aligned) reads, pair unmapped reads and long distance pairs (>500 bases, an average distance was 290 bases with 25 as a standard deviation) in the mapping result. The ReviSeq pipeline shows the smallest percentages for all types of problematic reads, which suggests that it is the most reliable among all compared pipelines (Fig. 2).
Figure 2. Percentages of problematic reads.
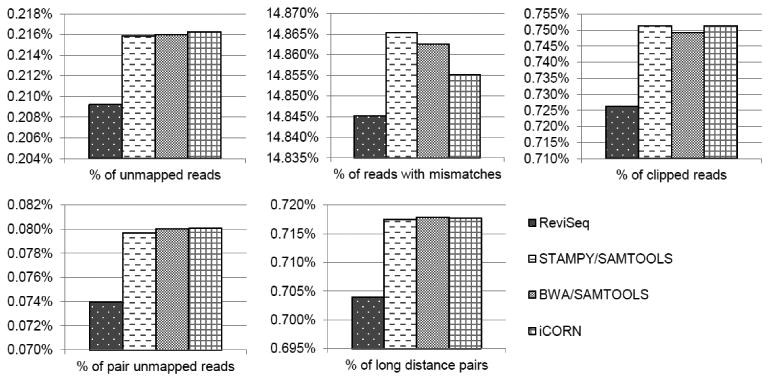
Sequence reads from sample 13 were re-mapped by BWA to the each consensus sequence, in which all variants identified by the corresponding pipeline from sample 13 replaced the reference bases. The graphs show the percentages of problematic reads including unmapped reads, reads with mismatches, clipped reads, pair unmapped reads and long distance pairs (>500 bases) in the mapping result for each consensus sequence. The ReviSeq pipeline shows the smallest percentage of the problematic reads in all comparisons.
Average numbers of variants called by BWA/SAMtools, STAMPY/SAMtools pipelines and iCORN were 78, 74 and 72, respectively (Table 1). Compared to the results of ReviSeq, STAMPY/SAMtools, BWA/SAMtools and iCORN predicted on average of 4, 2 and 3 more variants, and called on average of 13, 15 and 19 less variants per sample respectively. Each variant region inconsistent with the results of ReviSeq and its aligned reads were visually inspected. All three pipelines showed limitations in identifying long INDELs, and called several incorrect SNVs and short INDELs mainly in long INDELs loci (Fig. 3). We also tested the ABYSS [20] assembly/BWASW [21] mapping/SAMtools pipeline and the BWA/SNVer [17] pipeline along with the local alignment program of GATK [18] applied to the BWA/SAMtools pipeline but did not observe significant improvement (Supplemental Table 2).
Table 1.
Comparison to other resequencing analysis pipelines in identifying variants (SNVs:INDELs)
| Sample no. | ReviSeq | STAMPY/SAMTOOLS | BWA/SAMTOOLS | iCORN | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| com. | - | + | total | com. | - | + | total | com. | - | + | total | ||
| 13 | 89 (60:29) |
78 (57:21) |
11 (3:8) |
6 (1:5) |
84 (58:26) |
76 (57:19) |
13 (3:10) |
4 (1:3) |
80 (58:22) |
69 (57:12) |
20 (3:17) |
7 (5:2) |
76 (62:14) |
| 17 | 85 (64:21) |
74 (62:12) |
11 (2:9) |
2 (1:1) |
76 (63:13) |
73 (62:11) |
12 (2:10) |
1 (1:0) |
74 (63:11) |
70 (60:10) |
15 (4:11) |
2 (1:1) |
72 (62:10) |
| 22 | 84 (57:27) |
75 (56:19) |
9 (1:8) |
4 (3:1) |
79 (59:20) |
70 (56:14) |
14 (1:13) |
3 (3:0) |
73 (59:14) |
68 (56:12) |
16 (1:15) |
4 (4:0) |
72 (60:12) |
| 29 | 88 (63:25) |
73 (60:13) |
15 (3:12) |
3 (2:1) |
76 (62:14) |
71 (60:11) |
17 (3:14) |
1 (1:0) |
72 (61:11) |
71 (60:11) |
27 (3:14) |
2 (1:1) |
73 (61:12) |
| 34 | 87 (62:25) |
71 (59:12) |
16 (3:13) |
4 (2:2) |
75 (61:14) |
70 (59:11) |
17 (3:14) |
1 (1:0) |
71 (60:11) |
70 (59:11) |
17 (3:14) |
0 (0:0) |
70 (59:11) |
| 35 | 87 (64:23) |
73 (62:11) |
14 (2:12) |
4 (2:2) |
77 (64:13) |
71 (62:9) |
16 (2:14) |
2 (1:1) |
73 (63:10) |
71 (62:9) |
16 (2:14) |
0 (0:0) |
71 (62:9) |
com. : commonly identified variants by ReviSeq and another pipeline.
- : (count of variants identified by ReviSeq) – (count of commonly identified variants)
+ : (count of variants identified by another pipeline) – (count of commonly identified variants)
Figure 3. Invalid variant calls due to a long insertion.

Long insertions in a test sample frequently cause misalignments and invalid variant calls. The sequences in the first lines of A) and B) are the published reference sequence and a newly assembled sequence of sample 17, respectively. The locus is the same as the second sequence in Figure 1. A) illustrates reads misaligned to the reference sequence. Lower case bases at the ends of reads are bases clipped by a mapping program. B) shows reads correctly aligned to the newly assembled sequence.
INDELs predicted by PINDEL [19], a program to search for structural variations, were also compared with our results. The mapping results of BWA were used as input data to PINDEL. To reduce false positive calls in the PINDEL results, INDELs supported by at least 10 reads were chosen for comparison. Compared to the results of the ReviSeq pipeline, PINDEL predicted an average of 19 more variants and called an average of 9 less variants per sample (Table 2). Many of insertions and deletions falsely called by PINDEL were predicted from mis-alignments around long INDELs. Interestingly, a few insertions and deletions attributable to possible chimeric DNA, mutants, reads from different strains or contaminants existing in the samples were predicted by PINDEL but not by our method. Since the loci were unique regions in the genome and the frequency of reads contributing to these INDEL predictions were much lower than that of the average read depth (lower than 20% of average depth), but consistent in all variant calling results, we concluded that they were not derived from mis-mapping or mis-alignment (mapped to correct position but partially misaligned) and were therefore appropriately not called by our method.
Table 2.
Comparison with PINDEL in identifying insertions and deletions
| Sample no. | ReviSeq | PINDEL | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| INS | DEL | all | |||||||||||||
| INS | DEL | total | com. | - | + | total | com. | - | + | total | com. | - | + | total | |
| 13 | 15 | 14 | 29 | 11 | 4 | 32 | 43 | 12 | 2 | 1 | 13 | 23 | 6 | 33 | 56 |
| 17 | 14 | 7 | 21 | 8 | 6 | 16 | 24 | 6 | 1 | 0 | 6 | 14 | 7 | 16 | 30 |
| 22 | 12 | 15 | 27 | 8 | 4 | 8 | 16 | 12 | 3 | 3 | 15 | 20 | 7 | 11 | 31 |
| 29 | 15 | 10 | 25 | 7 | 8 | 18 | 25 | 6 | 4 | 1 | 7 | 13 | 12 | 19 | 32 |
| 34 | 15 | 10 | 25 | 9 | 6 | 17 | 26 | 6 | 4 | 2 | 8 | 15 | 10 | 19 | 34 |
| 35 | 12 | 11 | 23 | 7 | 5 | 14 | 21 | 7 | 4 | 3 | 10 | 14 | 9 | 17 | 31 |
com. : commonly identified variants by ReviSeq and PINDEL.
- : (count of variants identified by ReviSeq) – (count of commonly identified variants)
+ : (count of variants identified by PINDEL) – (count of commonly identified variants)
We additionally sequenced four 8-mer tandem repeat loci and two C homopolymer loci for samples 17, 22, 29, 34 and 35 using the Sanger method to confirm the variants called by our method and the other pipelines, which were different (Supplemental Table 3). Peaks in chromatograms for two C homopolymer loci at all samples were not clearly identified due to possible heterozygous alleles (Supplemental Fig. 1). ReviSeq could correctly identify alleles of 9 loci among 16 loci containing variants (4 in sample 17, 3 in sample 22, 3 in sample 29, 3 in sample 34 and 3 in sample 35), while STAMPY/SAMtools, BWA/SAMtools and iCORN pipelines identified 1, 0 and 0 loci. ReviSeq could not correctly identify alleles from seven tandem repeat loci of which allele lengths were close to or longer than the read lengths (76 bases), and this limitation could be reduced with longer reads.
2.3. Comparison of variants among field isolates
All six field isolates have a similar number of variants when compared with the revised reference. Among them, the number of common variants in all isolates totaled 39, including 32 SNVs and 7 INDELs. The pairwise comparison of their sequence variants is illustrative of the number of common variants of each pair (Supplemental Table 4), which reveals the evolutionary relationship of the samples (Fig. 4). Specifically, samples 13 and 22 have an additional insertion site of an IS711 element at position 1,578,904 of chromosome 1, which has not yet been reported in any of the previously sequenced Brucella species. The insertion site immediately follows the stop codon of a protein coding locus annotated as a ribose ABC transporter at the Brucella suis 1330 reference.
Figure 4. Phylogenic tree constructed from variation data illustrates possible evolutionary relationships of the samples.
The distances in the tree were measured by counting the number of different variants between samples.
As samples 29 and 34 were isolated from bovine tissue derived from the same herd, variants in their genomes were almost identical except for one SNV and the lengths of three 8-mer tandem repeat loci. Interestingly, although samples 13 and 22 were isolated from different hosts (samples 13 from equine tissue and 22 from bovine milk), their genomes have more common variants than seen in the other genomes. None of the variation in the genes in the six isolates have been reported to affect the host preferences of the Brucella genus, but approximately 24% of the genes have unknown functions which may be related to host preferences (Supplemental Table 5).
3. Discussion
Since next-generation sequencing (NGS) technologies were invented, resequencing followed by mapping to a reference genome has become one of the most widely used approaches for comparative genomic analysis. Even though this advanced methodology has enabled robust comparison of multiple individuals in a time and cost efficient manner, the traditional resequencing analysis methods using a simple mapping and variant calling pipeline were susceptible to falsely calling variants in the vicinity of repeat sequences and structural variants. Here, we have employed an iterative remapping and local assembly approach to improve variant calling from sequencing data and illustrated its effectiveness via analysis of six Brucella suis field isolates whose genomes contain several IS711 transposon elements and 8-mer tandem repeats which are highly variable. As variation analysis results using a resequencing/mapping approach can affect the quality of downstream studies, it is important to correctly identify variants. Using the reliable results from this approach, we identified a number of interesting variants, some common, among the field isolates, which are helping to understand the role that these variants may play in important issues such as transmission, pathogenicity and host preference shifting. For example, we have identified sequence differences within a large number of genes which have unknown functions (Supplemental Table 5), so, for example, host preference of the isolates still remain unexplained, and accurate viariant calls may help target appropriate mechanistic studies necessary to explain this aspect of the host-pathogen behavior of this species. To further enhance the utility of this approach in bacterial genome research, inversion and rearrangement of a genome sequence with respect to the backbone, which are also common variations in bacterial genomes and remain challenging, will be addressed in future versions.
4. Materials and Methods
4.1. Sample preparation and sequencing
The Texas Animal Health Commission (TAHC) provided Brucella samples from six field isolates that exhibited characteristics highly similar to Brucella suis 1330 in standard antibody and biochemical diagnostic tests [8] for carbon dioxide utilization, production of hydrogen sulphide and dye (thionin and basic fuschin) sensitivity. The samples were collected from equine tissue (sample 13), porcine tissue (sample 17), bovine milk (sample 22), and bovine tissue (sample 29, 34 and 35). Genomic DNA for each sample was sequenced via the Illumina GAIIx sequencer and data acquired using the standard Illumina 101 (sample 13) and 76 (sample 17, 22, 29, 34, and 35) cycle paired-end protocols generating approximately 23,500,000 sequencing read pairs (47,000,000 reads) per sample.
4.2. Iterative remapping and local assembly to generate correct consensus sequences
The Iterative remapping and local assembly approach used by ReviSeq is illustrated in Figure 5. This process was performed on each sample independently. To begin the analysis, we trimmed all low quality base calls from the ends of sequencing reads to remove base calls that have a high probability of being incorrect. Trimming at each read started from the end base of each read and stopped when a high quality base call (≥ Q20, Phred quality score) was encountered. If one of two reads in a pair were shorter than 20 bases after removing the low quality base calls, both reads were excluded. We then mapped the reads to the revised reference genome, S0, using BWA. This approach resulted in more than 99.9% of the reads mapping to the reference, yielding average sequence coverage of 1,460× and 1,048× for the 101- and 76-cycle data, respectively.
Figure 5. Iterative remapping and local assembly approach.
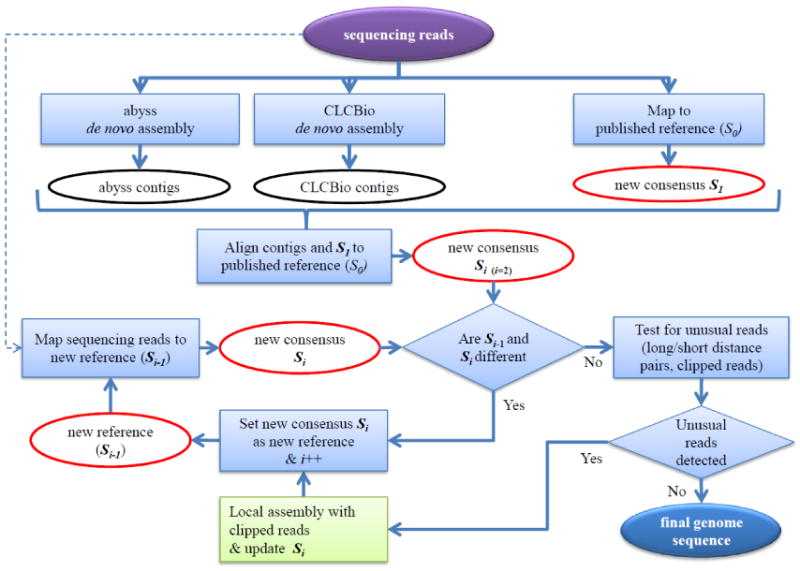
BWA was used for mapping, and ABYSS and CLCbio genomics workbench were used for de novo assembly. The iteration of mapping and local assembly continued until the two consensus sequences Si-1 and Si converged.
A new consensus sequence, S1, was generated using SAMtools from the read mapping results. Concurrent with mapping, we also performed two independent de novo assemblies of the data using ABYSS (with k-mer 55) [20] and the CLC genomics workbench (with default options) to enable detection of long insertion and deletion events, and possible genomic re-arrangements. After assembly, we aligned the resulting contig sequences and the new consensus sequence, S1, to the reference sequence, S0, using BWASW [21] which is a mapping tool to align long sequences to a reference. A new consensus sequence, S2, was generated using the simple majority voting method from the mapping results of the de novo assembly contigs and S1. For this step, we gave higher priority to S1 than contigs. As a result, if S1 and contig sequences were different at a locus, S1 was chosen unless two de novo assembly contig sequences were consistent. This step allows for detection of large INDEL variants that are not correctly detected by a simple mapping method.
We next mapped the sequencing reads to the new consensus sequence, S2, using BWA to generate another consensus sequence, S3. If consensus sequences S2 and S3 were different, the reads were mapped to S3 to test whether all reads were correctly aligned to the same positions and a new consensus, S4, was generated from the newly aligned reads. The mapping/comparing was iterated until Si-1 and Si converged. The purpose of this iteration was to remap incorrectly mapped reads which would otherwise cause incorrect variant calling at IS711 transposon elements.
Next, partially mapped reads (clipped reads) to Si were counted to search for long INDELs which were not detected in prior alignment steps. When the number of partially mapped reads and abnormal pairs (distance of a pair > μ + 3σ or < μ - 3σ, where μ is the mean and σ is the standard deviation of the distance between pairs) was more than 10% of total mapped reads at a position in Si, the partially mapped reads were locally assembled by searching for exact matches only and their contig was subsequently aligned to Si. If a long INDEL was detected, Si was modified and used as a new reference sequence and resubmitted to the iterative remapping process until there was no change.
4.3. Variants calling from consensus sequences
The final consensus sequence of each sample was aligned to the reference sequence by BWASW. Due to the varying lengths of the 8-mer tandem repeat loci and other long INDELs such as a new insertion of an IS711 element in the sample genome, BWASW could not map the whole query sequence to the reference sequence as a single alignment. Instead, it fragmented the query sequence into several pieces and aligned them to the reference separately. We extended the partial alignments adding INDEL information to merge them into one alignment and called the variants from the alignment. Further, the length variants (from 40 bases deletions to 80 bases insertions) of the 8-mer tandem repeat loci in field isolate genomes were fixed and confirmed by Sanger sequencing.
Supplementary Material
Highlights.
Sequence analysis remains limiting and problematic due to repeat sequences.
We used an iterative remapping/local assembly approach for variation calling.
We demonstrated the method using sequencing data from six B. suis field isolates.
Our results are more reliable than that of the traditional analysis pipelines.
Acknowledgments
This work was supported by the Medical Informatics and Systems Division director's funds; the U.S. Department of Homeland Security through the National Center of Excellence for Foreign Animal and Zoonotic Disease Defense at Texas A&M University [subaward 570636 from DHS 2007-ST-061-000002]; and the National Institute of Health, National Human Genome Research Institute, 1000 Genomes Project Dataset Analysis Grant [grant number 1U01HG-005719-01]. Genomic DNAs of Brucella field isolates were provided by TAHC (Texas Animal Health Commission) and Texas A&M University. The Core Laboratory Facility and the Data Analysis Core at the Virginia Bioinformatics Institute sequenced the genomic DNAs and assembled contigs, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drake JW, Charlesworth B, Charlesworth D, Crow JF. Rates of Spontaneous Mutation. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol İ. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otto TD, Sanders M, Berriman M, Newbold C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics. 2010;26:1704–1707. doi: 10.1093/bioinformatics/btq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glynn MK, Lynn TV. Brucellosis. J Am Vet Med Assoc. 2008;223:900–908. doi: 10.2460/javma.233.6.900. [DOI] [PubMed] [Google Scholar]
- 6.Young EJ. An overview of human brucellosis. Clin Infect Dis. 1995;21:283–289. doi: 10.1093/clinids/21.2.283. [DOI] [PubMed] [Google Scholar]
- 7.DelVecchio VG, Kapatral V, Redkar RJ, Patra G, Mujer C, Los T, Ivanova N, Anderson I, Bhattacharyya A, Lykidis A, Reznik G, Jablonski L, Larsen N, D'Souza M, Bernal A, Mazur M, Goltsman E, Selkov E, Elzer PH, Hagius S, O'Callaghan D, Letesson JJ, Haselkorn R, Kyrpides N, Overbeek R. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc Natl Acad Sci U S. 2002;99:443–448. doi: 10.1073/pnas.221575398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alton GG, Jones LM, Angus RD, Verger JM. Techniques for the brucellosis laboratory. INRA; Paris: 1988. [Google Scholar]
- 9.Paulsen IT, Seshadri R, Nelson KE, Eisen JA, Heidelberg JF, Read TD, Dodson RJ, Umayam L, Brinkac LM, Beanan MJ, Daugherty SC, Deboy RT, Durkin AS, Kolonay JF, Madupu R, Nelson WC, Ayodeji B, Kraul M, Shetty J, Malek J, Aken SEV, Riedmuller S, Tettelin H, Gill SR, White O, Salzberg SL, Hoover DL, Lindler LE, Halling SM, Boyle SM, Fraser CM. The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci U S. 2002;99:13148–13153. doi: 10.1073/pnas.192319099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lavigne JP, Vergunst AC, Bourg G, O'Callaghan D. The IncP island in the genome of Brucella suis 1330 was acquired by site-specific integration. Infect Immun. 2005;73:7779–7783. doi: 10.1128/IAI.73.11.7779-7783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wattam AR, Williams KP, Snyder EE, Almeida NF, Shukla M, Dickerman AW, Crasta OR, Kenyon R, Lu J, Shallom JM, Yoo H, Ficht TA, Tsolis RM, Munk C, Tapia R, Han CS, Detter JC, Bruce D, Brettin TS, Sobral BW, Boyle SM, Setubal JC. Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J Bacteriol. 2009;191:3569–3579. doi: 10.1128/JB.01767-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tae H, Shallom S, Settlage R, Preston D, Adams LG, Garner HR. Revised genome sequence of Brucella suis 1330. J Bacteriol. 2011;193:6410. doi: 10.1128/JB.06181-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halling SM, Tatum FM, Bricker BJ. Sequence and characterization of an insertion sequence, IS711, from Brucella ovis. Gene. 1993;133:123–127. doi: 10.1016/0378-1119(93)90236-v. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011;39:e132. doi: 10.1093/nar/gkr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.