

Abstract
Solid lipid nanoparticles (SLNs) are an alternative carrier system used to load the drug for targeting, to improve the bioavailability by increasing its solubility, and protecting the drug from presystemic metabolism. The avoidance of presystemic metabolism is due to the nano-metric size range, so that the liver cannot uptake the drug from the delivery system and is not metabolized by the liver. Montelukast sodium is an anti-asthmatic drug, because of its poor oral bioavailability, presystemic metabolism, and decreased half-life; it was chosen to formulate as the solid lipid nanoparticle (SLN) system by hot homogenization followed by an ultrasonication method, to overcome the above. Compritol ATO 888, stearic acid, and glyceryl monostearate were used as a lipid matrix and polyvinyl alcohol as a surfactant. The prepared formulations have been evaluated for entrapment efficiency, drug content, in vitro drug release, particle size analysis, scanning electron microscopy, Fourier transform-infrared studies (FT-IR), differential scanning calorimetry (DSC), and stability. Particle size analysis revealed that the SLN prepared from the higher melting point lipid showed a larger particle size and with increased carbon chain length of the fatty acids. Entrapment efficiency (EE) was ranging from 42% to 92%. In vitro release studies showed maximum cumulative drug release was obtained for F 1 (59.1%) containing stearic acid, and the lowest was observed for F 18 (28.1%) containing compritol ATO 888 after 12 h and all the formulations followed first-order release kinetics. FT-IR and DSC studies revealed no interaction between drug and lipids. Studies showed that increase in lipid concentration, increased particle size, EE, and maintained the sustained release of drug. Among all, compritol ATO 888 was chosen as the best lipid for formulating SLN because it had high EE and sustained the drug release.
Keywords: Entrapment efficiency, lipids, montelukast sodium, solid lipid nanoparticles, surfactant
INTRODUCTION
Solid lipid nanoparticles (SLNs) are one of the novel potential colloidal carrier systems as alternative materials to polymers which is identical to oil in water emulsion for parenteral nutrition, but the liquid lipid of the emulsion has been replaced by a solid lipid. They have many advantages such as good biocompatibility, low toxicity, and lipophilic drugs are better delivered by as the solid lipid nanoparticle (SLN), and the system is physically stable. SLN may be a promising sustained release and a drug targeting system for lipophilic drugs.[1]
SLNs are composed of physiological and compatible lipids with a high melting point as the solid core, which is coated by nontoxic amphiphilic surfactants as the outer shell. The nanoparticles are in the submicron size range (50–1000 nm) and in the solid state at both body and room temperatures. Studies have shown that the physiochemical characteristics and stability of drug-loaded SLNs depend on the properties of drug and ingredients.
Appropriate choice of lipids, surfactants, and their composition affects the particle size, long-term stability during storage, drug loading, and release behavior. It means that there is an optimal SLN formulation for each drug that can be obtained by investigating the effect of process variables on the characteristics of desired carriers.[2]
Montelukast sodium is a potent, selective, and orally active leukotriene receptor antagonist that acts by inhibiting physiological actions of the cysteinyl leukotrienes. It is used in the prophylaxis and treatment of asthma exercise-induced bronchospasm, allergic rhinitis, urticaria, and to relieve the symptoms of seasonal allergies. The main drawback of conventional montelukast formulation is that it undergoes hepatic first pass metabolism. Thus, it shows biological half-life of 2.5–5.5 h, thereby decreasing the bioavailability up to 64%. The short half-life, poor solubility in water, and low bioavailability of montelukast sodium make a promising candidate for formulation of a sustained-release dosage form. SLNs are an alternative carrier system used to load the drug for targeting, to improve the bioavailability by increasing its solubility and protecting drug from first pass metabolism. The avoidance of presystemic metabolism is due to the nano-metric size range, so that the liver cannot uptake the drug from the delivery system and is not metabolized by the liver.[1] Therefore, when montelukast sodium is formulated as SLN the first pass metabolism may be avoided.
In this work, montelukast sodium-loaded SLNs were prepared by using stearic acid, glyceryl monostearate (GMS), and compritol ATO 888 as the lipid matrix and polyvinyl alcohol as the surfactant. The effects of lipid type and its concentration and surfactant concentration on the entrapment efficiency (EE), particle size, thermal characteristics, and drug release behavior of the resulting nano drug delivery systems were investigated.
MATERIALS AND METHODS
Materials
Montelukast sodium and glyceryl behenate were obtained as a gift sample from Microlabs, Hosur (TN) and Orchid Pharma, Chennai (TN), India. Glyceryl monostearate [GMS] (CDH Pvt. Ltd., Mumbai, India), stearic acid (Nice chemicals, Kerala, India), dialysis membrane 50-LA 387 (Himedia, Mumbai, India) were purchased from the local market. All the other reagents and solvents used were of analytical grade.
Formulation of montelukast sodium loaded solid lipid nanoparticles
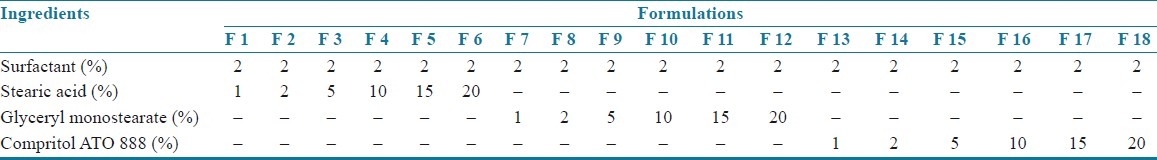
Montelukast sodium loaded SLNs[3–6] were prepared by using the method of hot homogenization followed by ultrasonication. Montelukast sodium and lipid of various concentrations, i.e. 1, 2, 5, 10, 15, and 20% (composition of the formulations is shown in Tables 1 and 2) were weighed and dissolved in ethanol. Organic solvents were completely removed using a rotary flash evaporator (super fit rotary flash evaporator). The embedded lipid layer was melted by heating 5–10°C above the melting point of the lipid. The aqueous phase was prepared by dissolving polyvinyl alcohol in distilled water (sufficient to produce 30 ml of preparation) and heated to the same temperature as the oil phase. The hot aqueous phase was added to the organic phase and homogenization was performed (at 2000 rpm) by using a mechanical stirrer for 60 min. The obtained coarse emulsion was allowed to cool to room temperature and stirred at 400 rpm for 30 min. Then, it was ultrasonicated using a probesonicator (Vibronics, Chennai) processor for 10 min. Then the prepared montelukast sodium loaded SLNs were finally stored in amber colored containers.
Table 1.
Composition of SLN formulations
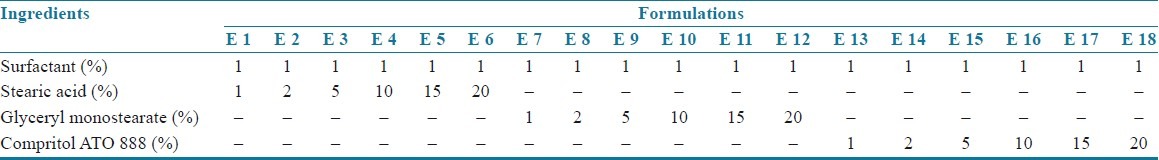
Table 2.
Composition of SLN formulations
Evaluation of montelukast sodium loaded SLN
Entrapment efficiency
Entrapment efficiency (EE)[7] of montelukast sodium loaded SLNs were determined by the centrifugation method. The nanoparticles were separated in a high speed cooling centrifuge (Eppendorf Centrifuge 5417 R) at 14,000 rpm for 90 min at 4°C. Then the supernatant liquid was made up to a desired volume with buffer to measure the absorbance at 345 nm (in the UV Spectrophotometer Pharmaspec 1607 Shimadzu, Japan) to estimate the unentrapped drug for the calculation of EE.
The percentage of entrapment efficiency (%EE) was calculated by following formula:
% EE = Total amount of drug taken - Unentrapped drug / Total amount of drug taken × 100.
Particle size analysis
The mean particle size[7–10] of the selected formulations was determined by using a laser light scattering instrument (LS230; COULTER) at a fixed angle of 90° at 25°C. The particle size analysis data were evaluated using the volume distribution.
Morphology
Surface morphology[8] of the selected formulation F 15 (5% compritol ATO 888) was evaluated using scanning electron microscopy. The sample was spread on an aluminium stub and allowed us to dry at room temperature. The dried sample then was sputter coated with gold for 40 s using Hitachi Ion-Sputter E-1010. The images were captured with the Hitachi S-3400 Scanning Electron Microscope.
In vitro release studies
In vitro release of montelukast sodium from SLN formulations were determined by dialysis bag diffusion technique.[4] The release studies[3–8] were performed using 0.5% sodium lauryl sulfate in distilled water as diffusion medium. A dialysis membrane having pore size of 2.4 nm and molecular weight cutoff, 14,000 Da (Himedia, Mumbai, India), was used. The aqueous nanoparticulate dispersion equivalent to 1 mg of drug was placed in a dialysis bag and sealed at both ends. The dialysis bag was immersed in a receptor compartment containing 250 ml of 0.5% SLS solution which was stirred at 100 rpm using a magnetic stirrer (MC Dalal, Chennai) and the receptor compartment was covered to prevent the evaporation of buffer. Samples were withdrawn at predetermined time intervals, and the same volume was replaced with fresh buffer to maintain the sink concentration. Samples were analyzed at 345 nm spectrophotometrically (UV Pharma Spec 1700, Shimadzu, Japan). All experiments were done in triplicate.
Release kinetics
In order to understand the release kinetics[9,11] of a drug, the results of in vitro drug release studies of nanoparticles were fit to various kinetic equations such as zero order (cumulative % release vs. time), first order (log % drug remaining vs. time), and Higuchi's model (cumulative % drug release vs. square root of time). Values of r2 and k were calculated for the linear curve obtained by regression analysis of the above plots. The exact mechanism of drug release was determined by the Korse-Meyer–Peppas model (log drug release vs. log time).
Fourier transform-infrared studies
The interaction between the lipids and drug was identified from the Fourier transform-infrared (FT-IR)[6–10] studies. The FT-IR spectrum of pure drug and combination of drug with lipids were obtained by using a Shimadzu FT-IR Spectrophotometer. The scanning range is 450–4000 cm–1 and the resolution is 4 cm–1. Samples were prepared as KBr pellets.
Differential scanning calorimetry
Differential scanning calorimetry (DSC)[6–10] was performed using a Perkin Elmer STA 6000 Thermal analyzer. The instrument was calibrated with an indium standard. Accurately weighed (it varies from 3—5 mg) samples were placed in an open type ceramic sample pan. Thermograms were obtained by heating the sample at a constant heating rate of 8°C/min. A dry purge of argon gas (60 ml/min) was used for all runs. Samples were heated from 37 to 400°C. DSC thermograms were recorded for pure drug, stearic acid, GMS, compritol ATO 888, and its physical mixture.
Stability studies
According to modified ICH guidelines, the stability studies[9–11] were performed for all the formulations. SLN formulations were stored at 25 ± 2°C and 60 ± 5% RH using a stability chamber (Inlab Equipments, India) and at 4 ± 2°C in a refrigerator (Kelvinator, India) and the EE was estimated periodically for one month period.
RESULTS AND DISCUSSION
Preparation of montelukast sodium loaded SLN
Homogenization followed by ultrasonication is a reliable, simple, and reproducible method for preparing SLN. The prepared SLN dispersion was found to be uniform and homogenous in appearance.
Physicochemical properties
The SLN dispersion was white in color, odorless, and fluid in nature. It was stable and did not show sedimentation even after centrifugation (2000 rpm for 30 min).
Entrapment efficiency
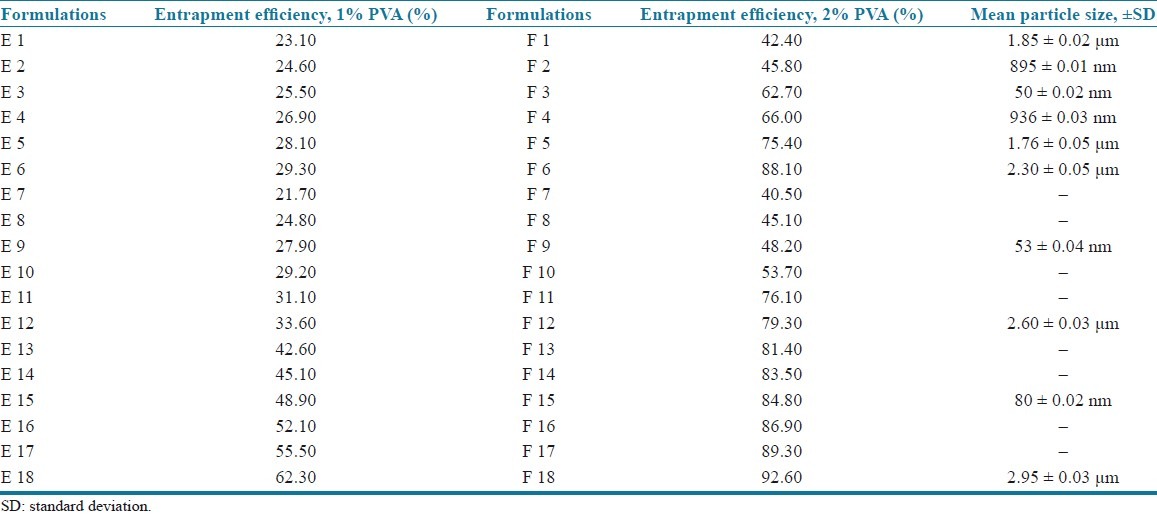
The EE of all the formulations is presented in Table 3.
Table 3.
Comparison of the particle size
Influence of surfactant on entrapment efficiency
Incorporation of 1% polyvinyl alcohol to all formulations showed EE of 23.1–62.3% (E 1–E 18), and when the concentration is increased to 2%, EE was found to be in the range of 42.4–92.6% (F 1–F 18). From the results, it was found that an increase in surfactant concentration increases EE,[6] independent of type and concentration of lipids as shown in Table 3. Hence 2% concentration containing formulations (F 1–F 18) was chosen for further evaluation.
Influence of lipid material on entrapment efficiency
The results indicated that increasing the lipid concentration also increases the EE [Table 3]. This may be due to the higher concentration of lipid would provide more space to increment of the lipid content and also reduces the escaping of drug into the external phase thus ensuring highest EE. The EE of the three lipids was in the order of compritol > GMS > stearic acid. Among the lipids used, compritol showed the highest EE when compared to GMS and stearic acid. This may be due to the fact that the presence of long chain fatty alcohol could lead to the creation of a less ordered solid lipid matrix and leaves enough space to accommodate drug molecules.[6,9–10]
Particle size analysis
The mean particle sizes of the empty SLN and the prepared formulations were measured by using a light scattering technique and the mean particle size of the selected formulations was shown in the Table 3. The particle size was found to vary depending upon the surfactant concentration and type and concentration of lipids.
Influence of surfactant on the particle size
The effect of surfactant concentration was determined by taking 1% and 2% concentrations of polyvinyl alcohol. Empty SLN containing 1% polyvinyl alcohol showed the mean particle size of 10.24 ± 0.05 μm and 2% showed the mean particle size of 50 ± 0.02 nm. This may be due to the fact that at low surfactant concentration, an optimal surface coverage of the surfaces may not be achieved resulting in less optimal stabilization of the particle dispersion. The results suggested that relatively high concentration of surfactant were needed to prevent particle aggregation.[4,9] Hence, 2% polyvinyl alcohol was selected for further studies.
Influence of lipid on the particle size
The results showed that the stearic acid containing formulations had the mean particle size of 1.85 μm, 895 nm, 50 nm, 936 nm, 1.76 μm, and 2.30 μm for 1%, 2%, 5%, 10%, 15%, and 20% lipid concentration (F1–F6), respectively. It was observed that at a particular surfactant concentration (2%), an increase in the lipid content decreased the particle size up to an optimum concentration (5%) after which the particle size was increased. This increase in the particle size may be due to the decreased emulsifying efficiency and therefore increased the particle agglomeration.[3–5] Therefore, 5% lipid concentration was selected and the particle sizes of other lipids at the same concentration were determined. From the results, it was further observed that the formulations containing 5% concentration of stearic acid had the particle size of 50 nm where as GMS had 53 nm and compritol ATO 888 had 80 nm particle sizes. In addition, the particle size of formulations containing 20% (highest) concentration of the three lipids were compared and the results indicated that stearic acid had 2.30 μm, GMS had 2.60 μm, and compritol had 2.95 μm. This increase in particle size in accordance with increase in lipid content may be due to the higher melting point of compritol than GMS and stearic acid. Hence, the higher the melting point of lipids the higher the particle size.[7]
The decreasing order of particle size for the three lipids is as follows:
compritol < GMS < stearic acid.
Scanning electron microscopy
The SEM photograph of the selected formulation (F15— 5% compritol ATO 888) is shown in Figure 1. The SLN dispersion showed the particle size was found to be less than 500 nm in size with the spherical shape and almost smooth surface.
Figure 1.

SEM photograph of the best formulation
In vitro release studies
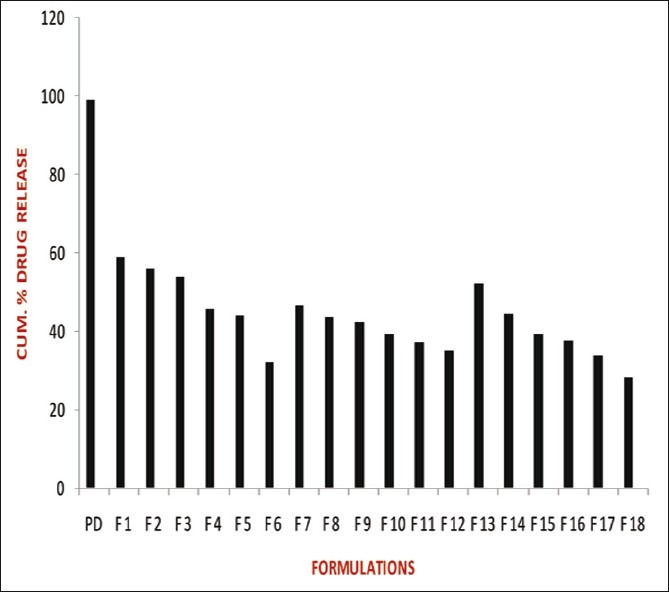
The release studies of all formulations compared with the pure drug is shown in Figures 2 (displaying burst release) and 3. The release of the best formulation was compared with the pure drug is shown in Figure 4. The release of the drug from the pure drug solution was much faster with nearly 100% of drug diffused into the release medium at fourth hour. SLN containing three different lipids displayed a similar biphasic drug release pattern with a burst release within 30 min followed by sustained release afterward.[7] Further, the initial burst effect of the formulations containing stearic acid varied from 7.5% to 21.9 % (F1–F6); for GMS 9–15.4% (F7–F12) and for compritol 9.3–16.3% (F13–F18). From the results, it was concluded that the higher lipid ratio leads to higher initial burst release.[7] The reason for burst release may be due to the drug associated with the surface of nanoparticles.
Figure 2.
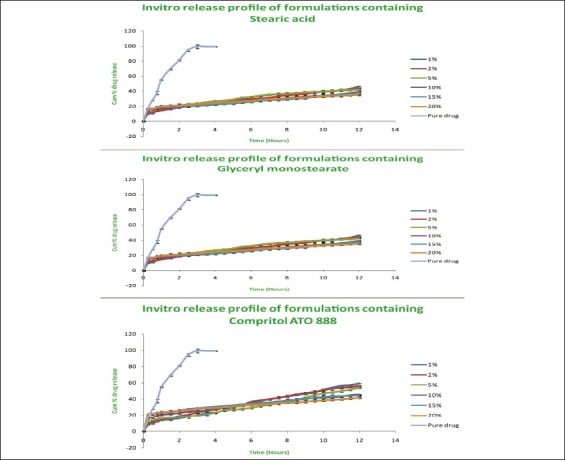
Comparison of release studies of all formulations with pure drug showing burst release
Figure 3.
Comparison of in vitro release profile of the best formulation with pure drug
Figure 4.
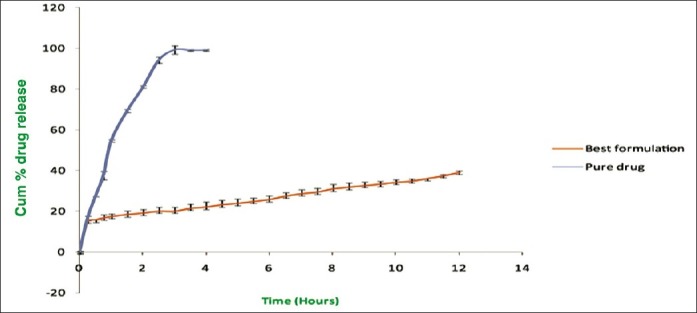
Release profile of best formulations compared with pure drug
The release of drug from the stearic acid containing formulations were found to be 59.1%, 56%, 54%, 45.6%, 44.5% and 41.6% (F1–F6); GMS containing formulations (F7–F12) showed 46.4%, 43.7%, 42.2%, 39.1%, 37.3%, and 35.2% of drug release; compritol containing formulations (F13–F18) showed 52.1%, 44.3%, 39.1%, 37.4%, 33.6%, and 28.1% of drug release after 12 h. The slow release of the drug in the later stage was attributed to the fact that the solubilized or dispersed drug can only be released slowly from the lipid matrices through dissolution and diffusion.[10]
The results displayed that the release was chiefly dependent on the type and concentration of the lipids. Increase in the lipid concentration decreased the release rate independent of the type of lipids.[10] Among the three lipids used, compritol showed more sustained release than stearic acid and GMS due to its longer carbon chain length than the other two lipids. The order of drug release from the three lipids as follows: compritol > GMS > stearic acid. Hence, the nature of fatty acids affected the release significantly. Thus, it was concluded that the higher the lipid concentration and the longer the carbon chain length of fatty acids, the slower the drug release.[4,8]
Release kinetics
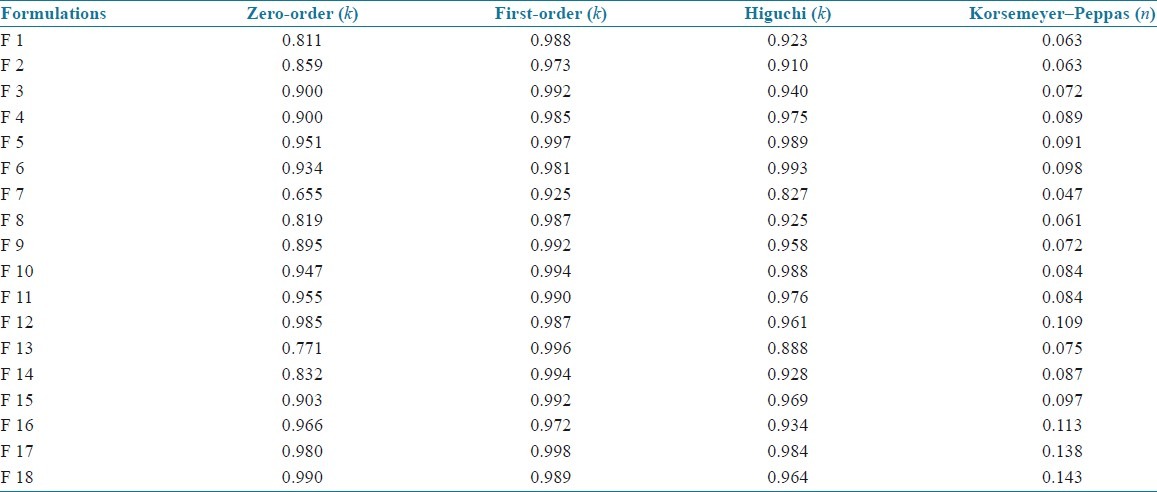
The kinetics values of all the SLN formulations are shown in Table 4. All the formulations showed first-order release which had higher linearity than the zero-order or Higuchi model. The exact mechanism of the release kinetics was determined by Korsemeyer–Peppas model. Results indicated that all the SLN formulations followed non-Fickian model of release kinetics.
Table 4.
Release kinetics values of all SLN formulations
FT-IR studies
The FT-IR spectra of pure drug, lipids, and its physical mixture are shown in Figure 5. Montelukast sodium pure drug shows the bond vibrations at 3396 cm–1 (COOH stretching), 3057 cm–1 (aromatic C–H stretching), 2925 cm–1 (aliphatic C–H stretching), and 1710 cm–1 (C–O stretching). From the physical mixtures of drug and lipids, there were no major shifting as well as no loss of any functional peaks between the spectra of drug, lipids, and its physical mixtures. Hence, it was confirmed that there was no interaction between the drug and lipids used.
Figure 5.
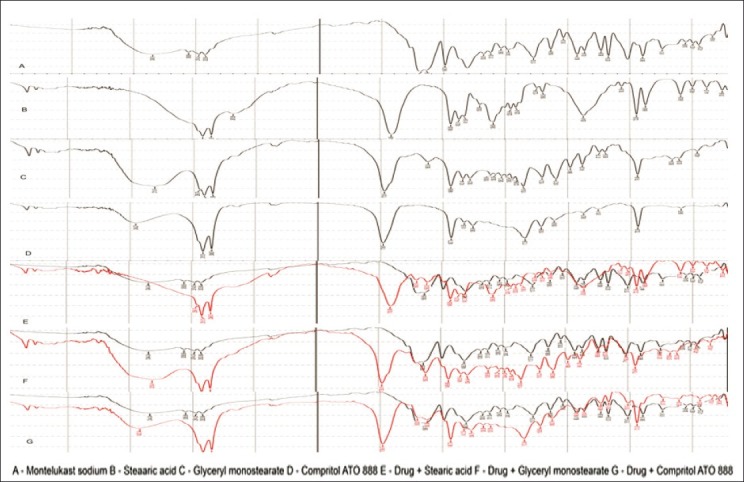
FT-IR spectra of a pure drug, lipids and its physical mixtures
Differential scanning calorimetry studies
DSC thermograms of pure drug, lipids, and its physical mixture are shown in Figure 6. From the results obtained, there was no obvious change in the endothermic peak around 125°C of the drug in physical mixtures. This suggested that there were no appearance of new peaks or disappearance of existing peak. Hence there was no considerable effect on the thermal behavior of drug with the lipid matrix under the experimental conditions.
Figure 6.
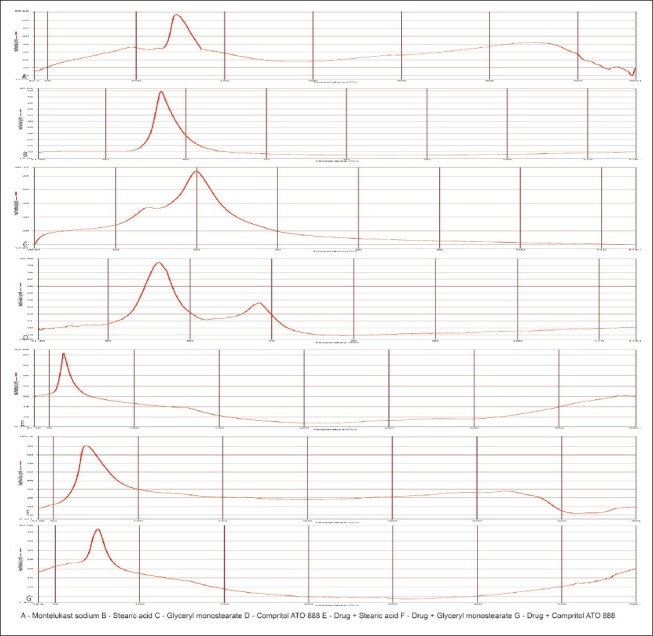
DSC thermograms of a pure drug, lipids and its physical mixtures
Stability studies of montelukast sodium-loaded SLN
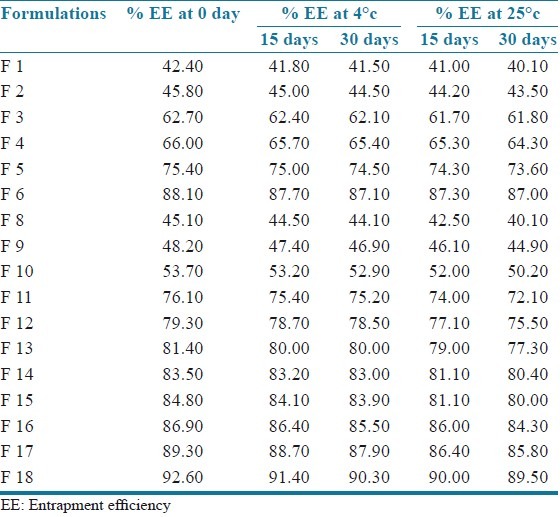
No significant change was observed in the EE of all formulations stored at temperature 4°C as well as at 25 ± 2°C and 60% ± 5% RH and the values are shown in Table 5. Hence montelukast sodium loaded SLN may be stored at both the temperatures.
Table 5.
Stability studies (percentage entrapment efficiency of all formulations stored at temperature 4°C and 25°C)
CONCLUSION
It was concluded that hot homogenization followed by ultrasonication is a feasible method for preparing montelukast sodium loaded SLN. The results of EE indicated that increasing the lipid concentration also increases the EE due to the presence of long chain fatty alcohol which could lead to the creation of a less ordered solid lipid matrix and leave enough space to accommodate drug molecules. Particle size analysis of SLNs revealed that the SLN prepared from the higher melting point lipid showed a larger particle size and with increased carbon chain length of the fatty acids; compritol had a higher melting point than the other two lipids and showed a larger particle size. SEM studies revealed that the SLN was spherical in shape and almost smooth surface. The nature of fatty acids affected the release significantly and the longer the carbon chain length of fatty acids, the slower the drug release. All the SLN formulations followed first-order release kinetics. FT-IR and DSC studies suggested that there is no interaction between drug and lipids. Even though the particle size of stearic acid containing formulations was smaller at all the concentrations than formulations containing GMS and compritol ATO 888, compritol ATO 888 may be considered for formulating SLN because it had a higher EE and sustained drug release profile than GMS and stearic acid.
ACKNOWLEDGMENTS
The authors are thankful to the Dean, and Department of Pharmaceutics, Madurai Medical College, Madurai, for providing the facilities to carry out the work. Also we extend our gratitude to Sastra University for estimating the particle size analysis and to Madras University, Chennai, where SEM studies were carried out.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Gohla S. Solid lipid nanoparticles for controlled drug delivery: A review of the state of art. Eur J Pharm Biopharm. 2000;50:161–77. doi: 10.1016/s0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 2.Kheradmandnia S, Vasheghani-Farahani E, Nosrati M, Atyabi F. Preparation and characterization of ketoprofen-loaded solid lipid nanoparticles made from beeswax and carnauba wax. Nanomedicine. 2010;6:753–9. doi: 10.1016/j.nano.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Rahman Z, Zidan AS, Khan MK. Non-destructive methods of characterization of risperidone solid lipid nanoparticles. Eur J Pharm Biopharm. 2010;76:127–37. doi: 10.1016/j.ejpb.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Yassin AE, Anwer MK, Mowafy HA, El-Bagory IM, Bayomi MA, Alsarra IA. Optimization of 5-fluorouracil solid-lipid nanoparticles: A preliminary study to treat colon cancer. Int J Med Sci. 2010;7:398–408. doi: 10.7150/ijms.7.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harivardhanreddy L, Murthy RS. Etoposide-loaded nanoparticles made from glyceride lipids: Formulation, characterization, in vitro drug release and stability evaluation. AAPS Pharm Sci Tech. 2005;6:E158–66. doi: 10.1208/pt060224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Misra A, Kalariya M, Padhi BK, Chougule M. Methotrexate-loaded solid lipid nanoparticles for topical treatment of psoriasis: Formulation and clinical implications. Drug Deliv Technol. 2002;5:1–13. [Google Scholar]
- 7.Doijad RC, Manvi FV, Deshmukh NV. Formulation and targeting efficiency of cisplatin engineered solid lipid nanoparticles. Int J Pharm. 2008;70:203–7. doi: 10.4103/0250-474X.41456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang QY, Wang MT, Chen F, Gong T, Jian YL, Zhang ZR, et al. Lung-targeting delivery of dexamethasone acetate loaded solid lipid nanoparticles. Arch Pharm Res. 2007;30:519–25. doi: 10.1007/BF02980228. [DOI] [PubMed] [Google Scholar]
- 9.Lv Q, Yu A, Xi Y, Li H, Song Z, Cui J, et al. Development and evaluation of penciclovir-loaded solid lipid nanooparticles for topical delivery. Int J Pharm. 2009;372:191–8. doi: 10.1016/j.ijpharm.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Sanna V, Caria G, Mariani A. Effect of lipid nanoparticles containing fatty alcohols having different chain length on the ex vivo skin permeability of Econazole nitrate. Powder Technol. 2010;201:32–6. [Google Scholar]
- 11.de Mendoza A Estella-Hermoso, Rayo M, Mollinedo F, Blanco-Prieto MJ. Lipid nanoparticles for alkyl lysophospholipidedelfosine encapsulation: Development and in vitro characterization. Eur J Pharm Biopharm. 2008;68:207–13. doi: 10.1016/j.ejpb.2007.06.015. [DOI] [PubMed] [Google Scholar]