

Abstract
Monoamine oxidase B (MAO-B) inhibitory potential of adenosine A2A receptor (AA2AR) antagonists has raised the possibility of designing dual-target–directed drugs that may provide enhanced symptomatic relief and that may also slow the progression of Parkinson's disease (PD) by protecting against further neurodegeneration. To explain the dual inhibition of MAO-B and AA2AR at the molecular level, molecular docking technique was employed. Lamarckian genetic algorithm methodology was used for flexible ligand docking studies. A good correlation (R2= 0.524 and 0.627 for MAO-B and AA2AR, respectively) was established between docking predicted and experimental Ki values, which confirms that the molecular docking approach is reliable to study the mechanism of dual interaction of caffeinyl analogs with MAO-B and AA2AR. Parameters for Lipinski's “Rule-of-Five” were also calculated to estimate the pharmacokinetic properties of dual-target–directed drugs where both MAO-B inhibition and AA2AR antagonism exhibited a positive correlation with calculated LogP having a correlation coefficient R2 of 0.535 and 0.607, respectively. These results provide some beneficial clues in structural modification for designing new inhibitors as dual-target–directed drugs with desired pharmacokinetic properties for the treatment of PD.
Keywords: Adenosine A2A antagonist, docking, dual-target–directed drugs, monoamine oxidase B
INTRODUCTION
Parkinson's disease (PD) is primarily a disorder of the nigrostriatal dopaminergic pathway that results in the cardinal motor symptoms of bradykinesia, tremor, and rigidity.[1] Nondopaminergic treatments are increasingly being recognized as part of the therapeutic armamentarium for PD,[2–7] because long-term treatment with dopamine replacement strategies is associated with drug-related complications, such as a loss of drug efficacy, the onset of dyskinesias, and the occurrence of psychosis and depression.[8] Therefore, development of suitable approach to the treatment of this devastating neurodegenerative disorder remains an indispensable component of research in medicinal chemistry. Nowadays, an emerging paradigm that proposes the targeting of multiple components of pathobiology through a single drug molecule is gaining increasing acceptance. Although the single-target or “silver bullet” approach currently remains the major drug discovery strategy in large pharmaceutical companies, there is increasing recognition of the limitations of such an approach for complex diseases.
Monoamine oxidase B (MAO-B) is an outer membrane–bound mitochondrial flavoenzyme that functions in the oxidative deamination of dopamine in the striatum.[9] Inhibition of MAO-B in the brain may slow the depletion of dopamine stores and elevate the levels of endogenous dopamine, and dopamine produced from exogenously administered levodopa.[10,11] Furthermore, inhibitors of the MAO-B may also exert a neuroprotective effect by decreasing the production of potentially hazardous byproducts of dopamine metabolism in the brain.[12] Adenosine A2A receptor (AA2AR) antagonists are another class of promising anti-Parkinsonian agents and a leading candidate class for the nondopaminergic treatment of symptomatic PD.[6] AA2AR antagonists may also possess neuroprotective properties and may prevent the development of dyskinesia that is usually associated with levodopa treatment.[13,14] Interestingly, it has been observed that AA2AR antagonists also inhibit MAO-B; therefore, they can be exploited in designing dual-target–directed drugs aimed at providing enhanced symptomatic relief in addition to slowing the progression of PD by protecting against further neurodegeneration.[15] In this regard, C8-substituted caffeinyl derivatives are becoming popular as dual-target–directed drugs that block MAO-B and AA2AR for the treatment of PD.
Significant progress has been made in computer-aided drug design by pharmaceutical companies at different stages of drug discovery, such as identifying new hits, enhancing molecule binding affinity in hit-to-lead, and lead optimization.[16] Moreover, in silico approaches are routinely used in modern drug design to help understand drug–receptor interactions. It has been shown in the literature that computational techniques can strongly support and help the design of novel, more potent inhibitors by revealing the mechanism of drug–receptor interactions.[17] However, so far, there has been no report concerning the application of molecular docking methodology for understanding the binding of dual-target–directed drugs that block MAO-B and AA2AR. To gain an insight into the structural requirements for the dual inhibition, we have used molecular docking studies to understand the mode of binding of C8-substituted caffeinyl analogs to MAO-B and AA2AR. In addition, we have also employed computational method for the determination of physicochemical parameters that are responsible for governing the pharmacokinetic properties of drug molecules. For the present study, AA2AR antagonists with MAO-B inhibitory properties were taken from the literature[15,18–24] and subjected to in silico studies. The results obtained from this study would be useful in both understanding the inhibitory mode of these derivatives as well as in rapidly and accurately predicting the activities of newly designed inhibitors. Some beneficial clues can also be inferred from these results that will be fruitful in designing novel inhibitors as dual-target–directed drugs with desired pharmacokinetic properties in the area of PD therapeutics.
MATERIALS AND METHODS
For the present study, crystal structures of human MAO-B (PDB code: 2V5Z)[25] and human AA2AR (PDB code: 3EML)[26] were downloaded from the protein databank (www.rcsb.org/pdb). A set of 18 inhibitors [Table 1] that inhibit MAO-B and antagonize AA2AR were taken from the literature[15,18–24] and docked onto the active site of MAO-B and AA2AR using AutoDock 4.2 (Release 4.2.2.1) program.
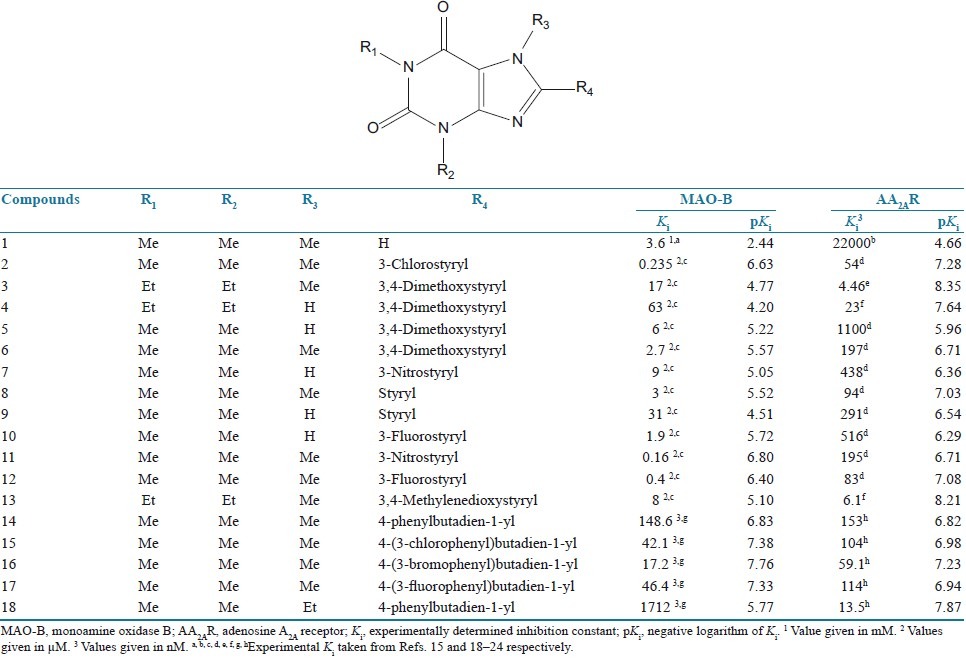
Table 1.
The structures, Ki and pKi values for MAO-B inhibition and AA2AR antagonism by 8-substituted caffeinyl analogs
Molecular docking studies
For docking experiments with AutoDock 4.2, ligand molecules were drawn in ChemBioDraw Ultra 12.0 and converted to their 3-dimensional structures in ChemBio3D Ultra 12.0, energy minimized by PM3 method using MOPAC Ultra 2009 program.[27] The prepared ligands were used as input files for AutoDock 4.2 in the next step. Lamarckian genetic algorithm method was employed for docking simulations.[28] The standard docking procedure was used for a rigid protein and a flexible ligand whose torsion angles were identified (for 10 independent runs per ligand). A grid of 60, 60, and 60 points in x, y, and z directions was built with a grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant were used for the calculation of the energetic map. The default settings were used for all other parameters. At the end of docking, the best poses were analyzed for hydrogen bonding/π–π interactions and root mean square deviation (RMSD) calculations using Discovery Studio Visualizer 2.5 program. From the estimated free energy of ligand binding (ΔGbinding, kcal/mol), the inhibition constant (Ki) for each ligand was calculated [Tables 2 and 3].
Table 2.
Results obtained after docking of 8-substituted caffeinyl analogs with human MAO-B
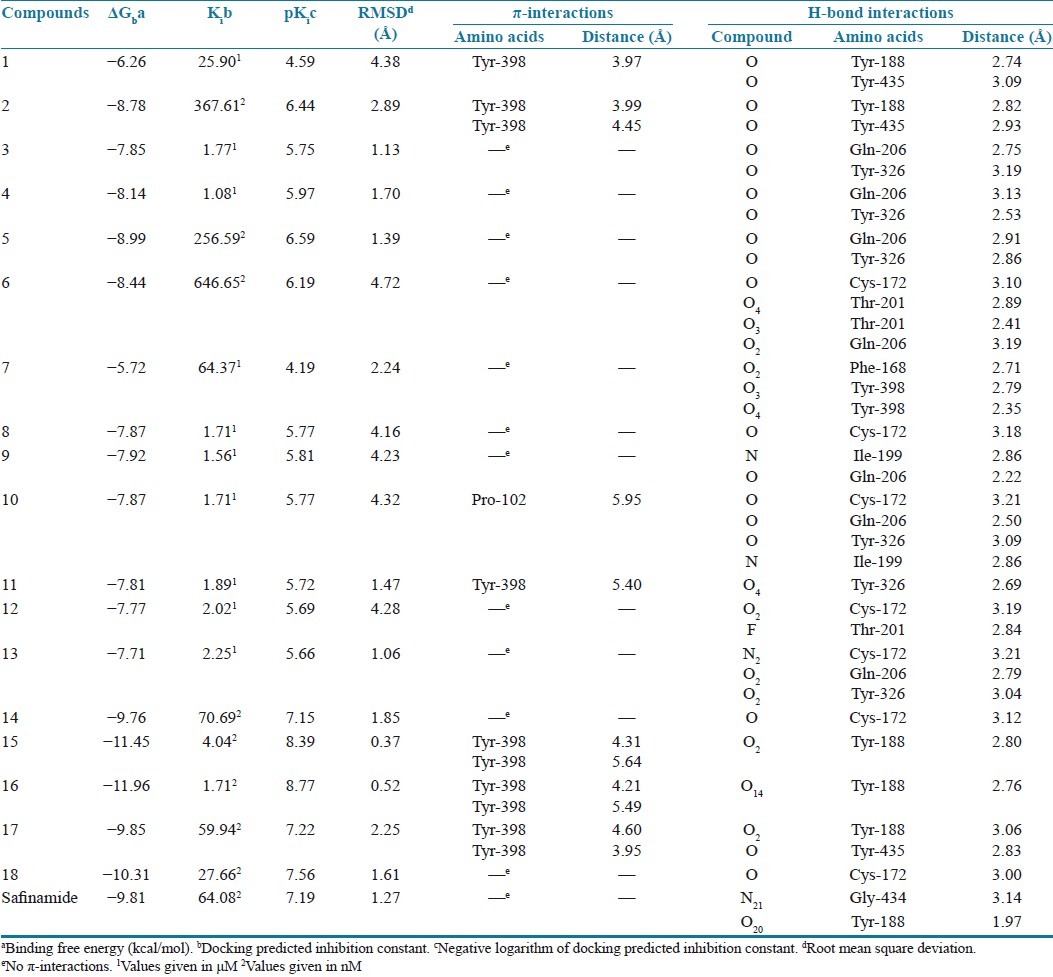
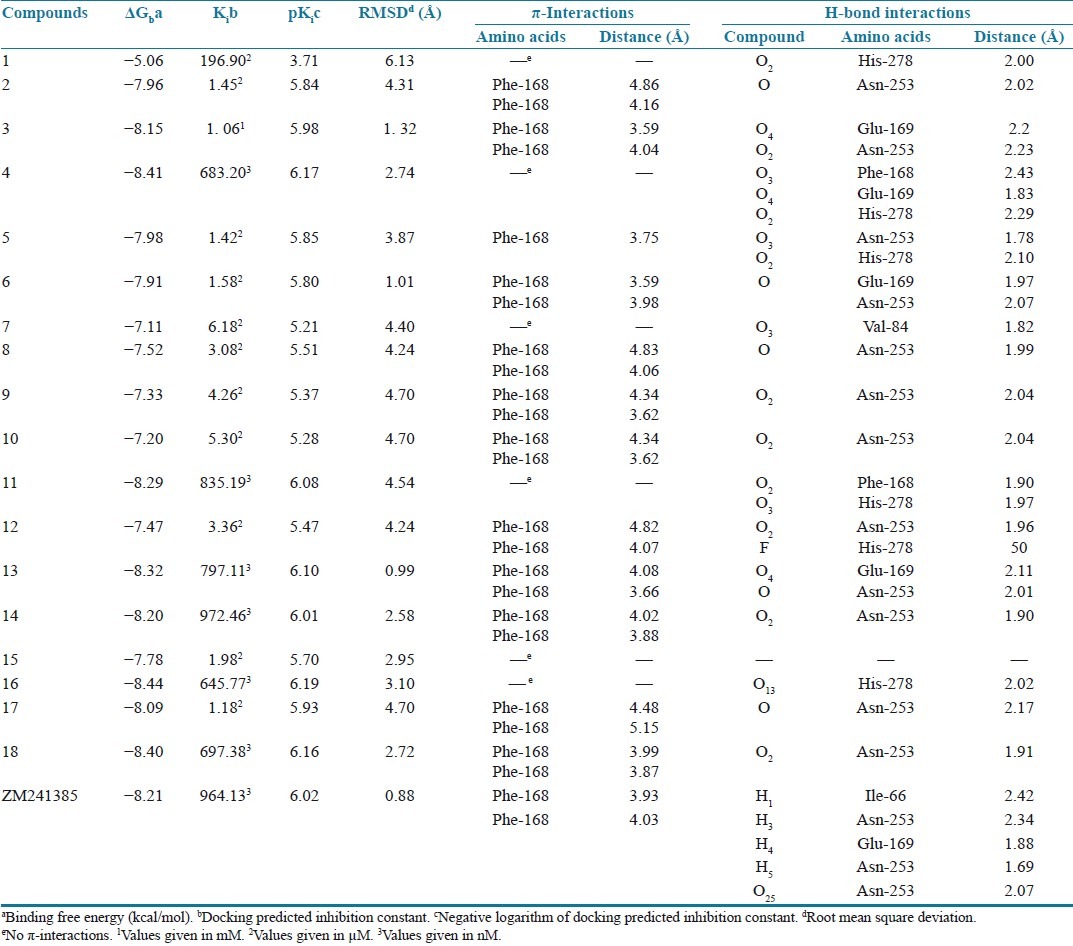
Table 3.
Results obtained after docking of 8-substituted caffeinyl analogs with human AA2AR
Calculation of physicochemical parameters
Absorption (%ABS) was calculated by: %ABS = 109 – [0.345 × topological polar surface area (TPSA)] according to the method of Zhao et al.[29] TPSA,[30] miLogP, number of rotatable bonds, and violations of Lipinski's “Rule-of-Five”[31] were calculated using Molinspiration online property calculation toolkit.[32]
RESULTS AND DISCUSSION
Molecular docking
Validation of the accuracy and performance of AutoDock 4.2
To validate the accuracy of AutoDock 4.2 as an appropriate docking tool for the present purpose, the co-crystallized ligands (Safinamide and ZM241385 for 2V5Z.pdb and 3EML.pdb, respectively) were docked within the inhibitor-binding cavity (IBC) of human MAO-B and human AA2AR, and the docked position was compared with the crystal structure position by calculating RMSD values (1.27 and 0.88 Å, respectively). As a general rule, if the best-docked conformation of a ligand resembles the bound native ligand in the experimental crystal structure, the used scoring function is said to be successful. According to the method of validation cited in the literature,[33] the successful scoring function is the one in which the RMSD of the best docked conformation is ≤2.0 Å from the experimental one. In this study, RMSD values of both MAO-B and AA2AR were within 2.0 Å [Figure 1], indicating our docking methods are valid for the given structures and AutoDock 4.2, therefore deemed reliable for docking dual-target–directed drugs into the IBC of MAO-B and AA2AR.
Figure 1.
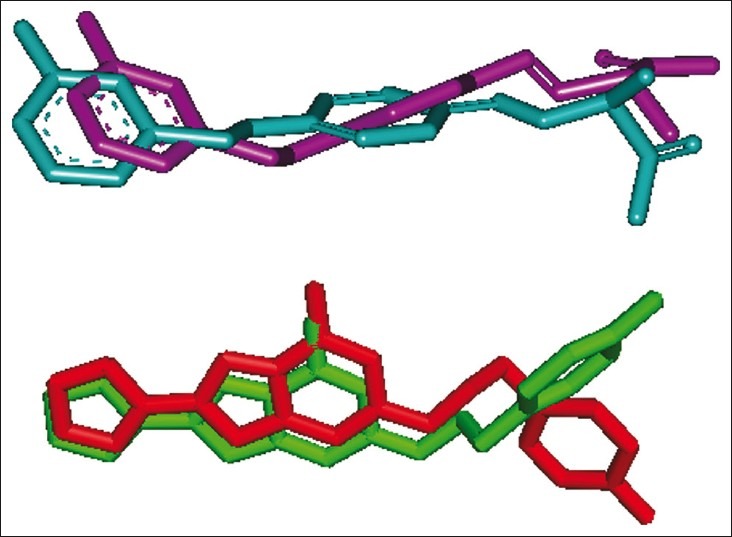
The validation of accuracy and performance of AutoDock 4.2. The docked safinamide (purple) and native safinamide (cyan) demonstrated a root mean square deviation of 1.27 Å, whereas the docked ZM241385 (red) and native ZM241385 (green) exhibited a root mean square deviation of 0.88 Å
Docking of the caffeinyl analogs into AA2AR
The co-crystallized AA2AR antagonist, ZM241385, is outlined by Leu-85, Phe-168, Glu-169, Met-177, Trp-246, Leu-249, His-250, Asn-253, His-264, Leu-267, and Met-270 residues, which constitute the active binding site.[26] The poor affinity of caffeine (compound 1) toward AA2AR in experimental studies can be clearly explained on the basis of our docking results as shown in Figure 2, where none of the residues of binding site was found to interact with caffeine neither in terms of hydrophobic nor hydrophilic interactions. However, in close proximity of the binding cavity, it interacted with His-278 by forming a hydrogen bond. It was interesting to note that the xanthine nucleus orients inside the binding cavity and interacts by both hydrophobic as well as hydrophilic interactions when C-8 position is substituted with (E)-styryl and 4-phenylbutadien-1-yl groups making these compounds fairly potent [Figure 3].
Figure 2.
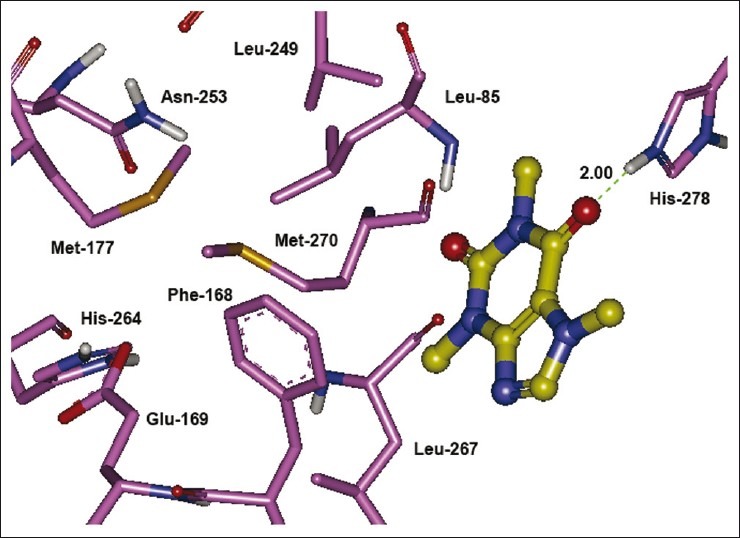
The lowest energy configuration of docking result of caffeine with binding pocket of human AA2AR. The residues of binding pocket are shown as stick in pink color, and caffeine is presented as ball and stick style in yellow color. Dashed lines in green indicate H-bonds. Nitrogens are in blue and oxygens are in red
Figure 3.
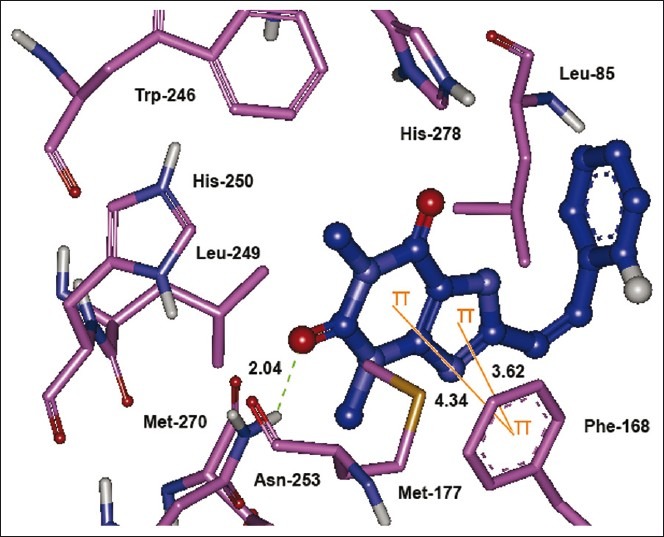
The lowest energy configuration of docking result of caffeinyl analog (Compound 10) with binding pocket of human AA2AR. The residues of binding pocket are shown as stick in pink color while compound 10 is presented as ball and stick style in blue color. Dashed lines in green indicate H-bonds while π–π stacking interaction are shown as orange lines. Sulfur is presented in dark yellow and oxygens in red
The bicyclic triazolotriazine core of ZM241385 is anchored by an aromatic stacking interaction with Phe-168,[34] an aliphatic hydrophobic interaction with Ile-274[13,35] and a hydrogen bonding interaction with Asn-253.[36,37] Likewise, the bicyclic xanthine ring was found to interact with aromatic ring of Phe-168 by π–π stacking interaction while Asn-253 contributed in hydrophilic interaction by forming a hydrogen bond. A report by Moro et al. has also proposed that the bicyclic ring of ZM241385 is anchored by hydrophobic interactions of Leu-249.[38] Adjacent to Phe-168, a polar residue Glu- 169 shares a hydrogen bond with the oxygen atom of 3,4-methylenedioxy group. Similar kind of interaction is also known between exocyclic amino group (N15 atom) linked to the bicyclic core of ZM241385.[34,39]
Docking of the caffeinyl analogs into MAO-B
The docked compounds 1–18 oriented into the IBC of MAO-B and AA2AR in a similar way as their native ligands safinamide and ZM241385 interact with MAO-B[25] and AA2AR,[26] respectively, exhibiting a reasonable RMSD values in the range of 0.37–6.17 Å. The reported and estimated inhibition constant (Ki) was converted to their respective pKi(–log Ki) and plotted as shown in Figure 4. A positive correlation was noted between docking predicted and experimentally reported pKi with a correlation coefficient R2 of 0.524 and 0.627 for MAO-B and AA2AR, respectively.
Figure 4.
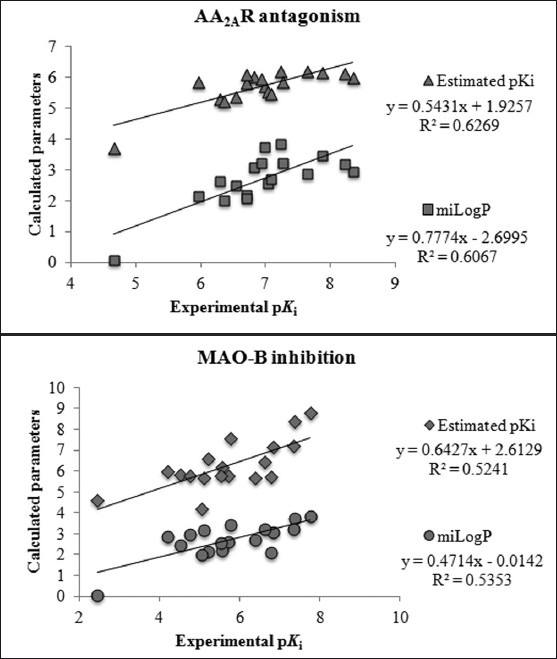
Experimental pKi is plotted against docking estimated pKi, and miLogP.
The caffeinyl derivatives docked into the IBC of human MAO-B was outlined by residues, such as Pro- 102, Leu- 171, Cys-172, Ile-198, Ile-199, Gln-206, Ile-316, Tyr- 326, Phe-343, Tyr-398, Tyr-435, and the isoalloxazine ring of FAD.[40] The major part of the IBC is hydrophobic, which allows for the tight binding of nonpolar substrates and inhibitors.[41] This is the reason why the calculated LogP presented in Table 4 bears a positive correlation with the MAO-B inhibitory activity exhibiting a correlation coefficient R2 of 0.535 [Figure 4]. However, the only hydrophilic portion is near the flavin and is required for recognition and directionality of the substrate amine functionality.[41] This hydrophilic region is located between Tyr-398 and Tyr-435, which, together with the flavin, form an aromatic cage for amine recognition.[42,43] Moreover, Gln-206 interacts by forming a hydrogen bond with the native co-crystallized ligand, safinamide. In a similar way, Gln-206 serves as hydrogen bond acceptor for most of the docked compounds [Figure 5].
Table 4.
Physicochemical parameters for good oral bioavailability of caffeinyl analogs
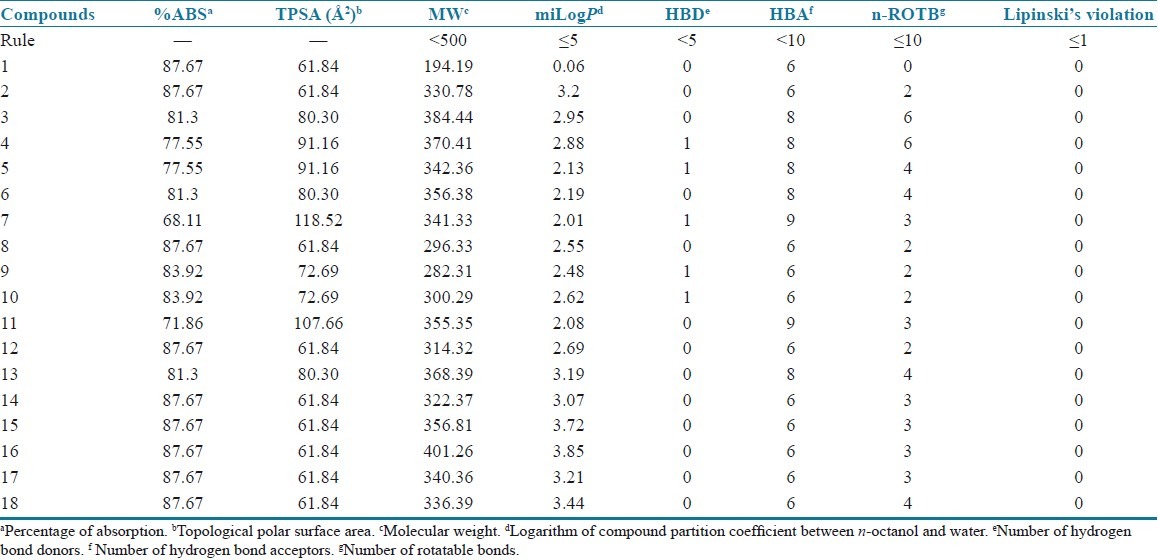
Figure 5.
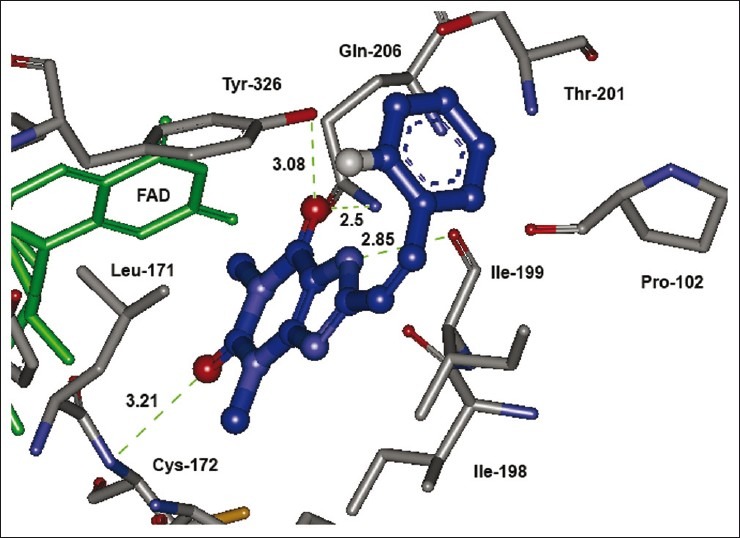
The lowest energy configuration of docking result of caffeinyl analog (Compound 10) with binding pocket of human MAO-B. The amino acids (gray) and FAD (green) are shown as stick while compound 10 is presented as ball and stick style in blue color. Dashed lines in green indicate H-bonds. Sulfur is presented in dark yellow and oxygens in red
In addition to contributing for hydrophobicity in the IBC, Phe-168, Cys-172, Ile-199, Thr-201, and Tyr-326 were also appeared to participate in hydrogen bond formation. Interestingly, (E)-8-(3-chlorostyryl)caffeine (CSC, compound 2) and compounds containing 4-phenylbutadien-1-yl groups at C-8 position of the caffeinyl moiety were observed to share a hydrogen bond with Tyr-188, a residue located at the distant site in the IBC. Likewise, 4-phenylbutadien-1-yl derivatives also interact with Tyr-435, a residue found in the hydrophilic region of the IBC [Table 2].
Caffeine, being a polar compound, is not able to accommodate well in the IBC and is a weak MAO-B inhibitor. However, substitution of the (E)-styryl and 4-phenylbutadien-1- yl groups at C-8 markedly decreases the polarity of the molecule as reflected by the high calculated LogP of these compounds [Table 4] appears to be beneficial for the MAO-B inhibitory activity. On the other hand, it is known that the active site of the MAO-B consists of an entrance connected to the substrate cavity where Ile-199 acts as a “gate” between the two cavities. When relatively large inhibitors, such as the reversible inhibitor 1,4-diphenyl-2-butene is bound, the side chain is rotated to a conformation such that the two cavities are no longer separated and are now fused forming a single cavity and such compounds demonstrate greater binding affinity.[41] Our docking results reflect that (E)-styryl and 4-phenylbutadien-1-yl groups at C-8 position of the caffeinyl moiety use both cavities as potential binding targets making them potent MAO-B inhibitors. Similarly, without the side chain at C-8, caffeine occupies only hydrophilic region leaving the hydrophobic region unoccupied and hence exhibits less binding affinity [Figure 6]. Based on these results, an overview of the structural requirements for antagonizing AA2AR and inhibiting MAO-B is presented in Figure 7.
Figure 6.
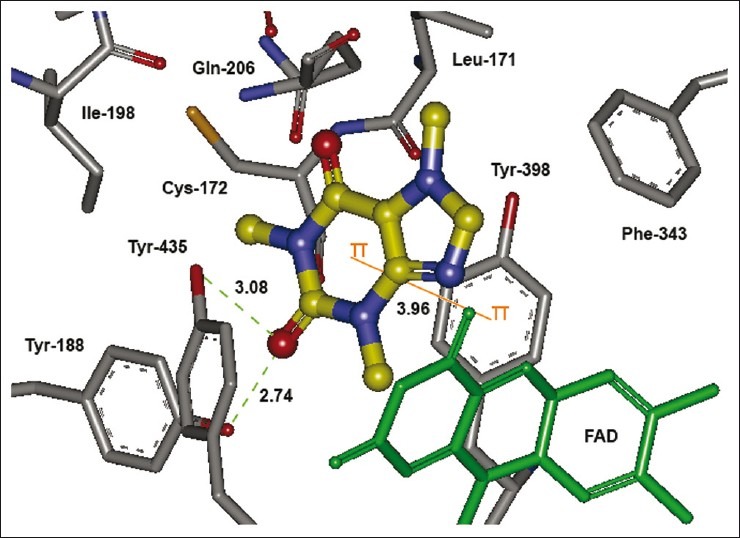
The lowest energy configuration of docking result of caffeine with binding pocket of human MAO-B. The amino acids (gray) and FAD (green) are shown as stick while caffeine is presented as ball and stick style in yellow color. Dashed lines in green indicate H-bonds while π–π stacking interaction is shown as orange line. Nitrogens are in blue and oxygens in red
Figure 7.
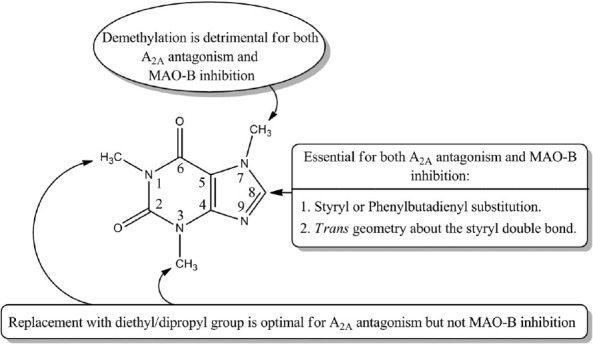
An overview of the structural requirements for antagonizing AA2AR and inhibiting MAO-B
Physicochemical parameters
Among xanthine-based AA2AR antagonists, poor water solubility is a considerable problem.[6] Lipinski's parameters[31] were calculated by using Molinspiration online property calculation toolkit[32] to estimate the pharmacokinetic properties of caffeinyl derivatives (1–18) and presented in Table 4. Topological polar surface area (TPSA), that is, surface belonging to polar atoms, is a descriptor that was shown to correlate well with passive molecular transport through membranes and, therefore, allows prediction of transport properties of drugs in the intestines and blood-brain barrier crossing.[30] TPSA was used to calculate the percentage of absorption (%ABS) according to the equation: %ABS = 109 – 0.345 × TPSA, as reported by Zhao et al.[29] Furthermore, according to Veber et al., good bioavailability is more likely for compounds with ≤10 rotatable bonds and TPSA of ≤140 Å2.[44] As the number of rotatable bonds increases, the molecule becomes more flexible and more adaptable for efficient interaction with a particular binding pocket. In the present study, compounds 1–18 exhibited % ABS ranging from 68% to 87%, which is an indication of good bioavailability by oral route. Moreover, nonviolations of Lipinski's “Rule-of-Five” and Veber's “criteria for good bioavailability” also confirm the suitability of these compounds to be used as a template for the design of dual-target–directed drugs.
CONCLUSION
In conclusion, these computational studies not only shed a light on understanding the dual mechanism of MAO-B inhibition as well as AA2AR antagonism, but also provide precious insight for the rational improvements of specificity and inhibitory potency of C-8 substituted caffeinyl analogs to be explored as novel anti-Parkinsonian drug candidates.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–19. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 2.Azam F. Synthesis of some urea and thiourea derivatives of naphtha[1,2-d]thiazol-2-amine as anti- Parkinsonian agents that cause neuroprotection against haloperidol-induced oxidative stress in mice. Med Chem Res. 2009;18:287–308. [Google Scholar]
- 3.Azam F. Therapeutic potential of free radical scavengers in neurological disorders. In: Kozyrev D, Slutsky V, editors. Handbook of Free radicals: Formation, Types and Effects. New York: Nova Publishers; 2010. pp. 57–97. [Google Scholar]
- 4.Azam F, Alkskas IA, Ahmed MA. Synthesis of some urea and thiourea derivatives of 3-phenyl/ethyl-2-thioxo-2,3-dihydrothiazolo [4,5-d]pyrimidine and their antagonistic effects on haloperidol-induced catalepsy and oxidative stress in mice. Eur J Med Chem. 2009;44:3889–97. doi: 10.1016/j.ejmech.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Azam F, Barodia SK, Anwer T, Alam MM. Neuroprotective effect of naphtha[1,2-d]thiazol-2-amine in an animal model of Parkinson's disease. J Enzyme Inhib Med Chem. 2009;24:808–17. doi: 10.1080/14756360802399183. [DOI] [PubMed] [Google Scholar]
- 6.Azam F, Ibn-Rajab IA, Alruiad AA. Adenosine A2A receptor antagonists as novel anti-Parkinsonian agents: A review of structure-activity relationships. Pharmazie. 2009;64:771–95. [PubMed] [Google Scholar]
- 7.Azam F, El-Gnidi BA, Alkskas IA, Ahmed MA. Design, synthesis and anti-Parkinsonian evaluation of 3-alkyl/aryl-8-(furan-2-yl)thiazolo[5,4-e][1,2,4]triazolo[1,5-c]pyrimidine-2(3H)-thiones against neuroleptic-induced catalepsy and oxidative stress in mice. J Enzyme Inhib Med Chem. 2010;25:818–26. doi: 10.3109/14756361003671052. [DOI] [PubMed] [Google Scholar]
- 8.Obeso JA, Olanow CW, Nutt JG. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 2000;23(Suppl 10):S2–7. doi: 10.1016/s1471-1931(00)00031-8. [DOI] [PubMed] [Google Scholar]
- 9.Singer TP. Perspectives in MAO: Past, present, and future. A review. J Neural Transm. 1987;23:1–23. doi: 10.1007/978-3-7091-8901-6_1. [DOI] [PubMed] [Google Scholar]
- 10.Di Monte DA, De Lanney LE, Irwin I, Royland JE, Chan P, Jakowec MW, et al. Monoamine oxidase-dependent metabolism of dopamine in the striatum and substantia nigra of L-DOPA-treated monkeys. Brain Res. 1996;738:53–9. doi: 10.1016/0006-8993(96)00761-5. [DOI] [PubMed] [Google Scholar]
- 11.Finberg JP, Wang J, Bankiewicz K, Harvey-White J, Kopin IJ, Goldstein DS. Increased striatal dopamine production from L-DOPA following selective inhibition of monoamine oxidase B by R(+)-N-propargyl-1-aminoindan (rasagiline) in the monkey. J Neural Transm. 1998;52:279–85. doi: 10.1007/978-3-7091-6499-0_28. [DOI] [PubMed] [Google Scholar]
- 12.Youdim MB, Bakhle Y. Monoamine oxidase: Isoforms and inhibitors in Parkinson's disease and depressive illness. Br J Pharmacol. 2006;147(Suppl 1):S287–96. doi: 10.1038/sj.bjp.0706464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson's disease. Trends Neurosci. 2006;29:647–54. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Bibbiani F, Oh JD, Petzer JP, Castagnoli N, Jr, Chen JF, Schwarzschild MA, et al. A2A antagonist prevents dopamine agonist-induced motor complications in animal models of Parkinson's disease. Exp Neurol. 2003;184:285–94. doi: 10.1016/s0014-4886(03)00250-4. [DOI] [PubMed] [Google Scholar]
- 15.Petzer JP, Castagnoli N, Schwarzschild MA, Chen J, Schyf CJ. Dual target–directed drugs that block monoamine oxidase B and adenosine A2A receptors for Parkinson's disease. Neurotherapeutics. 2009;6:141–51. doi: 10.1016/j.nurt.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun H, Scott DO. Structure-based drug metabolism predictions for drug design. Chem Biol Drug Des. 2010;75:3–17. doi: 10.1111/j.1747-0285.2009.00899.x. [DOI] [PubMed] [Google Scholar]
- 17.Azam F, Prasad MV, Thangavel N, Ali HI. Molecular docking studies of 1-(substituted phenyl)-3-(naphtha[1,2-d]thiazol-2-yl) urea/thiourea derivatives with human adenosine A2A receptor. Bioinformation. 2011;6:330–4. doi: 10.6026/97320630006330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller CE, Geis U, Hipp J, Schobert U, Frobenius W, Pawlowski M, et al. Synthesis and structure-activity relationship of 3,7-dimethyl- 1-propargylxanthine derivatives, A2A-selective adenosine receptor antagonists. J Med Chem. 1997;40:4396–405. doi: 10.1021/jm970515+. [DOI] [PubMed] [Google Scholar]
- 19.Petzer JP, Steyn S, Castagnoli KP, Chen JF, Schwarzschild MA, Schyf CJ, et al. Inhibition of monoamine oxidase B by selective adenosine A2A receptor antagonists. Bioorg Med Chem. 2003;11:1299–310. doi: 10.1016/s0968-0896(02)00648-x. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson KA, Gallo-Rodriguez C, Melman N, Fischer B, Maillard M, Van Bergen A, et al. Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine receptor antagonists. J Med Chem. 1993;36:1333–42. doi: 10.1021/jm00062a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada J, Koike N, Nonaka H, Shiozaki S, Yanagawa K, Kanda T, et al. Adenosine A2A antagonists with potent anti-cataleptic activity. Bioorg Med Chem Lett. 1997;7:2349–52. [Google Scholar]
- 22.Suzuki F, Shimada J, Koike N, Nakamura J, Shioazaki S, Ichikawa S, et al. Therapeutic agent for Parkinson's disease. United States Patent PN/5484920. 1996 [Google Scholar]
- 23.Pretorius J, Malan SF, Castagnoli N, Jr, Bergh JJ, Petzer JP. Dual inhibition of monoamine oxidase B and antagonism of the adenosine A2A receptor by (E,E)-8-(4-phenylbutadien-1-yl)caffeine analogues. Bioorg Med Chem. 2008;16:8676–84. doi: 10.1016/j.bmc.2008.07.088. [DOI] [PubMed] [Google Scholar]
- 24.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 25.Binda C, Wang J, Pisani L, Caccia C, Carotti A, Salvati P, et al. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. J Med Chem. 2007;50:5848–52. doi: 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- 26.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien YE, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–7. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart JJ. Stewart Computational Chemistry, Version 9.03CS. MOPAC 2009. Available from: http://OpenMOPAC.net .
- 28.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–62. [Google Scholar]
- 29.Zhao Y, Abraham MH, Lee J, Hersey A, Luscombe NC, Beck G, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res. 2002;19:1446–57. doi: 10.1023/a:1020444330011. [DOI] [PubMed] [Google Scholar]
- 30.Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–7. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 31.Lipinski CA, Lombardo L, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 32.Molinspiration Cheminformatics, Bratislava, Slovak Republic. [Last Accessed on 2010 Apr 22]. Available from: http://www.molinspiration.com/services/properties.html .
- 33.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46:2287–303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 34.Sawynok J, Liu XJ. Adenosine in the spinal cord and periphery: Release and regulation of pain. Prog Neurobiol. 2003;69:313–40. doi: 10.1016/s0301-0082(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 35.Yuzlenko O, Kiec-Kononowicz K. Molecular modeling of A1 and A2A adenosine receptors: Comparison of rhodopsin- and β2-adrenergic-based homology models through the docking studies. J Comput Chem. 2008;30:14–32. doi: 10.1002/jcc.21001. [DOI] [PubMed] [Google Scholar]
- 36.Shi Y, Liu X, Gebremedhin D, Falck JR, Harder DR, Koehler RC. Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J Cereb Blood Flow Metab. 2008;28:111–25. doi: 10.1038/sj.jcbfm.9600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Wess J, Van-Rhee AM, Schoneberg T, Jacobson KA. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J Biol Chem. 1995;270:13987–97. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moro S, Deflorian F, Spalluto G, Pastorin G, Cacciari B, Kim SK, et al. Demystifying the three dimensional structure of G protein-coupled receptors (GPCRs) with the aid of molecular modeling. Chem Commun. 2003;24:2949–56. doi: 10.1039/b303439a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, et al. A specific cholesterol binding site is established by the 2.8 Å structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Colibus L, Li M, Binda C, Lustig A, Edmondson DE, Mattevi A. Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc Natl Acad Sci U S A. 2005;102:12684–9. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Insights into the mode of inhibition of human mitochondrial monoamine oxidase B from high-resolution crystal structures. Proc Natl Acad Sci U S A. 2003;100:9750–5. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binda C, Mattevi A, Edmondson DE. Functional role of the “aromatic cage” in human monoamine oxidase B: Structures and catalytic properties of Tyr435 mutant proteins. J Biol Chem. 2002;45:4775–84. doi: 10.1021/bi051847g. [DOI] [PubMed] [Google Scholar]
- 43.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol. 2002;9:22–6. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 44.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–23. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]