

Abstract
Despite decades of research, cerebral malaria remains one of the most serious complications of Plasmodium infection and is a significant burden in Sub-Saharan Africa, where, despite effective antiparasitic treatment, survivors develop long-term neurological sequelae. Although much remains to be discovered about the pathogenesis of cerebral malaria, The American Journal of Pathology has been seminal in presenting original research from both human and experimental models. These studies have afforded significant insight into the mechanism of cerebral damage in this devastating disease. The present review highlights information gleaned from these studies, especially in terms of their contributions to the understanding of cerebral malaria.
Cerebral malaria (CM) is a severe and potentially fatal neurological manifestation of infection with Plasmodium species. P. falciparum, the major causative agent of human CM, accounts annually for approximately one million deaths of children in Sub-Saharan Africa alone.1 Despite effective antimalarial therapy, surviving individuals with CM may develop long-term neurological deficits,2–9 which indicates that elimination of the parasite does not completely resolve clinical consequences of infection. Over the past century, The American Journal of Pathology (AJP) has greatly advanced understanding of the pathogenesis and pathophysiology of human diseases. This review focuses on contributions to knowledge of the neurodegenerative disease CM in particular.
Until recently, observation regarding human CM has been somewhat limited, because it has relied mainly on examination of postmortem samples, which are not always accessible.3 This limitation has made animal models, despite their imperfections, necessary for longitudinal studies of disease pathogenesis.10 Studies with nonhuman primate models demonstrate several pathological features similar to those observed in humans, including sequestration of infected red blood cells (iRBCs) and vascular damage. However, these studies are difficult to conduct, because of their considerable ethical and cost constraints and thus are vastly underutilized.3,11,12 Furthermore, as with all animal models, studies on nonhuman primates present a number of reliability issues.3,4,11 Although the mouse model has many of the same limitations, it nonetheless serves as one of the most widely used animal models for in vivo CM research. Although some differences undoubtedly exist between human and murine CM, several studies highlight the range of similarities, including perturbations in the integrity of the blood-brain barrier (BBB), iRBC sequestration to the cerebral microvasculature, microvascular damage, and persistent cognitive impairment despite successful antimalarial therapy. These similarities support the use of the mouse model to characterize this human disease.10,13
Cerebral microcirculatory disturbances have been implicated as a contributing factor in the pathogenesis of CM. By a process known as sequestration, P. falciparum-infected RBCs (PfRBCs) adhere to the brain endothelium through binding of P. falciparum erythrocyte membrane protein 1 (PfEMP1), a specific cell-surface ligand expressed by iRBCs. There is no evidence to date of PfRBC entry into the brain parenchyma, suggesting that iRBCs remain in the vascular space when sequestered. PfRBC sequestration leads to multiple downstream vascular effects, including increased vasoconstriction, reduced cerebral blood flow, vascular obstruction, and disruption of BBB integrity.1,14,15
The BBB is a selectively permeable structure responsible for regulating ion and nutrient transport into the brain. It serves as a key interface between the central nervous system and the blood, restricting the free flow of physiological molecules between the bloodstream and parenchyma. The BBB is composed of specialized endothelial cells (ECs) that line cerebral blood vessels. These ECs are surrounded by a basal lamina and astrocyte end foot processes16 (Figure 1). Tight junctions are responsible for stitching ECs together for barrier maintenance and integrity, with astrocytes providing additional structural and physical support.16 Together, these cells serve as the gatekeepers of the BBB and help the barrier perform its critical function of maintaining proper brain homeostasis.
Figure 1.

Proposed mechanism of vascular damage and BBB dysfunction in CM. The BBB is composed of ECs (orange), the basal lamina (purple), and astrocyte end feet (blue). Left panel: The vasculopathy in CM is likely a multifactorial process involving an up-regulation of the host inflammatory response. Interaction among PfRBCs, monocytes, and endothelial cells results in the up-regulation of proinflammatory cytokines, including TNF-α and IL-1β, which activates the cerebral endothelium. Middle panel: Additionally, there is recruitment and sequestration of iRBCs, monocytes, and platelets; this leads to mechanical occlusion and potentiates endothelial cell activation, and the up-regulation of cell adhesion molecules (CAMs) to which PfRBCs, immune cells, and platelets can adhere and thus obstruct the blood vessels and disrupt cerebral blood flow. Right panel: These two sets of events are thought to lead to BBB dysfunction. Cells of the BBB are damaged and undergo apoptosis; this allows entry of foreign materials into the brain parenchyma, creating hemorrhagic lesions and edema of the brain.
The BBB is a highly complex structure, consisting of many cell types and possessing multiple functions, and we have simplified both our description and illustration of it (Figure 1) for clarity in this review. Perturbations to the BBB can lead to the passage of potentially harmful substances into the brain, which may subsequently cause disease. Vascular dysfunction with subsequent BBB damage is a major feature of CM17–19 (Figure 1) and has been observed both in pediatric CM and in experimental models. The mechanisms that mediate this vasculopathy are not fully characterized. Over the past 20 years, the damages to the cerebrovasculature and BBB that result from CM have been delineated in several seminal reports in AJP, broadening understanding of this aspect of disease and helping to identify important factors that might serve as future therapeutic targets. The present review focuses on the role of cerebrovascular dysfunction in CM, known mechanisms that mediate this dysfunction, and possible therapeutic options.
Pediatric CM
Of all populations at risk of acquiring malarial disease, children 5 years or younger in Sub-Saharan Africa are most susceptible to developing CM.20 This population is associated with a staggering 90% of CM-related fatalities.20 CM that occurs in young children, also termed pediatric CM, is characterized by impaired consciousness, severe anemia, hypoglycemia, fever, and neurological sequelae.20 It is clinically defined as the presence of P. falciparum parasitemia and coma with no other apparent causes of altered consciousness.21
A Vascular Approach to Diagnosing CM: Retinopathy
The diagnosis of CM is difficult because of the many nonspecific features characteristic of disease. The presence of parasite in comatose patients is a poor indicator of disease, because coma is a known consequence of a variety of neurological syndromes.22 Over the past 30 years, retinopathy has become one of the best diagnostic indicators of both pediatric and adult CM,22,23 because its severity correlates with the hallmark cerebrovascular PfRBC sequestration and because it helps distinguish CM coma from nonmalarial coma.23 The common features of retinopathy, which include whitening of the retina, hemorrhage, and abnormality-induced changes in vessel color, likely result in part from mechanical obstruction due to PfRBC sequestration and reduced perfusion.23 Funduscopic analysis on Malawian children with CM revealed that papilledema (ie, swelling of the optic disk that connects the optic nerve to the retina) is another frequent occurrence during CM and is associated with retinopathy-like characteristics.23 It is not specific to CM, however, because patients with papilledema do not always exhibit PfRBC sequestration to the cerebral vasculature.23 This observation is corroborated by the fact that the case-fatality rate of children with papilledema alone is higher than that of children with malarial retinopathy and papilledema, demonstrating that, like coma, papilledema is a nonspecific outcome of disease, a circumstance that can lead to misdiagnosis and thus to inappropriate treatment.23 The capacity of retinopathy to accurately diagnose CM, along with its high correlation with postmortem histopathological features of the neurological syndrome, establishes the importance of performing a funduscopic examination during evaluation of patients at risk of disease.
Histopathological Features
Postmortem studies of fatal cases of CM have allowed researchers to gain a better understanding of this disease. Dorovini-Zis et al20 demonstrated that, in brains of Malawian children with CM, the degree of PfRBC sequestration and microvascular pathology correlated with the severity of BBB disruption. Areas of fibrinogen extravasation into the brain parenchyma were observed near areas of thrombus, ring hemorrhage, ruptured capillaries, and cerebral blood vessels filled with PfRBCs20 (Figure 2). Additionally, myelin loss and axonal damage were increased in areas of severe vascular damage.20 These findings coincided with previous studies in Vietnamese adults with CM that also demonstrated axonal injury associated with hemorrhage and demyelination.24 Furthermore, patients with PfRBC sequestration and microvascular pathology demonstrated prominent gliosis, in contrast to those without sequestration and/or microvascular damage.24
Figure 2.

Patterns of BBB compromise in fatal pediatric CM. A and B: In young children with CM, fibrinogen leakage (arrows), signaling BBB breakdown, is observed immediately adjacent to capillaries densely packed with PfRBCs and leukocyte infiltration (arrowheads). C and D: Fibrinogen leakage (arrows) is also seen juxtaposed to a thrombosed vessel (C, arrowhead) and in ring hemorrhages in the white matter (D). Thrombosis and hemorrhage are two prominent features of CM.
Reprinted from Dorovini-Zis et al20 with permission of Elsevier.
The tight junctions that stitch ECs together are important for maintaining BBB integrity. Studies on Malawian children with CM revealed focal loss of EC tight junction proteins, such as zonular occludin-1 (ZO-1), occludin, and vinculin.25 Areas of loss of these tight junctions colocalized with areas of PfRBC sequestration.25 Loss of tight junctions likely contributes to the breakdown of the BBB and the subsequent downstream consequences that occur during CM.
Vasculopathy
Although the mechanisms of cerebrovascular dysfunction in CM remain incompletely characterized, several factors have been implicated, including increased levels of the vasoactive peptide endothelin-1 (ET-1), as well as the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ).1,26,27 Hyperinflammation has been observed in a murine model of CM, characterized by systemic activation of several cell types (ie, CD4+ and CD8+ T cells, macrophages, and platelets).28–31 Murine CM results from infection of mice with the mouse malarial strain P. berghei-ANKA (PbA); this model is here referred to as experimental CM (ECM). Enhanced expression of ET-1, TNF-α, and IFN-γ activates the endothelium, inducing increased recruitment of iRBCs, leukocytes, and platelets, as well as up-regulation of several endothelial cell adhesion molecules (CAMs), including intracellular adhesion molecule-1 (ICAM-1), vascular adhesion molecule-1 (VCAM-1), and E-selectin (CD62E).1,32,33
Cell Adhesion Molecules
Up-regulation of CAMs increases the potential for binding of recruited cells to the endothelial membrane, an event that results in cerebral microvascular sequestration.32,34 In this regard, ICAM-1 (CD54) and CD36 are two major binding partners for the PfEMP1 protein on PfRBCs and thus contribute to PfRBC sequestration. Enhanced PfRBC sequestration leads to vascular obstruction, hypoperfusion, and hypo-oxygenation, which are hallmark features of such characteristic CM conditions as acidosis, hypoxia, and ischemia.30,32,34,35 Sequestration results in both damage to and apoptosis of ECs and renders the BBB susceptible to dysfunction, thus increasing the possibility of vascular leakage into the brain tissue.32 Such vascular leakage likely contributes to the neuronal damage and brain edema observed during disease.32 Enhanced up-regulation of CAMs also increases the potential for adhesion of other inflammatory cell types to the endothelial surface, including leukocytes and platelets. Sequestration leads to the uncontrolled release of microparticles and inflammatory cytokines from ECs. This dysregulated inflammatory cascade further exacerbates BBB damage and permeability, increasing the likelihood of the hemorrhage characteristic of disease. In some cases of severe ECM, parasite-induced EC damage resulted in complete vessel collapse.14
Significant increases in both CAM expression and TNF-α and IL-1β levels have been observed in postmortem brains of children with CM, especially in the cerebellum.36 Because the cerebellum is important for motor coordination and for the learning of motor skills, damage to this brain region may contribute to the subsequent neurocognitive and motor coordination deficits commonly observed in children with CM.
Tripathi et al37 found that exposure of human brain microvascular ECs to PfRBCs induced the expression of ICAM-1. This is consistent with the postmortem observations of Turner et al,38 that iRBCs sequestered in the vasculature colocalized with ICAM-1. The importance of ICAM-1 in the development of CM is well established. A study by Favre et al39 showed that ICAM-1-deficient mice were protected from CM. Although PbA-infected wild-type mice survived to 6 to 8 days after infection, ICAM-1-deficient mice demonstrated a resistance phenotype, surviving more than 15 days after infection.39 Furthermore, ICAM-1-deficient mice exhibited decreased serum TNF-α levels, no apparent BBB breakdown, and no macrophage sequestration in the cerebral blood vessels, in contrast to wild-type infected mice.39 These findings demonstrate that ICAM-1 up-regulation during CM may be associated with macrophage and iRBC entrapment, as well as with immune dysregulation.
Angiopoietin-Tie-2 Signaling
CM has been shown to result in dysregulation of other elements of the cerebral vasculature, including the angiopoietin-Tie-2 system, a key regulator of EC function and vascular integrity.33 Under normal conditions, angiopoietin-1 (Ang-1) regulates the survival and activation of ECs through interactions with the EC receptor Tie-2, whereas angiopoietin-2 (Ang-2) opposes the effects of Ang-1.33 The antagonistic properties of Ang-2 sensitizes the endothelium to inflammation, which then contributes to increased expression of ICAM-1, to which PfRBCs and immune cells can adhere. Interestingly, in cases of severe malaria, low levels of Ang-1 and high levels of Ang-2 have been observed to correlate with the severity of malarial infection.33,40–42 Conroy et al33 examined brains of children with CM to determine whether a correlation exists between peripheral blood plasma levels of Ang-1, Ang-2, and the soluble form of Tie-2 (sTie-2) and retinopathy and mortality. Increased levels of Ang-2 and sTie-2, which binds angiopoietins to regulate their function, were associated with fatal cases; these molecules thus have potential as key biomarkers of disease.33 Retinopathy-positive CM children exhibited decreased Ang-1 and increased Ang-2 and sTie-2, compared with retinopathy-negative children with malaria.33 Ang-2 and sTie-2 levels correlated with the severity of retinopathy in children with CM.33 Interestingly, in retinopathy-positive CM children, Ang-2 was significantly higher in fatal cases.33
Inflammation
A hyperactive immune response is one of the major contributors to CM vasculopathy. Dysregulation of TNF-α is most commonly associated with disease.43 Circulating levels of this inflammatory cytokine are significantly higher in ECM mice, compared with mice that do not develop the neurological syndrome.43 TNF-α is thought to induce activation of ECs, thereby up-regulating expression of several CAMs, including ICAM-1 (CD54), CD36, P-selectin (CD62P), and VCAM-1 (CD106).43,44 LMP-420, a transcriptional inhibitor of TNF-α, has been shown to reduce TNF-α-mediated EC activation, ICAM-1 and VCAM-1 up-regulation, and PfRBC cytoadherence in an in vitro CM model.45 This molecule also inhibits lymphotoxin-α (LT-α; alias TNF-β), a TNF receptor 2 (TNFR2) ligand that is up-regulated in the sera of human malaria patients, as well as in the brains of mice with ECM.43,45 Interestingly, studies showed that LT-α-deficient mice were protected from developing ECM, but blocking TNF-α alone did not protect mice from developing disease. These findings point to a more specific role for LT-α in pathogenesis.43 PbA-infected mice lacking LT-α demonstrated no perivascular hemorrhage; these mice regulated ICAM-1 expression and exhibited resistance to ECM-induced early death.46 LT-α binds TNFR2 on ECs to induce its downstream inflammatory effects, as demonstrated by the observation that blocking this receptor also prevented mice from developing ECM.43 Additionally, the soluble form of TNFR2 is elevated in both human and murine CM; however, the importance of this for disease pathology is unclear.10,43
Another inflammatory mediator associated with CM is platelet-activating factor (PAF). PAF is an important factor in leukocyte recruitment and activation, cytokine/chemokine release, and vascular permeability.47,48 PAF signals through its cognate receptor, platelet-activating factor receptor (PAFR), to perform its functions. In a recent study by Teixeira and colleagues,47 PbA-infected mice lacking PAFR exhibited survival benefits, with reduced cerebrovascular occlusion and brain hemorrhage. ECM mice lacking PAFR also demonstrated a decrease in cell apoptosis, BBB breakdown, and inflammation.
Beyond Inflammation: Cerebral Microcirculation and Vasomodulatory Agents
In recent years, it has become apparent that alteration of the cerebral microcirculation is an important factor in the pathogenesis of CM. The cerebral microvasculature is essential for central nervous system homeostasis and normal brain activity. Breach of the BBB during CM, once thought to be primarily a manifestation of ECM, is increasingly recognized as a contributor to the development of human CM.20,49 During disease, sequestered PfRBCs interact closely with the cerebrovascular lining, enhancing permeability, endothelial activation, and vascular obstruction and thus contributing to cerebral microcirculatory alterations.17,18,50 Nonetheless, the role of vascular dysfunction in CM remains poorly understood, and this paucity of knowledge hampers development of adjunctive therapies targeting vasculopathy.
Endothelin-1
Studies of murine ECM demonstrate elevated levels of ET-1, a powerful vasoconstrictor, in whole brains of PbA-infected mice.26 These findings correlate with decreased cerebral blood flow and immunopathology.15,26 Enhanced expression of ET-1 is known to result in activation of the endothelium, thereby up-regulating CAM expression,26 which suggests that ET-1 may be involved in potentiating the increased recruitment of iRBCs, leukocytes, and platelets to the cerebral vasculature.51 These events may enhance sequestration, leading to vascular obstruction, hypoperfusion and hypo-oxygenation, and eventually ischemia.
NO and Vascular Dysfunction
The role of nitric oxide (NO) during CM development has been a matter of great debate. Although NO has been primarily associated with protection against CM, studies reveal that it may also contribute to pathogenesis. Elevated levels of NO can result in weight loss, interfere with neurotransmission, and contribute to intracranial pressure through vasodilatory effects.52 Conversely, depleted levels of the agent have been linked to endothelial dysfunction and damage in CM.53,54 Several factors may contribute to NO depletion during disease, including both hypoargininemia (leading to decreased NO production by central nervous system NO synthases) and the NO-scavenging activity of cell-free hemoglobin, which results from iRBC destruction.35,53 Through a mechanism similar to that underlying the effects of ET-1, TNF-α, and IFN-γ on CM pathogenesis, decreased NO is thought to mediate vascular obstruction and reduction of cerebral blood flow via endothelial activation and increased leukocyte and platelet sequestration.27,53,55–61 Hemorrhage from vessel collapse has also been associated with deficits in NO.1,14,27,53
Carvalho and colleagues27 demonstrated that treatment of PbA-infected mice with NO reduced endothelial activation and the ensuing up-regulation of ICAM-1, VCAM-1, and P-selectin. Reduced up-regulation of these cell-surface proteins correlated with reduced sequestration of leukocytes and platelets to the brain endothelial lining, which subsequently induced vasodilation, resulting in improved microvascular blood flow and decreased potential for vascular leakage and subsequent hemorrhage.27,53 In Asian adult CM patients, as well as in PbA-infected mice, treatment with exogenous arginine has been shown to increase NO production and subsequently ameliorate endothelial function, thus reducing CM-associated BBB rupture.53,54,62,63
Neuroimaging
Neuroimaging has become an important tool in elucidating the underlying mechanisms of CM. Various brain scanning modalities have been used to enhance the clinical characterizations of disease, including magnetic resonance imaging (MRI), computerized tomography (CT), positron emission tomography (PET), and intravital microscopy. Imaging studies have allowed malaria researchers to look beyond postmortem samples and to perform longitudinal studies, thus improving understanding of the development of the neurological syndrome.
Visualization Through MRI and CT
MRI is a powerful diagnostic tool for monitoring and understanding key features involved in the progression of central nervous system diseases such as CM. Using this noninvasive imaging device in ECM models, researchers have demonstrated that ischemia and edema are complications associated with murine CM.19 For example, MRI studies have demonstrated cranial nerve damage as an initial lesion of ECM, depicting areas of axonal damage and swelling of the trigeminal and optic nerves.19 MRI also demonstrated decreased cerebral blood flow, as well as neuronal and axonal injury, in mice infected with PbA.15 Such applications of MRI allow for more thorough longitudinal studies, which are difficult to achieve with humans.
Imaging techniques have been used in the setting of human CM. For example, one group performed head CT scanning on children diagnosed with CM.22 Images obtained from children with prolonged coma demonstrated large vessel infarcts with diffuse cerebral swelling in the brainstem, whereas survivors of CM with neurological sequelae demonstrated areas of atrophy corresponding to regions affected by seizures.22 Recent evidence indicates that retinopathy-positive children with CM have markedly increased cerebral volume, with abnormal T2 signal intensity in the basal ganglia, white matter, and corpus callosum, compared to retinopathy-negative children with CM.64 These findings highlight the advantage of incorporating these imaging modalities in the study of the pathogenesis of this neurodegenerative syndrome.
FDG-PET: Imaging Brain Metabolism
Perhaps as a result of the cerebral microvascular disruptions associated with CM, cerebral metabolic activity is found to be altered as disease progresses. Fluorodeoxyglucose positron emission tomography (FDG-PET) is a noninvasive imaging tool used to quantify metabolic activity in various organs, including the brain. FDG is a glucose analog that is taken up by cells but is not metabolized, allowing researchers to visualize its uptake in vivo as disease progresses. Sugiyama et al65 demonstrated decreased cerebral metabolic activity in a nonhuman primate model of ECM by FDG-PET. They observed a diffuse and heterogeneous reduction of metabolic activity in the frontal and temporal lobes before any evidence of neuropathological findings, suggesting that cerebral metabolic changes occur before parenchymal damage in primate ECM models.65 Diffuse reduction in activity may result from impaired circulation due to sequestration.65
Intravital Microscopy
Intravital microscopy enables studies to go beyond brain histochemical analyses, viewing the live brain through a cranial window. This technique allows for long-term imaging of a single area in the brain for comparison of histopathological alterations and behavioral performances with microvascular changes.66 Intravital microscopy has emerged as an important tool in determining the underlying pathological features contributing to ECM. Carvalho and colleagues used this modality to explore the cerebral microvasculature during disease progression (Figure 3).14,27,53 One of their intravital microscopic studies revealed increased leukocyte adhesion and rolling in brains of PbA-infected mice, which resulted in reduced cerebral blood flow, arteriole diameter, and RBC velocity.14
Figure 3.
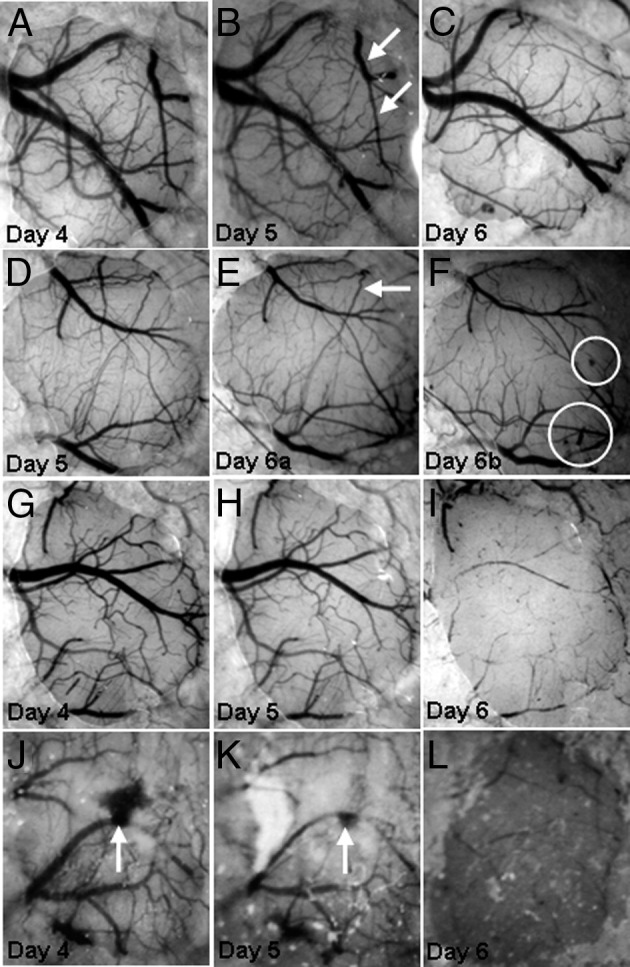
The murine model ECM is associated with vascular collapse. Mice were infected with P. berghei-ANKA strain. A–C: In mice with ECM, collapse of a major vessel and branches (arrows, B) is observed on day 6 of infection. D–F: There is collapse of two branches (arrow, E) of a vessel and microhemorrhages (circles, F). Vessel collapse occurred between two observations on the same day (day 6 of infection); the mouse developed clinical CM during the interval between observations. G–I: Collapse of virtually the entire vascular network is observed on day 6 of infection. J–L: Collapse of the vascular network on day 6 after a major hemorrhage (arrows, J and K) on days 4 and 5 of infection.
Reprinted from Cabrales et al14 with permission of Elsevier.
Intravital microcopy was used to visualize the microvasculature after administration of a calcium channel blocker nimodipine to PbA-infected mice. Nimodipine not only reversed the cerebral vascular disturbances during infection, but also improved survival and motor coordination.14 Intravital microscopy was also used to demonstrate the benefits of treating ECM with exogenous NO, which reduced leukocyte and platelet adherence and also reduced BBB permeability.27,53 The importance of intravital imaging is evident in its ability to visualize in vivo brain events that will contribute to a greater understanding of microcirculatory hemodynamics and vascular pathology during the pathogenesis of CM. This tool can also allow researchers to visually assess the function of specific vascular genes through knock-out studies, and to determine the efficacy of various therapeutic treatments.
Looking Forward
Bridging the Gap: Human Disease versus Experimental Models
Despite controversy regarding their validity, animal models have greatly improved understanding of the disease process of CM and its pathogenesis. For instance, findings from mouse studies demonstrating the diagnostic value of MRI, and its potential for therapeutic applications, have been an important rationale for the increased use of MRI in patients with cerebral malaria in endemic areas.64 In addition, recent investigations in Ghanaian children have deepened understanding of the role of endothelial progenitor cells in the maintenance of the cerebrovasculature during infection, as well as of the involvement of these cells in the development of CM. These findings corroborate evidence from animal models delineating the role of these cells in studies involving endothelial cell injury.67 Although only a few other recent investigations demonstrate correlation between experimental models and human CM,18,20,24,49 such studies underscore the need for further research linking the two, as well as the need for collaborations between those who investigate human CM and those who study experimental models, to further understanding of the disease and to derive adjuvant therapies that may be neuroprotective.
Adjunctive Therapies: Repairing the Cerebrovasculature
Although early adjuvant therapy is the subject of ongoing debate,12 agents targeting the endothelium have been established as neuroprotective in animal models and may also prove to be important both in decreasing mortality with CM and decreasing morbidity in survivors who experience persistent neurocognitive impairment after the disease. In this regard, compounds that function in the regulation and repair of cerebrovascular dysfunction hold promise for both mechanistic discoveries and therapeutic options for the treatment of CM. The peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist rosiglitazone, the calcium-channel blocker nimodipine, and erythropoietin fall into this category of cerebrovascular modulators, and preliminary evidence demonstrates that these agents decrease mortality due to ECM.1,68,69 These compounds mediate their effects primarily by increasing levels of progenitor ECs and inducing their differentiation.1,70 Because damage to the endothelium is critical to the development of CM, restoring normal levels of ECs could ameliorate or prevent negative outcomes of this neurodegenerative disease.
Calcium-Channel Blockade
Mice with ECM that received both the calcium-channel blocker nimodipine and the antimalarial agent artemether showed a marked improvement in vascular function, characterized by increased vasodilation/vasorelaxation and enhanced cerebral blood flow.1,14 This demonstrates a possible role for dysregulated calcium flux in the pathogenesis of CM. Combination treatment with artemether and the calcium-channel blocker also improved neurocognitive function in mice with disease.1,14
Peroxisome Proliferator-Activated Receptors
The balance between inflammation and immunosuppression is critical to a successful defense against infectious agents. Given that CM is a syndrome characterized by hyperinflammation, regulating the host immune response may ameliorate disease pathology. PPARs have several immunoregulatory properties, including suppression of both the expression of specific CAMs and Toll-like receptors and the secretion of various chemokines; PPARs thus hold promise for reversing the deleterious effects of CM-associated inflammation.71,72 Although knowledge of the role of PPARs in CM is limited, agonists have been shown to ameliorate many of the features typically found during disease in ECM by, for example, regulating CAM activation, the secretion of inflammatory cytokines, and the recruitment of leukocytes to sites of inflammation.71 PPARs also possess neuroprotective capabilities, an exciting characteristic in the face of the neuronal damage and neurological deficits that normally result from CM.71
Targeting Hypoxia
Oxygen is essential for normal cellular function, homeostasis and metabolism.73,74 Inadequate oxygenation of tissues can lead to hypoxia during CM.73 Therapies that reverse hypoxia during disease may thus be beneficial to patients. Oxygen treatment of mice with ECM has been shown to significantly reduce disease pathology by improving survival and BBB integrity and by reducing parasitemia, leukocyte sequestration, and mRNA levels of TNF-α and IFN-γ.48,73,75 Grau and colleagues73 demonstrated that treatment of CM-susceptible mice with the hypoxia-responsive hormone erythropoietin (EPO) both reduced the number of hypoxia-positive central nervous system cells and restored body temperature to normal levels. In the same report, they also corroborated a previous finding on the survival benefits of EPO in ECM.73,76 Additionally, EPO has been shown to decrease neuronal apoptosis and regulate expression of TNF-α, IFN-γ, and IL-1β in ECM, thus demonstrating a simultaneous neuroprotective and immunoregulatory role, which further establishes EPO as a possible vasoregulatory therapeutic for CM.76
Endothelin-1
A recent report by Dai et al77 demonstrated the role of endothelin on the vasculopathy of cerebral malaria via actions through the endothelin receptor type A (ETA). These investigators demonstrated that blockade of the ETA receptor not only significantly reduced hemorrhage in the brain, it seemed also to synergistically result in a survival benefit when used as an adjunct to artemisinin. This finding highlights the fact that aborting activation of the endothelium and properly maintaining endothelial homeostasis are vital in preventing downstream vascular and BBB damage in ECM.
Conclusions
Over the decades, AJP has been an important contributor in advancing understanding of CM pathogenesis, especially in the area of vasculopathy. We celebrate the contributions of individuals such as Terrie E. Taylor, Katerina Dorovini-Zis, Leonardo José de Moura Carvalho, Isabelle M. Medana, Georges E.R. Grau, and countless others to the Journal. Through their work and that of others, we now have a greater appreciation of the pathogenesis of CM in both humans and mice. Dorovini-Zis, Taylor, and colleagues20 provided a comprehensive postmortem analysis of brains from Malawian children with CM, which demonstrated the correlation between PfRBC sequestration to the cerebrovasculature and such pathological consequences as BBB disruption, hemorrhage, damage to neuronal axons, demyelination, vascular thrombosis, and monocyte accumulation. Carvalho and colleagues14 characterized the damage to the cerebrovascular system which occurs in ECM, specifically emphasizing the constricting effects on vessel diameter and the reduction in blood flow which results, in part, from sequestration of accumulating leukocytes. Their work not only elucidated potential vascular components to target therapeutically, such as calcium flux, but also highlighted the value of intravital microscopy as an imaging tool to study this neurological syndrome.14 Grau and colleagues73 further established hypoxia as one of the major complications which results from ECM and illuminated the hypoxia-reversing hormone EPO as another possible therapeutic with which to treat disease. Publication in AJP of work by these prominent scientists, and by many others, exemplifies the role of the Journal as a major conduit for changing concepts of the pathogenesis of CM. Such contributions will, one can reasonably hope, eventually lead to improved therapy.
Acknowledgments
We thank Dr. David C. Spray and Minxian Dai for their assistance with collegial conversation and constructive review of the manuscript.
Footnotes
Supported by the NIH (Training Grant in Molecular Neuropathology T32 NS007098-31 to H.J.S; NS069577 to M.S.D.; AI076248 to H.B.T.), the Burroughs-Wellcome Fund (Career Awards for Medical Scientists to M.S.D.), and the Einstein-Montefiore Institute for Clinical and Translational Research (Career Development Award to M.S.D. and B.F.).
H.J.S. and B.F. contributed equally to this work.
M.P.L. is the current Editor-in-Chief, and H.B.T. is the current Senior Associate Editor of The American Journal of Pathology.
A guest editor acted as editor-in-chief for this manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition of this article.
References
- 1.Desruisseaux M.S., Machado F.S., Weiss L.M., Tanowitz H.B., Golightly L.M. Cerebral malaria: a vasculopathy. Am J Pathol. 2010;176:1075–1078. doi: 10.2353/ajpath.2010.091090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desruisseaux M.S., Gulinello M., Smith D.N., Lee S.C., Tsuji M., Weiss L.M., Spray D.C., Tanowitz H.B. Cognitive dysfunction in mice infected with Plasmodium berghei strain ANKA. J Infect Dis. 2008;197:1621–1627. doi: 10.1086/587908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newton C.R., Krishna S. Severe falciparum malaria in children: current understanding of pathophysiology and supportive treatment. Pharmacol Ther. 1998;79:1–53. doi: 10.1016/s0163-7258(98)00008-4. [DOI] [PubMed] [Google Scholar]
- 4.Lou J., Lucas R., Grau G.E. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin Microbiol Rev. 2001;14:810–820. doi: 10.1128/CMR.14.4.810-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boivin M.J., Bangirana P., Byarugaba J., Opoka R.O., Idro R., Jurek A.M., John C.C. Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics. 2007;119:e360–e366. doi: 10.1542/peds.2006-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boivin M.J. Effects of early cerebral malaria on cognitive ability in Senegalese children. J Dev Behav Pediatr. 2002;23:353–364. doi: 10.1097/00004703-200210000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Carter J.A., Mung'ala-Odera V., Neville B.G., Murira G., Mturi N., Musumba C., Newton C.R. Persistent neurocognitive impairments associated with severe falciparum malaria in Kenyan children. J Neurol Neurosurg Psychiatry. 2005;76:476–481. doi: 10.1136/jnnp.2004.043893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al Serouri A.W., Grantham-McGregor S.M., Greenwood B., Costello A. Impact of asymptomatic malaria parasitaemia on cognitive function and school achievement of schoolchildren in the Yemen Republic. Parasitology. 2000;121:337–345. doi: 10.1017/s0031182099006502. [DOI] [PubMed] [Google Scholar]
- 9.Kihara M., Carter J.A., Newton C.R. The effect of Plasmodium falciparum on cognition: a systematic review. Trop Med Int Health. 2006;11:386–397. doi: 10.1111/j.1365-3156.2006.01579.x. [DOI] [PubMed] [Google Scholar]
- 10.Hunt N.H., Grau G.E. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends in immunology. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 11.Martin C. Experimental use of nonhuman primates is not a simple problem. Nat Med. 2008;14:1011–1013. doi: 10.1038/nm1008-1011a. [DOI] [PubMed] [Google Scholar]
- 12.Langhorne J., Buffet P., Galinski M., Good M., Harty J., Leroy D., Mota M.M., Pasini E., Rénia L., Riley E., Stins M., Duffy P. The relevance of non-human primate and rodent malaria models for humans. Malar J. 2011;10:23. doi: 10.1186/1475-2875-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor-Robinson A.W. Validity of modelling cerebral malaria in mice: argument and counter argument. J Neuroparasitol. 2010;1:1–5. [Google Scholar]
- 14.Cabrales P., Zanini G.M., Meays D., Frangos J.A., Carvalho L.J.M. Murine cerebral malaria is associated with a vasospasm-like microcirculatory dysfunction, and survival upon rescue treatment is markedly increased by nimodipine. Am J Pathol. 2010;176:1306–1315. doi: 10.2353/ajpath.2010.090691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kennan R.P., Machado F.S., Lee S.C., Desruisseaux M.S., Wittner M., Tsuji M., Tanowitz H.B. Reduced cerebral blood flow and N-acetyl aspartate in a murine model of cerebral malaria. Parasitol Res. 2005;96:302–307. doi: 10.1007/s00436-005-1349-z. [DOI] [PubMed] [Google Scholar]
- 16.Abbott N.J., Rönnbäck L., Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 17.Rénia L., Howland S.W., Claser C., Gruner A.C., Suwanarusk R., Teo T.H., Russell B., Ng L.F.P. Cerebral malaria: mysteries at the blood-brain barrier. Virulence. 2012;3:193–201. doi: 10.4161/viru.19013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medana I.M., Turner G.D. Human cerebral malaria and the blood-brain barrier. Int J Parasitol. 2006;36:555–568. doi: 10.1016/j.ijpara.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Penet M.F., Viola A., Confort-Gouny S., Le Fur Y., Duhamel G., Kober F., Ibarrola D., Izquierdo M., Coltel N., Gharib B., Grau G.E., Cozzone P.J. Imaging experimental cerebral malaria in vivo: significant role of ischemic brain edema. J Neurosci. 2005;25:7352–7358. doi: 10.1523/JNEUROSCI.1002-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorovini-Zis K., Schmidt K., Huynh H., Fu W., Whitten R.O., Milner D., Kamiza S., Molyneux M., Taylor T.E. The neuropathology of fatal cerebral malaria in Malawian children. Am J Pathol. 2011;178:2146–2158. doi: 10.1016/j.ajpath.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.2000. Severe falciparum malaria: World Health Organization, Communicable Diseases Cluster, Transactions of the Royal Society of Tropical Medicine and Hygiene. 94 Suppl 1:S1-S90. [PubMed] [Google Scholar]
- 22.Potchen M.J., Birbeck G.L., Demarco J.K., Kampondeni S.D., Beare N., Molyneux M.E., Taylor T.E. Neuroimaging findings in children with retinopathy-confirmed cerebral malaria. Eur J Radiol. 2010;74:262–268. doi: 10.1016/j.ejrad.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beare N.A., Lewallen S., Taylor T.E., Molyneux M.E. Redefining cerebral malaria by including malaria retinopathy. Future Microbiol. 2011;6:349–355. doi: 10.2217/fmb.11.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medana I.M., Day N.P., Hien T.T., Mai N.T., Bethell D., Phu N.H., Farrar J., Esiri M.M., White N.J., Turner G.D. Axonal injury in cerebral malaria. Am J Pathol. 2002;160:655–666. doi: 10.1016/S0002-9440(10)64885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown H., Rogerson S., Taylor T., Tembo M., Mwenechanya J., Molyneux M., Turner G. Blood-brain barrier function in cerebral malaria in Malawian children. Am J Trop Med Hyg. 2001;64:207–213. doi: 10.4269/ajtmh.2001.64.207. [DOI] [PubMed] [Google Scholar]
- 26.Machado F.S., Desruisseaux M.S., Nagajyothi, Kennan R.P., Hetherington H.P., Wittner M., Weiss L.M., Lee S.C., Scherer P.E., Tsuji M., Tanowitz H.B. Endothelin in a murine model of cerebral malaria. Exp Biol Med (Maywood) 2006;231:1176–1181. [PubMed] [Google Scholar]
- 27.Zanini G.M., Cabrales P., Barkho W., Frangos J.A., Carvalho L.J. Exogenous nitric oxide decreases brain vascular inflammation, leakage and venular resistance during Plasmodium berghei ANKA infection in mice. J Neuroinflammation. 2011;8:66. doi: 10.1186/1742-2094-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grau G.E., Piguet P.F., Engers H.D., Louis J.A., Vassalli P., Lambert P.H. L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. J Immunol. 1986;137:2348–2354. [PubMed] [Google Scholar]
- 29.Belnoue E., Kayibanda M., Vigario A.M., Deschemin J.C., van Rooijen N., Viguier M., Snounou G., Rénia L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol. 2002;169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 30.Yañez D.M., Manning D.D., Cooley A.J., Weidanz W.P., van der Heyde H.C. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol. 1996;157:1620–1624. [PubMed] [Google Scholar]
- 31.van der Heyde H.C., Nolan J., Combes V., Gramaglia I., Grau G.E. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Bauer P.R., van der Heyde H.C., Sun G., Specian R.D., Granger D.N. Regulation of endothelial cell adhesion molecule expression in an experimental model of cerebral malaria. Microcirculation. 2002;9:463–470. doi: 10.1038/sj.mn.7800159. [DOI] [PubMed] [Google Scholar]
- 33.Conroy A.L., Glover S.J., Hawkes M., Erdman L.K., Seydel K.B., Taylor T.E., Molyneux M.E., Kain K.C. Angiopoietin-2 levels are associated with retinopathy and predict mortality in Malawian children with cerebral malaria: a retrospective case-control study. Crit Care Med. 2012;40:952–959. doi: 10.1097/CCM.0b013e3182373157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beare N.A., Harding S.P., Taylor T.E., Lewallen S., Molyneux M.E. Perfusion abnormalities in children with cerebral malaria and malarial retinopathy. J Infect Dis. 2009;199:263–271. doi: 10.1086/595735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gramaglia I., Sobolewski P., Meays D., Contreras R., Nolan J.P., Frangos J.A., Intaglietta M., van der Heyde H.C. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–1422. doi: 10.1038/nm1499. [DOI] [PubMed] [Google Scholar]
- 36.Armah H., Dodoo A.K., Wiredu E.K., Stiles J.K., Adjei A.A., Gyasi R.K., Tettey Y. High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Ann Trop Med Parasitol. 2005;99:629–647. doi: 10.1179/136485905X51508. [DOI] [PubMed] [Google Scholar]
- 37.Tripathi A.K., Sullivan D.J., Stins M.F. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262–3270. doi: 10.1128/IAI.01625-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner G.D., Morrison H., Jones M., Davis T.M., Looareesuwan S., Buley I.D., Gatter K.C., Newbold C.I., Pukritayakamee S., Nagachinta B., White N.J., Berendt A.R. An immunohistochemical study of the pathology of fatal malaria: Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am J Pathol. 1994;145:1057–1069. [PMC free article] [PubMed] [Google Scholar]
- 39.Favre N., Da Laperousaz C., Ryffel B., Weiss N.A., Imhof B.A., Rudin W., Lucas R., Piguet P.F. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect. 1999;1:961–968. doi: 10.1016/s1286-4579(99)80513-9. [DOI] [PubMed] [Google Scholar]
- 40.Yeo T.W., Lampah D.A., Gitawati R., Tjitra E., Kenangalem E., Piera K., Price R.N., Duffull S.B., Celermajer D.S., Anstey N.M. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci USA. 2008;105:17097–17102. doi: 10.1073/pnas.0805782105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lovegrove F.E., Tangpukdee N., Opoka R.O., Lafferty E.I., Rajwans N., Hawkes M., Krudsood S., Looareesuwan S., John C.C., Liles W.C., Kain K.C. Serum angiopoietin-1 and -2 levels discriminate cerebral malaria from uncomplicated malaria and predict clinical outcome in African children. PLoS One. 2009;4:e4912. doi: 10.1371/journal.pone.0004912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conroy A.L., Lafferty E.I., Lovegrove F.E., Krudsood S., Tangpukdee N., Liles W.C., Kain K.C. Whole blood angiopoietin-1 and -2 levels discriminate cerebral and severe (non-cerebral) malaria from uncomplicated malaria. Malar J. 2009;8:295. doi: 10.1186/1475-2875-8-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haldar K., Murphy S.C., Milner D.A., Taylor T.E. Malaria: mechanisms of erythrocytic infection and pathological correlates of severe disease. Annu Rev Pathol. 2007;2:217–249. doi: 10.1146/annurev.pathol.2.010506.091913. [DOI] [PubMed] [Google Scholar]
- 44.Wassmer S.C., Moxon C.A., Taylor T., Grau G.E., Molyneux M.E., Craig A.G. Vascular endothelial cells cultured from patients with cerebral or uncomplicated malaria exhibit differential reactivity to TNF. Cell Microbiol. 2011;13:198–209. doi: 10.1111/j.1462-5822.2010.01528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wassmer S.C., Cianciolo G.J., Combes V., Grau G.E. Inhibition of endothelial activation: a new way to treat cerebral malaria? PLoS Med. 2005;2:e245. doi: 10.1371/journal.pmed.0020245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engwerda C.R., Mynott T.L., Sawhney S., De Souza J.B., Bickle Q.D., Kaye P.M. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J Exp Med. 2002;195:1371–1377. doi: 10.1084/jem.20020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacerda-Queiroz N., Rodrigues D.H., Vilela M.C., Rachid M.A., Soriani F.M., Sousa L.P., Campos R.D., Quesniaux V.F., Teixeira M.M., Teixeira A.L. Platelet-activating factor receptor is essential for the development of experimental cerebral malaria. Am J Pathol. 2012;180:246–255. doi: 10.1016/j.ajpath.2011.09.038. [DOI] [PubMed] [Google Scholar]
- 48.Blanco Y.C., Farias A.S., Goelnitz U., Lopes S.C., Arrais-Silva W.W., Carvalho B.O., Amino R., Wunderlich G., Santos L.M., Giorgio S., Costa F.T. Hyperbaric oxygen prevents early death caused by experimental cerebral malaria. PLoS One. 2008;3:e3126. doi: 10.1371/journal.pone.0003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams S., Brown H., Turner G. Breaking down the blood-brain barrier: signaling a path to cerebral malaria? Trends Parasitol. 2002;18:360–366. doi: 10.1016/s1471-4922(02)02353-x. [DOI] [PubMed] [Google Scholar]
- 50.Grab D.J., Chakravorty S.J., van der Heyde H., Stins M.F. How can microbial interactions with the blood-brain barrier modulate astroglial and neuronal function? Cell Microbiol. 2011;13:1470–1478. doi: 10.1111/j.1462-5822.2011.01661.x. [DOI] [PubMed] [Google Scholar]
- 51.McCarron R.M., Wang L., Stanimirovic D.B., Spatz M. Endothelin induction of adhesion molecule expression on human brain microvascular endothelial cells. Neurosci Lett. 1993;156:31–34. doi: 10.1016/0304-3940(93)90432-k. [DOI] [PubMed] [Google Scholar]
- 52.Clark I.A., Rockett K.A., Cowden W.B. Proposed link between cytokines, nitric oxide and human cerebral malaria. Parasitol Today. 1991;7:205–207. doi: 10.1016/0169-4758(91)90142-b. [DOI] [PubMed] [Google Scholar]
- 53.Cabrales P., Zanini G.M., Meays D., Frangos J.A., Carvalho L.J. Nitric oxide protection against murine cerebral malaria is associated with improved cerebral microcirculatory physiology. J Infect Dis. 2011;203:1454–1463. doi: 10.1093/infdis/jir058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeo T.W., Lampah D.A., Gitawati R., Tjitra E., Kenangalem E., McNeil Y.R., Darcy C.J., Granger D.L., Weinberg J.B., Lopansri B.K., Price R.N., Duffull S.B., Celermajer D.S., Anstey N.M. Impaired nitric oxide bioavailability and L-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007;204:2693–2704. doi: 10.1084/jem.20070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lefer D.J., Jones S.P., Girod W.G., Baines A., Grisham M.B., Cockrell A.S., Huang P.L., Scalia R. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am J Physiol. 1999;276:H1943–H1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- 56.Davenpeck K.L., Gauthier T.W., Lefer A.M. Inhibition of endothelial-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology. 1994;107:1050–1058. doi: 10.1016/0016-5085(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 57.Luvara G., Pueyo M.E., Philippe M., Mandet C., Savoie F., Henrion D., Michel J.B. Chronic blockade of NO synthase activity induces a proinflammatory phenotype in the arterial wall: prevention by angiotensin II antagonism. Arterioscler Thromb Vasc Biol. 1998;18:1408–1416. doi: 10.1161/01.atv.18.9.1408. [DOI] [PubMed] [Google Scholar]
- 58.Scalia R., Appel J.Z., 3rd, Lefer A.M. Leukocyte-endothelium interaction during the early stages of hypercholesterolemia in the rabbit: role of P-selectin, ICAM-1, and VCAM-1. Arterioscler Thromb Vasc Biol. 1998;18:1093–1100. doi: 10.1161/01.atv.18.7.1093. [DOI] [PubMed] [Google Scholar]
- 59.Schäfer A., Bauersachs J. Endothelial dysfunction, impaired endogenous platelet inhibition and platelet activation in diabetes and atherosclerosis. Curr Vasc Pharmacol. 2008;6:52–60. doi: 10.2174/157016108783331295. [DOI] [PubMed] [Google Scholar]
- 60.Hiratsuka M., Katayama T., Uematsu K., Kiyomura M., Ito M. In vivo visualization of nitric oxide and interactions among platelets, leukocytes, and endothelium following hemorrhagic shock and reperfusion. Inf Res. 2009;58:463–471. doi: 10.1007/s00011-009-0011-0. [DOI] [PubMed] [Google Scholar]
- 61.Kubes P., Suzuki M., Granger D.N. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeo T.W., Lampah D.A., Tjitra E., Gitawati R., Kenangalem E., Piera K., Granger D.L., Lopansri B.K., Weinberg J.B., Price R.N., Duffull S.B., Celermajer D.S., Anstey N.M. Relationship of cell-free hemoglobin to impaired endothelial nitric oxide bioavailability and perfusion in severe falciparum malaria. J Infect Dis. 2009;200:1522–1529. doi: 10.1086/644641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bertinaria M., Guglielmo S., Rolando B., Giorgis M., Aragno C., Fruttero R., Gasco A., Parapini S., Taramelli D., Martins Y.C., Carvalho L.J. Amodiaquine analogues containing NO-donor substructures: synthesis and their preliminary evaluation as potential tools in the treatment of cerebral malaria. Eur J Med Chem. 2011;46:1757–1767. doi: 10.1016/j.ejmech.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 64.Potchen M.J., Kampondeni S.D., Seydel K.B., Birbeck G.L., Hammond C.A., Bradley W.G., Demarco J.K., Glover S.J., Ugorji J.O., Latourette M.T., Siebert J.E., Molyneux M.E., Taylor T.E. Acute brain MRI Findings in 120 Malawian children with cerebral malaria: new insights into an ancient disease. AJNR Am J Neuroradiol. 2012 doi: 10.3174/ajnr.A3035. [Epub ahead of press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sugiyama M., Ikeda E., Kawai S., Higuchi T., Zhang H., Khan N., Tomiyoshi K., Inoue T., Yamaguchi H., Katakura K., Endo K., Suzuki M. Cerebral metabolic reduction in severe malaria: fluorodeoxyglucose-positron emission tomography imaging in a primate model of severe human malaria with cerebral involvement. Am J Trop Med Hyg. 2004;71:542–545. [PubMed] [Google Scholar]
- 66.Cabrales P., Carvalho L.J. Intravital microscopy of the mouse brain microcirculation using a closed cranial window. J Vis Exp. 2010;45:2184. doi: 10.3791/2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gyan B., Goka B.Q., Adjei G.O., Tetteh J.K., Kusi K.A., Aikins A., DoDoo D., Lesser M.L., Sison C.P., Das S., Howard M.E., Milbank E., Fischer K., Rafii S., Jin D., Golightly L.M. Cerebral malaria is associated with low levels of circulating endothelial progenitor cells in African children. Am J Trop Med Hyg. 2009;80:541–546. [PMC free article] [PubMed] [Google Scholar]
- 68.Serghides L., Patel S.N., Ayi K., Lu Z., Gowda D.C., Liles W.C., Kain K.C. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. J Infect Dis. 2009;199:1536–1545. doi: 10.1086/598222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bienvenu A.L., Ferrandiz J., Kaiser K., Latour C., Picot S. Artesunate-erythropoietin combination for murine cerebral malaria treatment. Acta Trop. 2008;106:104–108. doi: 10.1016/j.actatropica.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 70.Aicher A., Zeiher A.M., Dimmeler S. Mobilizing endothelial progenitor cells. Hypertension. 2005;45:321–325. doi: 10.1161/01.HYP.0000154789.28695.ea. [DOI] [PubMed] [Google Scholar]
- 71.Balachandar S., Katyal A. Peroxisome proliferator activating receptor (PPAR) in cerebral malaria (CM): a novel target for an additional therapy. Eur J Clin Microbiol Infect Dis. 2011;30:483–498. doi: 10.1007/s10096-010-1122-9. [DOI] [PubMed] [Google Scholar]
- 72.Straus D.S., Glass C.K. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 73.Hempel C., Combes V., Hunt N.H., Kurtzhals J.A., Grau G.E. CNS hypoxia is more pronounced in murine cerebral than noncerebral malaria and is reversed by erythropoietin. Am J Pathol. 2011;179:1939–1950. doi: 10.1016/j.ajpath.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gotsch U., Jäger U., Dominis M., Vestweber D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha in vivo. Cell Adhes Commun. 1994;2:7–14. doi: 10.3109/15419069409014198. [DOI] [PubMed] [Google Scholar]
- 75.Fink M.P. Bench-to-bedside review: cytopathic hypoxia. Crit Care. 2002;6:491–499. doi: 10.1186/cc1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wiese L., Hempel C., Penkowa M., Kirkby N., Kurtzhals J.A. Recombinant human erythropoietin increases survival and reduces neuronal apoptosis in a murine model of cerebral malaria. Malar J. 2008;7:3. doi: 10.1186/1475-2875-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dai M., Freeman B., Bruno F.P., Shikani H.J., Tanowitz H.B., Weiss L.M., Reznick S.E., Stephani R.A., Desruisseaux M.S. The novel ETA receptor antagonist HJP-272 prevents cerebral microvascular hemorrhage in cerebral malaria and synergistically improves survival in combination with an artemisinin derivative. Life Sci. 2012 doi: 10.1016/j.lfs.2012.07.006. pii: S0024-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]