

Abstract
The common variant rs12608932, located within an intron of UNC13A gene on chromosome 19p13.3, has been suggested to influence susceptibility to ALS, as well as survival, in patients of north European descent. To examine this possibility further, we evaluated the association of rs12608932 with susceptibility and survival in a population-based cohort of 500 Italian ALS patients and 1,457 Italian control samples. Although rs12608932 was not associated to ALS susceptibility in our series (p=0.124), it was significantly associated with survival under the recessive model (median survival for AA/AC genotypes = 3.5 years [IQR 2.2–6.4]; CC = 2.5 years [IQR 1.6–4.2]; p=0.017). Furthermore, rs12608932 genotype remained an independent prognostic factor in Cox multivariable analysis adjusting for other factors known to influence survival (p=0.023). Overall, minor allele carrier status of rs12608932 was strongly associated with an ~1-year reduction of survival in ALS patients, making it a significant determinant of phenotype variation. The identification of UNC13A as a modifier of prognosis among sporadic ALS patients potentially provides a new therapeutic target aimed at slowing disease progression.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder, clinically characterized by a progressive paralysis at the bulbar, respiratory and limb levels of the CNS. Although considerable progress has been made in understanding this rapidly fatal disorder, much about this disease remains unknown. In particular, the genetic factors influencing important phenotype parameters, such as survival, are unclear. Certainly, genetic mutations, such as missense mutations of the FUS gene and pathogenic repeat expansions of the C9ORF72 gene adversely influence survival (Byrne et al, 2012; Chiò et al, 2012; Sabatelli et al, 2012; Millecamps et al, 2012). However, each of these mutations affect relatively small proportions of the ALS population, rather than exerting an influence across all patients diagnosed with this fatal neurodegenerative disease.
Using the genome wide association screening (GWAS) approach, the common variant rs12608932, located within an intron of UNC13A gene on chromosome 19p13.3, was found to be significantly associated with risk of developing ALS (van Es et al, 2009). Three subsequent studies did not replicate this finding (Daoud et al, 2010; Laaksovirta et al, 2010; Kwee et al, 2012), while one larger study reported the same finding (Shatunov et al, 2010). The same group that initially proposed UNC13A as a susceptibility factor, have recently extended their observations by publishing data that the minor allele of rs12608932 is associated with shorter survival of ALS patients of Dutch, Belgian, or Swedish descent (Diekstra et al, 2012). Such a finding, if true, would be of great interest to the ALS research community, as it would represent an initial attempt to identify a final common pathway of motor neuron neurodegeneration. Consequently, such a pathway would be relevant across the entire gamut of ALS patients, and would be an attractive therapeutic target aimed at slowing disease progression.
The aim of the current study is to evaluate the association of rs12608932 with susceptibility and survival of ALS in a population-based cohort of Italian ALS patients collected through the Piemonte and Valle d’Aosta Registry for ALS (PARALS). The key advantage of this study is that it was collected in a population-based manner, and consequently is more representative of the ALS population, compared to a clinic referral-based cohort. Such patient ascertainment considerations are particularly relevant in designing genetic studies to dissect phenotype aspects of this fatal neurodegenerative disease.
2. Methods
Cases and controls
A total of 504 cases diagnosed with probable, probable laboratory-supported or definite ALS according to the El Escorial criteria were included in the study (Brooks et al, 2000). These cases were ascertained through PARALS, an ongoing population-based epidemiological study of ALS performed in two regions of northwestern Italy (Chiò et al, 2009). All cases were classified as sporadic, as they did not report first, second or third degree relatives with ALS. Written informed consent for genetic analysis was obtained from each individual, and appropriate institutional review board approval was obtained concerning human subjects.A total of 1,457 healthy Italian subjects were included in the study as controls. Of these, 247 samples were collected in Piemonte, and 1,210 samples were obtained from the InChianti study, a population-based cohort of older persons living in the Chianti geographic area (Tuscany, Northern Italy) (Ferrucci et al, 2000).
Genotyping
1,714 samples were genotyped on Infinium HumanHap550 beadchips (Illumina Inc, San Diego, CA, USA), and 236 samples were genotyped on Infinium HumanHap610-Quad beadchips (Illumina) according to the manufacturer’s specifications (Traynor et al, 2010; Melzer et al, 2008). These data are publicly available on the dbGAP repository (www.ncbi.nlm.nih.gov/gap, phs000101.v3.p1 and phs000215.v1.p1). 535,468 SNPs were common across both platforms, including rs12608932 on chromosome 19p13.3. Information about rs12608932 was missing for four cases.
Statistical analysis
Continuous variables (i.e. age at symptom onset) were compared with t-test and discrete variable (i.e. gender, site of onset) with χ-square. Survival was calculated with Kaplan-Meier curves and compared using the log-rank test (all events weighted equally) and the Peto & Peto Generalized Wilcoxon test (earlier events weighted more heavily), using the date of onset as day 0 and death or tracheostomy as primary end point. For censored cases, the last day of follow-up was November 1, 2011.
Rs12608932 were tested for association with patient survival under the following genetic models; additive (AA vs. AC vs. CC genotypes), dominant (AA vs. AC and CC), and recessive (AA and AC vs. CC), where base pair A is the major allele and C the minor. Association of rs12608932 with altered risk of developing disease was analyzed using logistic regression adjusted for age, gender, and population (based on the top five principal components calculated using genome-wide genotype data). A cohort of 500 cases and 1,457 controls has 80% power to detect association of a variant with an odds ratio of 1.23 under the additive model at a significance level of 0.05, assuming a minor allele frequency of 0.33 and complete linkage disequilibrium between the genotyped and causative SNPs. All analyses were performed using the PLINK toolset (Purcell et al, 2007).
Multivariable analysis was performed with the Cox’s proportional hazards model (stepwise backward). The following variables were included in the model: age (included as continuous variable), gender (male vs. female), site of onset (bulbar vs. spinal), C9ORF72 status (expanded vs. not expanded), non-invasive ventilation (NIV, yes vs. no) and enteral nutrition (EN, yes vs. no). Non-invasive ventilation and enteral nutrition were included as time-dependent variables. A p-value <0.05 was considered significant. All calculation were made with SPSS (IBM corporation, version 18).
3. Results
Our case cohort consisted of 271 (54.2%) men and 229 (45.8%) women. Of these, 366 (73.2%) had limb-onset symptoms, whereas the remaining 134 (26.8%) presented with bulbar-onset disease. Median age at onset was 63.3 years (interquartile range [IQR] 54.3–69.3; range=20.5–87.3), and 390 (78.5%) patients were dead or had undergone tracheostomy at the time of last follow-up. Median survival from symptom onset of the whole cohort was 3.4 years (IQR 2.2–6.1). Nineteen (3.8%) patients carried the C9ORF72 hexanucleotide repeat expansion.
For our SNP of interest in UNC13A (rs12608932), 236 (47.2%) Italian patients carried the AA genotype, 203 (40.6%) the AC genotype and 61 (12.2%) the CC genotype. The corresponding frequencies among controls were 731 for AA (50.2%), 620 for AC (42.5%), and 106 for CC (7.3%). Rs12608932 genotype did not alter risk of developing ALS (logistic regression p-value = 0.124).
Clinical and demographic characteristics of patients with different rs12608932 genotypes are shown in Table 1. There was no significant association with gender, age at onset or site of onset for any of the other tested genetic models (data not shown).
Table 1.
Clinical and demographic characteristics of patients, according to patients’ rs12608932 in a recessive model
| AA/AC n=439 | CC n=61 | p value | |
|---|---|---|---|
| Median age at onset (years, IQR) | 63.1 (54.2–69.2) | 62.6 (54.7–68.4) | 0.68 |
| Gender (female, %) | 200 (45.6%) | 29 (47.5%) | 0.89 |
| Site of onset (bulbar, %) | 113 (25.7%) | 21 (34.4%) | 0.17 |
IQR, interquartile range
Rs12608932 was associated to survival in our population-based cohort of 497 Italian under the recessive model (median survival for AA/AC genotypes = 3.5 years [IQR 2.2–6.4]; CC = 2.5 years [IQR 1.6–4.2]; log-rank p-value = 0.017, Peto p-value = 0.003, Figure 1), but not under the additive (median survival for AA genotype = 3.5 years [IQR 2.3–6.4]; AC = 3.4 years [IQR 2.2–6.5]; CC = 2.5 years [IQR 1.6–4.2]; log-rank p-value = 0.056, Peto p-value = 0.012) or the dominant models (median survival for AA genotype = 3.5 years [IQR 2.3–6.4]; AC/CC = 3.3 years [IQR 2.1–5.6]; log-rank p-value = 0.385, Peto p-value = 0.197, Figure not shown). This effect was more significant when excluding the 19 patients carrying the C9ORF72 hexanucleotide repeat expansion (median survival for AA/AC genotypes = 3.6 years [IQR 2.2–6.6]; CC = 2.4 years [IQR 1.5–4.1]; log-rank p-value = 0.010 under the recessive model). In Cox multivariable analysis rs12608932 genotype remained an independent prognostic factor (hazard ratio [HR] 1.40, 95% confidence interval [c.i.], 1.05–1.87) (Table 2).
Figure 1.
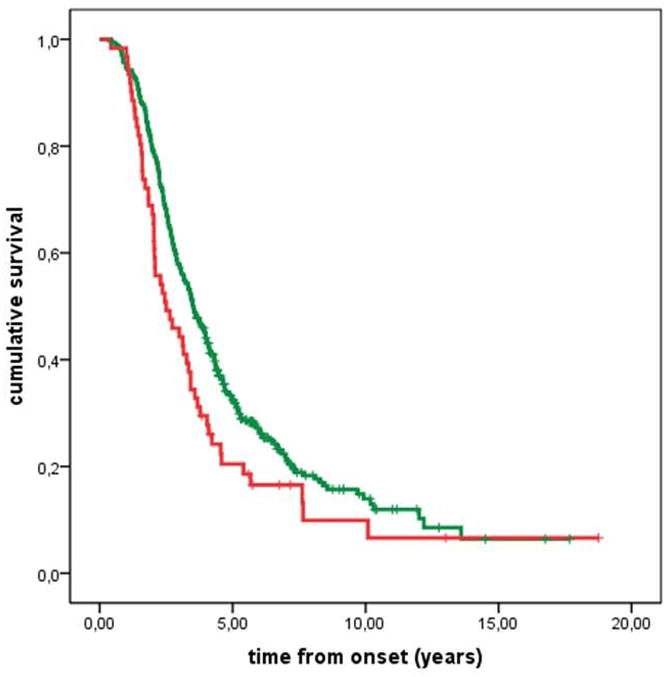
Tracheostomy-free survival of patients according to a recessive model (AA/AC vs. CC). Green line = AA/AC; red line = CC. Ticks are censored patients.
Table 2.
Cox multivariable analysis
| Factor | Hazard ratio | 95% confidence interval | p |
|---|---|---|---|
| Age at onset | 1.03 | 1.02–1.04 | 0.000 |
| Site of onset (bulbar) | 1.36 | 1.09–1.70 | 0.006 |
| Non-invasive ventilation (no) | 3.12 | 1.62–4.55 | 0.000 |
| Enteral nutrition (no) | 2.29 | 1.43–4.12 | 0.000 |
| UNC13A (minor allele) | 1.40 | 1.05–1.87 | 0.023 |
Age at onset, hazard ratio for each year of age after 18.
4. Discussion
In our population-based series of Italian ALS patients, rs12608932 genotype did not alter the risk of developing ALS, but it was strongly related to ALS survival with the minor allele carrier status displaying a ~1-year reduction of survival under the recessive model. This effect was independent from age at onset, site of onset and other variables known to affect survival, including therapeutic inventions such as non-invasive ventilation and enteral nutrition.
Our findings confirm the recent findings of Diekstra et al (2012). In that paper, analysis of survival in a population-based series of Dutch ancestry and a larger clinical-based series of Dutch, Belgian and Swedish descent showed an unadjusted HR of 1.6 (95% c.i. 1.2–2.3) and 1.2 (95% c.i. 1.1–1.4) respectively. These data were tested with multivariable analysis, including clinico-demographic variables but not treatment (NIV and enteral nutrition) and C9ORF72 genetic status, since this gene was not known at the time of their publication. In our study, we confirmed this finding, also demonstrating that the worst outcome related to the minor allele of rs12608932 was independent from a large set of relevant prognostic variables, thereby strengthening the observation.
This paper, along with that by Diekstra et al (2012), demonstrates the importance of using population-based cohorts of incident cases instead of prevalent cases in studying genetic risks factors that may modify outcome of ALS. This may be a consequence of the fact that prevalent populations tend not to include patients with a more rapid course and are typically younger than incident populations (O’Toole et al, 2007; Chiò et al, 2009).
The possible mechanism of the effect of UNC13A on ALS survival is still unclear. The UNC13A gene encodes protein unc-13 homolog A, a member of a large family of presynaptic brain proteins. Interestingly, the mouse homolog of the human protein, Munc13-1, is involved in neurotransmission, including glutamate release and presynaptic long term potentiation (augustin et al, 1999; Yang & Calakos, 2011). Impairment of glutamatergic transmission has been recognized as a pathogenetic mechanism in ALS (Rothstein et al, 1990), and the only FDA-approved drug for ALS, riluzole, is a glutamate release inhibitor (Lacomblez et al, 1996). Munc13-1 also regulates neurite outgrowth (Broeke et al, 2010), a process that is impaired in ALS (Jokic et al, 2006). There are therefore increasing evidence that UNC13A is related to several processes that are pathogenically relevant in ALS.
Homozygosity for the minor allele of rs12608932 shortens survival by ~12 months, which is clinically relevant in ALS where median survival is approximately three years (Chiò et al, 2009). The identification of UNC13A as a determinant of progression rate of sporadic ALS provides a new therapeutic target to slow disease progression and gives insight into possible pathways involved in the degenerative process in this devastating disease.
Acknowledgments
Adriano Chiò had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. We thank the patients and their families for having collaborated to this study.
This work was funded in part by Federazione Italiana Giuoco Calcio, Fondazione Vialli e Mauro per la Sclerosi Laterale Amiotrofica onlus, Ministero della Salute (Ricerca Sanitaria Finalizzata, 2007) (and Regione Piemonte, Progetti Finalizzati (A. Chiò). The research leading to these results has received funding from the European Community’s Health Seventh Framework Programme (FP7/2007–2013) (grant agreements no. 259867 and 278611) (A. Chiò). This work was supported in part by the Intramural Research Programs of the NIH, National Institute on Aging (Z01-AG000949-02) (B.J.T.). Funding organizations had no role in design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Footnotes
Disclosure statement
Adriano Chiò serves on scientific advisory boards for Biogen Idec and Cytokinetics. Bryan Traynor has a patent pending on the discovery of the C9ORF72 hexanucleotide repeat expansion. All other authors disclose no conflicts. Appropriate Ethical Committee approvals were in place for this work.
Author Contributions: Study concept and design: Chiò, Mora, Restagno, Traynor. Acquisition of data: Brunetti, Ossola, Calvo, Moglia, Fuda, Ferrucci, Canosa, Manera. Analysis and interpretation of data: Chiò, Mora, Restagno, Ferrucci, Calvo, Traynor. Drafting of the manuscript: Chiò and Traynor. Critical revision of the manuscript for important intellectual content: Chiò, Mora, Restagno, Brunetti, Calvo, Traynor. Obtained funding: Chiò and Traynor. Administrative, technical, and material support: Chiò, Brunetti, Ossola, Fuda, Canosa, Manera. Study supervision: Chiò, Mora and Traynor.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Broeke JH, Roelandse M, Luteijn MJ, Boiko T, Matus A, Toonen RF, Verhage M. Munc18 and Munc13 regulate early neurite outgrowth. Biol Cell. 2010;102:479–488. doi: 10.1042/BC20100036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Miller RG, Swash M, Munsat TL World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, McLaughlin RL, Iyer PM, O’Brien C, Phukan J, Wynne B, Bokde AL, Bradley DG, Pender N, Al-Chalabi A, Hardiman O. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9ORF72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11:232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R PARALS. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology. 2009;72:725–731. doi: 10.1212/01.wnl.0000343008.26874.d1. [DOI] [PubMed] [Google Scholar]
- Chiò A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, Murru MR, Marrosu MG, Marrosu F, Marinou K, Mandrioli J, Sola P, Caponnetto C, Mancardi G, Mandich P, La Bella V, Spataro R, Conte A, Monsurrò MR, Tedeschi G, Pisano F, Bartolomei I, Salvi F, Lauria Pinter G, Simone I, Logroscino G, Gambardella A, Quattrone A, Lunetta C, Volanti P, Zollino M, Penco S, Battistini S, Renton AE, Majounie E, Abramzon Y, Conforti FL, Giannini F, Corbo M, Sabatelli M the ITALSGEN Consortium. Clinical characteristics of familial ALS patients carrying the pathogenic GGGGCC hexanucleotide repeat expansion of the C9ORF72 gene. Brain. 2012;135:784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, Traynor BG. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–323. doi: 10.3109/17482960802566824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoud H, Belzil V, Desjarlais A, Camu W, Dion PA, Rouleau GA. Analysis of the UNC13A gene as a risk factor for sporadic amyotrophic lateral sclerosis. Arch Neurol. 2010;67:516–517. doi: 10.1001/archneurol.2010.46. [DOI] [PubMed] [Google Scholar]
- Diekstra FP, van Vught PWJ, van Rheenen W, Koppers M, Pasterkamp RJ, van Es MA, Schelhaas HJ, de Visser M, Robberecht W, Van Damme P, Andersen PM, van den Berg LH, Veldink JH. UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:630.e3–630.e8. doi: 10.1016/j.neurobiolaging.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Bandinelli S, Benvenuti E, Di Iorio A, Macchi C, Harris TB, Guralnik JM. Subsystems contributing to the decline in ability to walk: bridging the gap between epidemiology and geriatric practice in the InCHIANTI study. J Am Geriatr Soc. 2000;48:1618–1625. doi: 10.1111/j.1532-5415.2000.tb03873.x. [DOI] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar J-L, Dimou L, Lin S, Fergani A, Ruegg MA, Schwab ME, Dupuis L, Loeffler J-P. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwee LC, Liu Y, Haynes C, Gibson JR, Stone A, Schichman SA, Kamel F, Nelson LM, Topol B, van Den Eeden SK, Tanner CM, Cudkowicz ME, Grasso DL, Lawson R, Muralidhar S, Oddone EZ, Schmidt S, Hauser MA. A high-density genome-wide association screen of sporadic als in US veterans. PlosOne. 2012;7:e32768. doi: 10.1371/journal.pone.0032768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai S-L, Myllykangas L, Sulkava R, Jansson L, Hernandez DG, Gibbs JR, Nalls MA, Heckerman D, Tienari PJ, Traynor BJ. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: A genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347:1425–1431. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- Melzer D, Perry JR, Hernandez D, Corsi AM, Stevens K, Rafferty I, Lauretani F, Murray A, Gibbs JR, Paolisso G, Rafiq S, Simon-Sanchez J, Lango H, Scholz S, Weedon MN, Arepalli S, Rice N, Washecka N, Hurst A, Britton A, Henley W, van de Leemput J, Li R, Newman AB, Tranah G, Harris T, Panicker V, Dayan C, Bennett A, McCarthy MI, Ruokonen A, Jarvelin MR, Guralnik J, Bandinelli S, Frayling TM, Singleton A, Ferrucci L. A genome-wide association study identifies protein quantitative trait loci (pQTLs) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Boillée S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, Moigneu C, Vandenberghe N, Danel-Brunaud V, Corcia P, Pradat PF, Le Forestier N, Lacomblez L, Bruneteau G, Camu W, Brice A, Cazeneuve C, Leguern E, Meininger V, Salachas F. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49:258–263. doi: 10.1136/jmedgenet-2011-100699. [DOI] [PubMed] [Google Scholar]
- O’Toole O, Traynor BJ, Brennan P, Sheenan C, Frost E, Corr B, Hardiman O. Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J Neurol Neurosurg Psychiatry. 2007;79:30–32. doi: 10.1136/jnnp.2007.117788. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:8–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Sabatelli M, Conforti FL, Zollino M, Mora G, Monsurrò MR, Volanti P, Marinou K, Salvi F, Corbo M, Giannini F, Battistini S, Penco S, Lunetta C, Quattrone A, Gambardella A, Logroscino G, Simone I, Bartolomei I, Pisano F, Tedeschi G, Conte A, Spataro R, La Bella V, Caponnetto C, Mancardi G, Mandich P, Sola P, Mandrioli J, Renton AE, Majounie E, Abramzon Y, Marrosu F, Marrosu MG, Murru MR, Sotgiu MA, Pugliatti M, Rodolico C, Moglia C, Calvo A, Ossola I, Brunetti M, Traynor BJ, Borghero G, Restagno G, Chiò A. C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, Johnson L, Veldink JH, van Es MA, van den Berg LH, Robberecht W, Van Damme P, Hardiman O, Farmer AE, Lewis CM, Butler AW, Abel O, Andersen PM, Fogh I, Silani V, Chiò A, Traynor BJ, Melki J, Meininger V, Landers JE, McGuffin P, Glass JD, Pall H, Leigh PN, Hardy J, Brown RH, Powell JF, Orrell RW, Morrison KE, Shaw PJ, Shaw CE, Al-Chalabi A. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor BJ, Nalls M, Lai SL, Gibbs RJ, Schymick JC, Arepalli S, Hernandez D, Van Der Brug MP, Johnson JO, Dillman A, Cookson M, Moglia C, Calvo A, Restagno G, Mora G, Chiò A. Kinesin-associated protein 3 (KIFAP3) has no effect on survival in a population-based cohort of ALS patients. PNAS. 2010;107:12335–12338. doi: 10.1073/pnas.0914079107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Es MA, Veldink JH, Saris CGJ, Blauw HM, Van Vught PWJ, Birve A, Lemmens R, Schelhaas HJ, Groen EJN, Huisman MHB, Van Der Kooi AJ, De Visser M, Dahlberg C, Estrada K, Rivadeneira F, Hofman A, Zwarts MJ, Van Doormaal PTC, Rujescu D, Strengman E, Giegling I, Muglia P, Tomik B, Slowik A, Uitterlinden AG, Hendrich C, Waibel S, Meyer T, Ludolph AC, Glass JD, Purcell S, Cichon S, Nöthen MM, Wichmann H-E, Schreiber S, Vermeulen SHHM, Kiemeney LA, Wokke JHJ, Cronin S, McLaughlin RL, Hardiman O, Fumoto K, Pasterkamp RJ, Meininger V, Melki J, Leigh PN, Shaw CE, Landers JE, Al-Chalabi A, Brown RH, Robberecht W, Andersen PM, Ophoff RA, Van Den Berg LH. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Munc13-1 is required for presynaptic long-term potentiation. J Neurosci. 2011;31:12053–12057. doi: 10.1523/JNEUROSCI.2276-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]