

Abstract
So far, the annotation of translation initiation sites (TISs) has been based mostly upon bioinformatics rather than experimental evidence. We adapted ribosomal footprinting to puromycin-treated cells to generate a transcriptome-wide map of TISs in a human monocytic cell line. A neural network was trained on the ribosomal footprints observed at previously annotated AUG translation initiation codons (TICs), and used for the ab initio prediction of TISs in 5062 transcripts with sufficient sequence coverage. Functional interpretation suggested 2994 novel upstream open reading frames (uORFs) in the 5′ UTR, 1406 uORFs overlapping with the coding sequence, and 546 N-terminal protein extensions. The TIS detection method was validated on the basis of previously published alternative TISs and uORFs. Among primates, TICs in newly annotated TISs were significantly more conserved than control codons, both for AUGs and near-cognate codons. The transcriptome-wide map of novel candidate TISs derived as part of the study will shed further light on the way in which human proteome diversity is influenced by alternative translation initiation and regulation.
In eukaryotic cells, enormous proteome diversity is generated from a limited number of genes through several different mechanisms, including alternative splicing and the activation of alternative promoters, polyadenylation sites, and translation initiation sites (TISs) (Nabeshima et al. 1984; Zavolan et al. 2003; Carninci et al. 2005; Nilsen and Graveley 2010). While a significant contribution of alternative splicing to proteome diversity is well established, the influence upon genome plasticity of the recruitment of alternative TISs, or the presence of upstream open reading frames (uORFs), has only recently been recognized (Kochetov 2008; Sonenberg and Hinnebusch 2009).
In the canonical “scanning model” of ribosomal function, the 43S preinitiation complex containing the initiator tRNA in ternary complex with the GTP-bound form of EIF2 attaches to the 5′ cap of the mRNA and migrates in the 3′ direction until it reaches the AUG codon nearest to the 5′ end of the mRNA (Kozak 2005; Lorsch and Dever 2010). There, AUG recognition triggers the irreversible hydrolysis of the GTP bound to EIF2 by EIF5, and a stable 48S preinitiation complex is formed. Following release of EIF2-GDP and several other eIFs, EIF5B catalyzes joining of the 60S ribosomal subunit, thereby forming an 80S ribosome ready to elongate (Lorsch and Dever 2010; Hinnebusch 2011). The first AUG encountered in the mRNA will usually be used as the translation initiation codon (TIC), provided that it is surrounded by a suitable consensus sequence (Miyasaka et al. 2002; Nakagawa et al. 2008; Volkova and Kochetov 2010) called the “Kozak sequence.” However, different AUGs may also interact with the translational machinery so as to lead to ribosome binding without classical AUG scanning (Reigadas et al. 2005; Fernández-Miragall et al. 2006). In addition, translation initiation at non-AUG codons has been reported for both vertebrate and viral mRNAs (Sugihara et al. 1990; Helsens et al. 2011; Ingolia et al. 2011; Ivanov et al. 2011). The mechanisms by which ribosomes select non-AUG codons for translation initiation, however, are largely unknown.
A control of gene expression at the level of translation is also known to be exerted by uORFs. Bioinformatic analysis identified uORFs in 35%–50% of rodent and human transcripts (Iacono et al. 2005; Matsui et al. 2007), and uORFs tend to be conserved between species (Neafsey and Galagan 2007). From the analysis of >11,000 matched mRNA and protein level measurements, it was estimated that the activation of uORFs may reduce protein expression by up to 80%, and uORF-activating mutations in disease-associated genes have been found to lead to complete silencing of the main ORF (Calvo et al. 2009). However, the regulatory effect of uORFs may be more complex than this as is evidenced by the recent finding that the translation of uORFs may also lead to a more efficient translation of the main ORF, in this case in yeast (Brar et al. 2012).
Until recently, systematic searches for TISs and uORFs primarily relied upon in silico approaches, including protein prediction from transcript sequences and evolutionary conservation analysis (Iacono et al. 2005; Volkova and Kochetov 2010; Bazykin and Kochetov 2011; Ivanov et al. 2011). While in silico techniques have been important for improving proteome understanding, their predictive capability is nevertheless hampered by complex phenomena such as internal ribosome entry, initiation at non-AUG codons, and nonsense read-through (Komar et al. 1999; Touriol et al. 2003). As regards the analysis of the transcriptome, the advent of systematic cDNA sequencing has facilitated an experimental assessment of the extent and origin of mRNA variation (Tomb et al. 1997).
Recently, a ribosomal footprinting technique based upon high-throughput DNA sequencing has been developed that allows systematic monitoring of protein translation in yeast and mammalian cells (Ingolia et al. 2009; Guo et al. 2010). This technique was successfully adapted to the specific identification of translation-initiating ribosomes and was used for this purpose in mouse embryonic stem cells, employing harringtonine as the peptide elongation inhibitor (Ingolia et al. 2011). In the present study, we pretreated a human monocyte cell line with puromycin (Allen and Zamecnik 1962; Nathans 1964) and subsequently applied cycloheximide in order to release peptide-elongating ribosomes from the respective transcripts and to block elongation during the first steps after initiation. Compared with harringtonine (Ingolia et al. 2011), puromycin may yield a less precise localization of TISs but at the same time allows better detection of non-AUG TICs because it does not interfere with the assembly of the elongation complex at near-cognate TICs (Ingolia et al. 2011). Our approach to identifying human TISs was then validated using previously published N-terminal protein extensions and uORFs that were established by classical experimental methodology. As a bottom line, the present study provides a transcriptome-wide map of TISs that may further highlight the role of alternative translation initiation in generating and regulating human proteome diversity.
Results
Ribosomal footprint library enrichment for TISs by puromycin treatment
Ribosomal footprint libraries from THP-1 cells were enriched for TISs through puromycin treatment, leading to the release of peptide-elongating ribosomes, followed by the arrest of elongation using cycloheximide (Methods; Supplemental Fig. 1A). Sucrose gradient fractionation of lysates revealed a loss of polysomal peaks in the ribosomal profile of puromycin-treated cells, compared with native cells, thereby providing indirect evidence for the successful release of peptide-elongating ribosomes (Supplemental Fig. 1B). Ribosomal footprints were then generated in triplicate using the standard cycloheximide protocol (Guo et al. 2010) either alone or in combination with prior puromycin treatment. The pooled read length distribution in cycloheximide- and puromycin-treated samples peaked at 30 bp, as was described before for mammalian ribosomal footprints (Supplemental Fig. 1C; Guo et al. 2010). Reads were aligned against the human genome assembly (hg19), and unique matches to annotated RefSeq sequences were observed for 5–18 million reads per sample (Supplemental Table 1). Sequence-derived ribosomal binding patterns were found to be highly reproducible over biological replicates, as was confirmed by both visual inspection (Supplemental Fig. 2A) and statistical analysis. In particular, the pairwise correlation coefficients of the read coverage per nucleotide ranged from 0.82 to 0.84 in the case of puromycin-treated cells (Supplemental Fig. 2B). Read data were therefore pooled over replicates for further analysis.
An enrichment of the ribosomal footprints with genuine TISs after puromycin treatment was confirmed using annotated AUG TICs from human RefSeq sequences, as is exemplified in Figure 1A for the TPP1 gene. Inspection of the pooled read density for the 500 most abundantly translated transcripts also corroborated the TIS-enriching effect of puromycin (Fig. 1B). Moreover, a 3-bp periodicity of the read coverage became apparent at the 5′ end of the analyzed coding sequences (CDS), which provides additional evidence for an enrichment with TISs of the aligned reads (Fig. 1C). However, the observed coverage distribution also highlights the fact that hindrance of peptide elongation by puromycin is less than perfect and that some ribosomes will have undergone a few steps of protein synthesis before elongation of the nascent peptide was stopped.
Figure 1.

Enrichment of THP-1 cell ribosomal footprint data for TISs, following puromycin treatment. (A) Polysome profile of the TPP1 gene in control and puromycin-treated THP-1 cells. (B) Pooled read coverage for the 500 most highly expressed genes. Transcript-specific coverage values were normalized to the total number of reads for each gene and the transcript length was scaled to 1000 bp for all RefSeq sequences. (C) Pooled read coverage around the annotated AUG TICs of the 500 most highly expressed genes in puromycin-treated cells.
Detection of TISs using a neural network
Since the above experiments were intended to detect novel uORFs and N-terminal protein extensions, only the 5′ UTR and the first 30 bp of the CDS of the analyzed RefSeq sequences were considered for further study. Moreover, to ensure sufficient sequence data quality, at least one nucleotide position with at least 20-fold (“20×”) coverage was required in the region of interest, a criterion met by a total of 5062 transcripts. In order to facilitate systematic searching for potential TISs in the ribosomal footprint data, a neural network was trained on a manually curated set of 604 annotated AUG TICs. The latter were chosen so as to reflect the whole range of read coverage pertinent to the available sequencing data, namely 158 AUGs with 100–8390× coverage, 250 AUGs with 30–99× coverage, and 196 AUGs with 10–29× coverage. Ten neural networks were trained on different random selections of two thirds of each data set, and the corresponding ROC curves were derived from the remaining third (Supplemental Fig. 3). In order to attenuate the possible effects of chance overtraining, the neural network with the median ROC (AUC 0.97) was selected for further analysis. With this network, a signal of 0.001 (from the possible range of values between 0 and 1) was used as a cutoff for TIS identification (“positive signal”). As a result, 93% of the true TISs in the validation set were predicted correctly (i.e., the sensitivity equaled 93%) whereas 1.4% of the controls yielded a false positive signal (specificity: 98.6%).
TIS identification by means of the neural network thus gave binary, nucleotide position-specific results (“positive” or “negative”), and in order to reduce both noise and redundancy, positive TIS signals were also merged over adjacent nucleotides, combining up to two base-pair positions at a time.
The neural network was next applied to the complete ribosomal footprint data from puromycin-treated cells, including the transcripts of the respective training set. In total, 14,464 individual positive TIS signals and 10,386 merged positive TIS signals were obtained (Supplemental Table 2). Following the original experimental design, putative TISs downstream from the annotated TIC were not considered further, leading to a total of 8710 merged positive TIS signals for further analysis (Supplemental Table 2). To validate the use of the pooled puromycine data set, the neural network was next applied to all three replicates individually. In this analysis, 1720 transcripts met the minimum expression criteria (20× coverage for at least one nucleotide in the region of interest) in at least one replicate. A total of 3336 TISs were identified in these transcripts (Supplemental Table 3), 2365 of which (71%) were also detected in the pooled data. Only 13 of the 1639 TISs (0.8%) that were identified in all three replicates were not found in the pooled data. Similarly, only 20 of the 2385 TISs identified in the pooled data (0.8%) were not found in at least one replicate (Supplemental Table 3). The individual analyses are provided as separate annotation tracks electronically (http://gengastro.1med.uni-kiel.de/suppl/footprint/).
Functional interpretation of neural network-predicted TISs
No TIS was predicted by the neural network in 698 transcripts (14%; Fig. 3A, see below). Translation of these sequences may have initiated at downstream TISs not covered by the present study. In the remaining 4364 RefSeq sequences, neural network-predicted TISs were next classified according to their position in the actual transcript sequence (Fig. 2). First, those network-predicted TISs that were located within 3 bp of an annotated TIC (N = 2305) were interpreted as indicators of the corresponding translation-initiating ribosome. Then other AUGs followed by near-cognate TICs were considered iteratively in the vicinity (i.e., ±3 bp) of network-defined TISs upstream of annotated TISs, each time starting with the TIS with the highest aligned sequence count among the remaining unclassified putative TIS for each transcript. Neither an AUG nor a near-cognate codon was present in the vicinity of 220 network-predicted TISs, a figure that further highlights the specificity of the network prediction tool (Supplemental Table 4). Unclassified TISs occurred in combination with all transcript categories in Table 1, and 42 transcripts only contained unclassified TIS.
Figure 3.

Codon usage and functional classification of neural network-defined TISs: (A) Distribution of the number of putative TISs per transcript. (B) Functional classification of putative TISs. (C) TIC usage in putative TISs, either including AUG (upper row) or for near-cognate codons only (bottom row). (D) Average codon frequency over all three reading frames in the analyzed 5′ UTRs, considering either all possible codons (left), the 10 TIS-relevant codons identified in our study only (middle), or near-cognate codons only (right).
Figure 2.

Algorithm used for the functional classification of neural network-predicted TISs as either “annotated TIS,” “N-terminal protein extension,” “upstream ORF” (uORF),” or “CDS-overlapping uORF.” The respective AUG or non-cognate codon was searched for in a ±3-bp window around the merged positive TIS signal emitted by the neural network. For each transcript, the depicted algorithm is applied until no further network-predicted TISs are available for classification.
Table 1.
Putative functional classification and TIC usage in neural network-predicted TISs
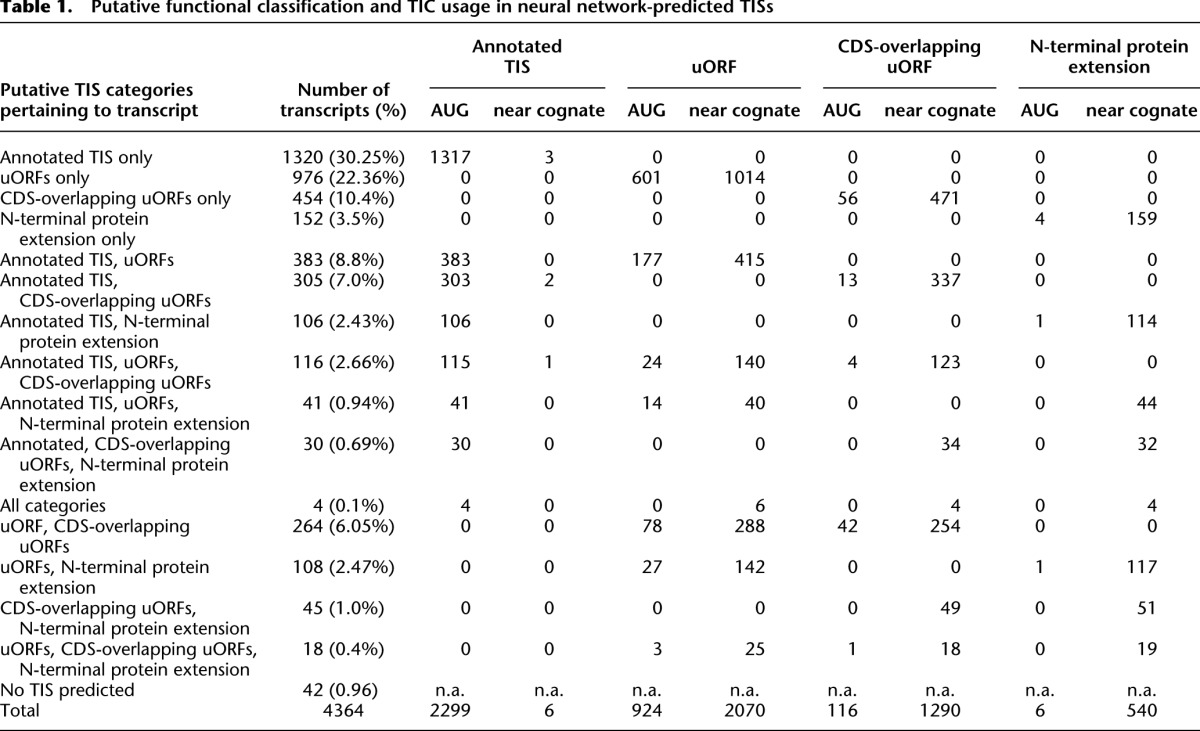
As the overall result, 2305 of the 7251 classified network-predicted TISs (31%) coincided with an annotated TIS (Figs. 2, 3B). Another 2994 putative TISs (40%) predicted in the 5′ UTR were associated with a downstream stop codon and were therefore classified as defining a uORF (Figs. 2, 3B). Yet another 1406 putative TISs (19%) were interpreted as belonging to a uORF overlapping with the respective CDS, because the TIS was out of frame with the annotated ORF but was associated with a downstream stop codon within the CDS. A minority comprising 546 putative TISs (7%) was classified as leading to an N-terminal extension of the encoded protein. Additional, downstream network-predicted TISs within the newly annotated N-terminal protein extensions or uORFs were interpreted as evidence of ribosomal pausing and/or incomplete suppression of elongation by puromycin. They were thus assigned to the respective annotated sequence feature and not classified independently (N = 1239).
The read coverage around newly identified TISs displayed a pronounced 3-bp periodicity, irrespective of their classification, which lent additional support to the presence of true TICs at these positions (Supplemental Figs. 4, 5). A higher read coverage of the second codon at TISs with an AUG TIC rather than a near-cognate TIC (Supplemental Fig. 5) might be indicative of a delayed initiation kinetic pertaining to the type of TIS.
Analysis of the overall TIC usage at the classified putative TISs (N = 7251) revealed an abundance of consensus sequence NUG (N = 6364, 85%), with AUG being the most frequent TIC (N = 3345, 47%) (Fig. 3C). At the level of the individual functional category, usage of AUG in uORFs, CDS-overlapping uORFs, and N-terminal protein extensions equaled 30%, 8%, and 1%, respectively (Fig. 3C, top row). Interestingly, when only near-cognate (i.e., non-AUG) codons were considered, a very similar TIC usage was observed in all three functional categories (Fig. 3C, bottom row). Comparison of the codon usage at identified TICs and in the analyzed 5′ UTRs as a whole, considering all three reading frames, revealed an enrichment of CUG and GUG, and a depletion of AGG and AAG, among TICs (Fig. 3D). A similar pattern of TIC usage as observed for humans in our study was also evident in the previously published murine data (Supplemental Fig. 6; Ingolia et al. 2011).
A transcript-based analysis revealed that all possible combinations of functional TIS classification occurred (Table 1; Supplemental Fig. 7). A TIS was predicted exclusively at the annotated TIC for 1320 (30.3%) of the 4364 transcripts that harbored at least one neural network-predicted TIS. Exclusive translation of one or more uORFs in the 5′ UTR was predicted for 976 transcripts (22.4%), with initiation occurring at AUG for 601 uORFs and at a near-cognate codon for 1014 uORFs. N-terminal protein extensions alone were predicted for 152 transcripts, with putative translation starting at AUGs in four cases and at near-cognate codons in 159 cases. Sequences surrounding the newly identified TISs showed no particular enrichment of the Kozak consensus or any alternative consensus sequence, suggesting that these sites may be subject to more complex initiation mechanisms (Supplemental Figs. 8, 9).
The aligned reads, the network-predicted TIS positions, and their classification are available as UCSC style online material at http://gengastro.1med.uni-kiel.de/suppl/footprint/. An example is provided in Figure 4.
Figure 4.

Gene-based examples for the annotation of TIS for the AMD1 (A) and TOP2A (B) genes in the presented data set: Screenshots from the online resource (the annotation tracks at http://gengastro.1med.uni-kiel.de/suppl/footprint/) are provided. The network-identified TISs are marked in gray and are numbered consecutively along the genome assembly for each RefSeq sequence. The classification results of TIS according to the algorithm in Figure 2 are noted in red. In addition to the markings provided in the online resource, the open reading frames are highlighted with red arrows for uORFs, a blue arrow for the N-terminal protein extension of TOP2A, and green arrows for the annotated CDS of the two genes. (A) Previously known uORF at network-predicted TIS NM_001634:1. An additional internal network-predicted TIS is present in this uORF and was thus not annotated independently as noted in the results. (B) A novel uORF at network-predicted TIS NM_001067:3 and a novel N-terminal protein extension at network-predicted TIS NM_001067:2 are shown. The annotated AUG TISs are detected in both genes.
Literature-based validation of neural network-predicted TISs
Read coverage peaks in ribosomal footprint data provide experimental evidence for ribosome binding during translation initiation at the respective sites. However, both the original laboratory experiments and the subsequent TIS prediction by a neural network are potentially prone to errors, and therefore their joint outcome requires validation. To this end, we focused upon those genes for which experimental evidence for functional uORFs or non-AUG-initiated N-terminal protein extension has been reported before. We used information from the published literature (Tikole and Sankararamakrishnan 2006; Ivanov et al. 2011) and from the “database of mRNA sequences with non-AUG start codons” (http://bioinfo.iitk.ac.in/).
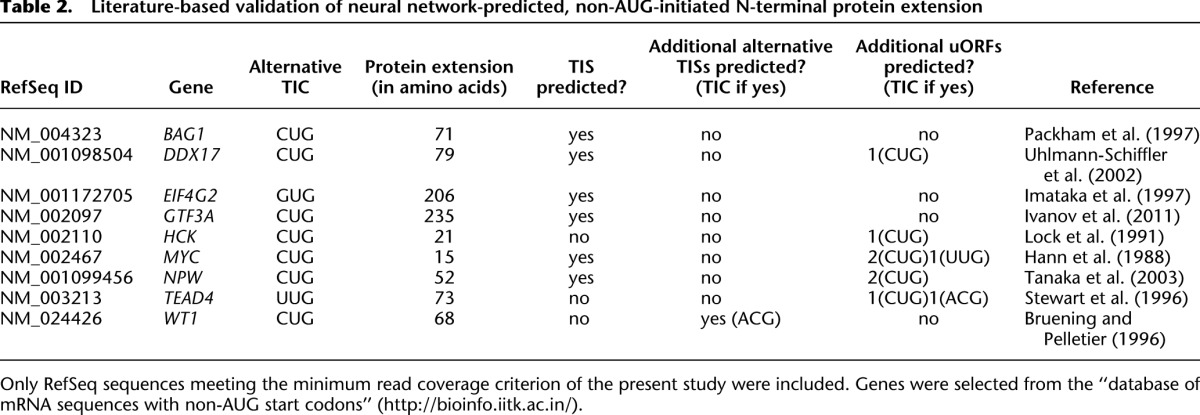
Of the 28 human genes for which the occurrence of N-terminal protein extension had been experimentally verified before, 18 genes were not sufficiently translationally active in the THP-1 cell line studied here (Supplemental Table 5). Another gene (SP3) was not represented in our data set by the appropriate isoform because, for each RefSeq sequence, we consistently analyzed only the mRNA splice variant with the longest 5′ extent (Supplemental Table 5). For six of the remaining nine genes (Table 2), namely BAG1, DDX17, EIF4G2, GTF3A, MYC, and NPW, our combined experimental and in silico approach correctly predicted the previously reported N-terminal protein extension. The apparent non-AUG TICs in the TEAD4 and HCK genes may have been silenced or obscured in our ribosomal footprinting experiment by the uORFs that were predicted by the neural network. Finally, while a previously verified CUG TIC in the WT1 gene (Bruening and Pelletier 1996) was not predicted, the neural network identified a putative upstream ACG TIC instead, activation of which would result in a WT1 protein isoform that is extended by an additional 20-amino acid residues.
Table 2.
Literature-based validation of neural network-predicted, non-AUG-initiated N-terminal protein extension
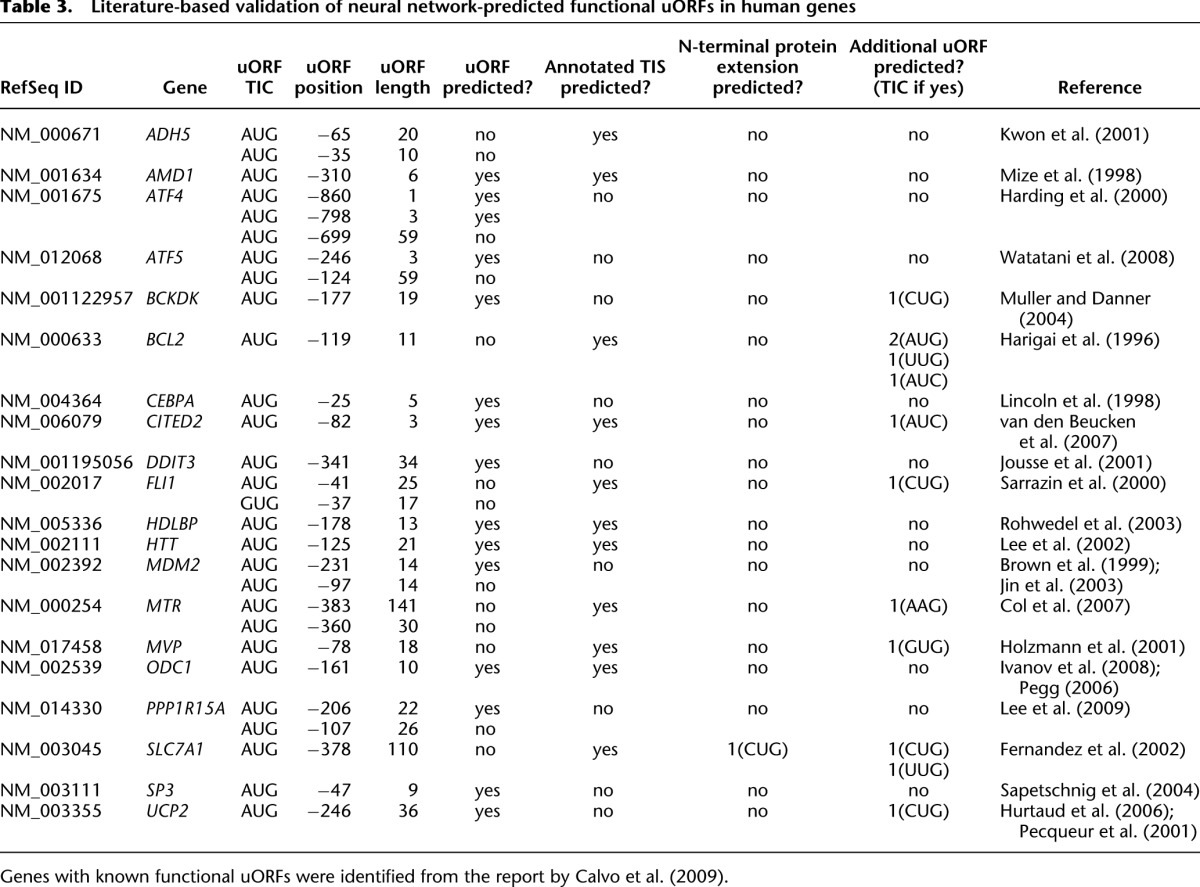
Next, all mammalian genes were examined for which functional uORFs had been experimentally verified before (Calvo et al. 2009). Out of the 50 genes identified, a majority of 25 did not meet the minimum read coverage criteria employed in the present study (Supplemental Table 5). The uORFs of another five genes could not be analyzed because the uORFs were not represented by the appropriate mRNA isoform in our data (Supplemental Table 4). Of the remaining 20 genes (Table 3), both a known uORF and the annotated TIS were predicted correctly for five genes (AMD1, CITED2, HDLBP, HTT, and ODC1). Only one or more of the known uORFs, but not the annotated TIS, was predicted for another nine transcripts (ATF4, ATF5, BCKDK, CEBPA, DDIT3, MDM2, PPP1R15A, SP3, UCP2). For the remaining six genes (ADH5, BCL2, FLI1, MTR, MVP, SLC7A1), translation initiation was only predicted at the annotated TIS whereas the respective uORFs were deemed inactive. In addition, 12 novel uORFs were predicted in the 19 genes analyzed (Table 3).
Table 3.
Literature-based validation of neural network-predicted functional uORFs in human genes
Sequence conservation at neural network-predicted TISs
Many functional uORFs are conserved between different species (Zimmer et al. 1994; Göpfert et al. 2003). To assess the level of recent evolutionary conservation pertinent to the TICs of neural network-predicted TIS, their orthologous positions in nine nonhuman primate species (chimpanzee, gorilla, orangutan, rhesus macaque, baboon, marmoset, tarsier, mouse lemur, bushbaby) were extracted from the PhastCons46Primates track in the UCSC Genome Browser (Pollard et al. 2010). The corresponding conservation score provided by UCSC for the predicted TICs was compared with that of control codons taken from the 5′ UTR of the analyzed RefSeq sequences (Supplemental Table 6), matched for their annotated nucleotide position. As was to be expected, annotated AUG TICs were highly conserved, with a mean conservation score of 0.58 compared with 0.20 in controls (t-test P < 10−10). However, the TICs of neural network-predicted TISs were also found to be significantly more conserved than control codons, both overall (ANOVA controlling for codon type: P < 10−10) and when stratified by codon type (AUG: t-test P < 10−10; non-AUG: ANOVA P = 3.8 × 10−3). The degree of TIC conservation varied significantly between different codons and between different functional TIS categories. The most highly conserved TICs were AUG and AUA, with significant conservation observed in both the uORF and the CDS-overlapping uORF categories (Fig. 5; Supplemental Table 6). The most consistent intra-category conservation across codons was observed for uORFs (Fig. 5; Supplemental Table 6). Generally similar results were obtained with SiPhy (Garber et al. 2009), which nevertheless revealed stronger conservation of AUG in the uORF categories and less conservation of near-cognate codons (Supplemental Table 7).
Figure 5.

Primate conservation analysis of TICs at neural network-predicted TISs. For each functional category and codon type, the difference in mean Conservation Score in nine primate species (with 95% confidence interval) is depicted between case and control TICs. For comparison, the difference in mean Conservation Score for the annotated AUG TICs (open box) is also included for each category. Numbers below boxes refer to the number of predicted TIS falling into the respective TIS by TIC category. TICs showing statistically significant conservation after Bonferoni correction (31 tests, P < 0.0016) are marked by an asterisk. Further details of the primate conservation analysis are provided in Supplemental Table 3.
To further analyze the degree of conservation of TICs across species, the human data set reported here was compared with experimentally defined TICs in mouse embryonic stem cells (Ingolia et al. 2011). In total, 2141 of the 3294 TICs that could be mapped from the human to the mouse genome (64%), and 4458 (60.3%) of the 7391 reciprocally “mappable” TICs in mice (60%), were conserved. Not surprisingly, 98% of the canonical TISs were found to be conserved between the two species whereas uORFs, overlapping uORFs, and N-terminal extensions showed less conservation (Supplemental Table 8). When comparing the TICs of the 1490 TISs that were shared between human and mouse, an almost identical TIC usage became evident (Supplemental Tables 9–11; Supplemental Fig. 6).
In a comparison of 2216 transcripts from humans and mice, the majority of canonical TISs in human were also found to be used in mouse embryonic stem cells. Usage of uORFs and overlapping uORFs seemed to be more conserved than usage of N-terminal extensions (Supplemental Table 12).
Discussion
In the present study, we derived the first transcriptome-wide map of alternative TISs in humans, using an adaption of a recently described ribosomal footprinting technique (Ingolia et al. 2009; Guo et al. 2010) to puromycin-treated cells. The newly annotated TISs will shed new light on the complexity of human proteome composition and regulation. For instance, novel uORFs were predicted for >44% of RefSeq sequences, and 28% of the analyzed transcripts were found to contain putative uORFs overlapping the respective CDS. This observation corroborates recent data on mouse embryonic stem cells where most of the newly identified TISs indeed mapped to uORFs and CDS-overlapping uORFs. Our results are also in agreement with bioinformatic analyses suggesting that ∼50% of human RefSeq sequences contain uORFs, although these analyses defined uORFs on the basis of sequence features alone, rather than experimental evidence for translation (Iacono et al. 2005; Matsui et al. 2007; Calvo et al. 2009). Individual analyses of the three biological replicates in our study provided compelling evidence for the robustness of the predicted TISs. Nevertheless, 1697 newly detected TISs were predicted in some, but not all, replicates. While one possible explanation for these less robust predictions may be lower read coverage, they may also point toward a combination of programmed initiation and stochastic positioning of ribosome binding.
The annotated TIS was also ascertained by our combined experimental and in silico approach for 2305 of the 4364 analyzed RefSeq transcripts (53%). In 1694 transcripts (39%), the annotated TIS has likely been silenced by the activation of uORFs because ribosome binding in these transcripts was only detected experimentally at uORFs and CDS-overlapping uORFs. This observation highlights the likely regulatory role of uORFs in the control of translation of the canonical CDS (Calvo et al. 2009).
While an abundance of sequence reads at predicted albeit not yet annotated TISs provides experimental evidence for the presence of translation-initiating ribosomes, independent validation of such TISs seemed necessary. We therefore screened the literature and public databases for reports of experimentally verified noncanonical TISs and uORFs. The number of such sites was found to be very limited: Only 28 N-terminal protein extensions and 50 functional uORFs could be found probably reflecting the lack of systematic means of experimental analysis before the establishment of ribosomal footprinting (Ingolia et al. 2009; Guo et al. 2010). The number of sites available for validation was reduced further by a lack of expression of the respective gene or by missing data for the reported isoforms. However, for eight out of nine instances of N-terminal protein extension, the outcome of our ribosomal footprint analysis was compatible with that of previous reports. Similarly, 14 out of 19 known human uORFs were confirmed by our approach. Only the annotated TISs were predicted in the remaining six cases, and this less-than-perfect coincidence may reflect that other cell lines and/or functional cellular states were investigated here compared with previous studies.
Ribosomal footprint cDNA libraries were enriched for TISs in the present study by the use of elongation termination agent puromycin. Elongation termination by puromycin is based upon the structural similarity of the latter to aminoacyl-tRNAs, which normally binds to the ribosome and expedites protein synthesis. Binding of puromycin, in contrast, disrupts elongation of the nascent peptide chain and leads to the release of the ribosome from the transcript (Allen and Zamecnik 1962; Nathans 1964). In a previous study geared to identifying alternative TISs, harringtonine was used to arrest ribosomes at the TIS (Ingolia et al. 2011). Harringtonine is known to bind free 60S ribosomal subunits and to inhibit elongation of ribosomes after joining of harringtonine-bound 60S subunits to an 80S ribosome (Fresno et al. 1977; Robert et al. 2009), leading to well-defined signals in ribosome-profiling experiments. The combined use of puromycin and cycloheximide in our study resulted in somewhat less sharply defined read coverage peaks, with elongation leakage downstream from the initiation site (Fig. 1C). However, puromycin has the advantage of a potentially better detection of near-cognate codons because harringtonine is resistant to near-cognate codons under certain circumstances (Ingolia et al. 2011). While any analysis of downstream translation initiation would have been affected seriously by this possible type of error, our specific goal was the identification of TISs upstream of the annotated TIS, and for this purpose, puromycin is a valid and potentially superior experimental agent.
Interestingly, the newly identified putative TISs showed significant evolutionary conservation of the respective TIC among primates, both for AUG and for near-cognate codons. Moreover, trans-species mapping of newly identified and annotated TICs in human and mice, respectively, suggests that this conservation may even extend to primates and rodents. Bearing in mind that conservation analysis of candidate TIS has been used in previous bioinformatics-based identifications of novel uORFs and TISs (Iacono et al. 2005), the strong level of conservation observed here provides further, albeit indirect evidence, for the validity of the newly predicted TISs. Moreover, the different degree of conservation seen for different TIC types and functional classes of TIS may be a direct consequence of the molecular mechanisms underlying TIS recognition, thereby providing a basis for future functional studies. A particularly interesting example in this respect is the different frequency of AUG TICs as observed in different classes of protein-coding sequences and uORFs, for which the pattern of near-cognate codon usage was virtually identical (Fig. 3C). In addition, transcripts containing a uORF in humans were shown to also contain a uORF in mouse in 57% of cases. Although a direct comparison between the two species and studies was difficult due to the differential developmental and metabolomics stages analyzed, and the difficulties of mapping human transcripts onto the mouse genome and vice versa, we found strong evidence for a shared TIC usage in the two species (Supplemental Tables 8–11; Ingolia et al. 2011).
In summary, we provide a transcriptome-wide map of previously non-annotated candidate TISs in a human monocytic cell line. The results, together with the underlying alignments and functional classification (http://gengastro.1med.uni-kiel.de/suppl/footprint/), will add another detail to our understanding of the translational regulation of human proteome diversity.
Methods
Cell culture
Human monocytic cell line THP-1 was obtained from the German Resource Center for Biological Material (DSMZ). Cells were maintained in RPMI 1640 (PAA Laboratories GmbH) supplemented with 10% (v/v) fetal calf serum (Biochrom AG) at 37°C under 5% CO2. Cells were seeded on 15-cm dishes and four plates were pooled for each subsequent biological replicate of the ribosome profiling experiment.
Ribosome profiling and cDNA library preparation
Ribosome footprint cDNA libraries were prepared as previously described with minor modifications (Ingolia et al. 2009; Guo et al. 2010). Release of elongating ribosomes was achieved by the addition of puromycin 48 h after seeding to a final concentration of 100 μg/mL and incubation for 15 min at 37°C. Translation-initiating ribosomes were arrested by subsequent treatment with cycloheximide at a final concentration of 100 μg/mL and subsequent incubation at 37°C. Treatment with puromycin was omitted in the control samples. Cells were washed two times in ice-cold PBS supplemented with cycloheximide (100 μg/mL) and resuspended in 1 mL ice-cold polysome lysis buffer (20 mM Tris pH 8.0, 140 mM KCl, 35 mM MgCl2, 1% [v/v] Triton X-100), supplemented with one Complete ULTRA Tablet (Roche) per 10 mL, and incubated for 10 min on ice after homogenization by pipetting the lysis mixture 10 times through a 19 gauge needle. After centrifugation for 8 min at 1300g, the supernatant was digested with 2000 units RNaseI (Ambion) for 60 min at 30°C with gentle mixing. The digested extracts and control samples were layered onto a 10%–50% sucrose density gradient, and centrifuged at 110,000g for 3 h at 4°C. After ultracentrifugation, the gradients were fractionated using an Isco gradient fractionation system (Teledyne Isco) by continually monitoring A254 nm extinction values at 30-sec intervals, resulting in 500 μL fractions. Monosome fractions were pooled and concentrated using Ultra-4 centrifugal filters (Millipore). The filtrate was treated and RNA extracted as previous described (Guo et al. 2010).
Sequence analysis
Libraries were sequenced on an Illumina HiSeq 2000 instrument using primer oNTI202, 5′-CGACAGGTTCAGAGTTCTACAGTCCGACGATC, to give ∼20 million single-end reads of 50 bp. The resulting FASTQ files were mapped by simultaneous elimination of adaptor sequences using the short-read alignment software Novoalign V2.07.13 (Novocraft). First, the contaminating reads from rRNAs were removed by mapping the reads against RefSeq sequences NR_023371, NR_003287, NR_003286, NR_003285, and NR_023363. The remaining reads were aligned to the human reference genome (UCSC GRCh37/hg19 release) and splice junctions from the UCSC RefSeq track. The position of the footprint in the ribosome was determined by adding 12 nucleotides (nt) to the 5′ end of the read. When the first nucleotide of the read failed to align to the reference sequence, 13 nt were added.
TIS prediction and interpretation
For the ab initio detection of TIS, a set of reference transcripts with one transcript per gene was compiled. If more than one transcript per gene was available, the transcript with the most 5′ location of the annotated TIS and the longest 5′ UTR was chosen. All subsequent analyses were limited to a region comprising the 5′ UTR and the first 30 bp downstream from the TIC of the annotated TIS of each RefSeq sequence. The number of aligned reads per nucleotide position was normalized for each transcript by the highest number observed in the above-mentioned region. Ten neural networks with five input neurons, three hidden layers with 11 neurons each, and one output neuron were then trained and validated on the normalized read coverage per nucleotide position from puromycine ribosomal footprints of a manually curated set of 604 transcripts. The different training sets comprised 403 randomly chosen transcripts per network, and network-specific ROC curves were estimated from the remaining 201 transcripts. The training and validation sets contained the annotated TISs of transcripts with different expression levels (see Results section) and the sites surrounding positions −6, −3, +3, +9, +15, +21, +45, +90, +180, +300, and +480, relative to the annotated TIC, as negative controls. The neural network that yielded the median area under the curve of 0.97 (Supplemental Fig. 3) was used for further analysis. After determining a threshold for the network-emitted signal, TIS identification gave binary, nucleotide position-specific results (“positive signal” or “negative signal”), and in order to reduce both noise and redundancy, positive signals were also merged over adjacent nucleotides, covering 2 bp at a time. The TISs predicted by the neural network were finally classified into one of four presumptive functional categories: “annotated TIS,” “N-terminal protein extension,” “upstream ORF,” or “CDS-overlapping uORF” (Fig. 2).
Statistical analysis
All statistical analyses were performed with R version 2.13.2 (www.r-project.org; R Development Core Team 2011). Control TISs from the 5′ UTRs of the analyzed transcripts were matched by both codon and annotated position, and were selected with the sample() function of R using a weighting scheme according to the spatial distribution of network-identified TISs in the respective 5′ UTRs. Pairwise differences between TIC categories in terms of their Evolutionary Conservation Scores (Pollard et al. 2010) were tested for statistical significance using the Student t-test. (R function t.test()). Differences in Conservation Score across TIC categories were tested for statistical significance using ANOVA as implemented in R function aov().
Data access
The sequence data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE39561.
Acknowledgments
This study was supported by the BmBF (German Federal Ministry of Education and Research) through the Colon Cancer Network (grant no. 01GS08182) and the “Virtual Liver” project (grant no. 0315763), and by the German Research Council (DFG) through the “LABIMI/F” archiving project (grant no. RI 1000/2-1).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.139568.112.
References
- Allen DW, Zamecnik PC 1962. The effect of puromycin on rabbit reticulocyte ribosomes. Biochim Biophys Acta 55: 865–874 [DOI] [PubMed] [Google Scholar]
- Bazykin GA, Kochetov AV 2011. Alternative translation start sites are conserved in eukaryotic genomes. Nucleic Acids Res 39: 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS 2012. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CY, Mize GJ, Pineda M, George DL, Morris DR 1999. Role of two upstream open reading frames in the translational control of oncogene mdm2. Oncogene 18: 5631–5637 [DOI] [PubMed] [Google Scholar]
- Bruening W, Pelletier J 1996. A non-AUG translational initiation event generates novel WT1 isoforms. J Biol Chem 271: 8646–8654 [DOI] [PubMed] [Google Scholar]
- Calvo SE, Pagliarini DJ, Mootha VK 2009. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci 106: 7507–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309: 1559–1563 [DOI] [PubMed] [Google Scholar]
- Col B, Oltean S, Banerjee R 2007. Translational regulation of human methionine synthase by upstream open reading frames. Biochim Biophys Acta 1769: 532–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J, Yaman I, Merrick WC, Koromilas A, Wek RC, Sood R, Hensold J, Hatzoglou M 2002. Regulation of internal ribosome entry site-mediated translation by eukaryotic initiation factor-2α phosphorylation and translation of a small upstream open reading frame. J Biol Chem 277: 2050–2058 [DOI] [PubMed] [Google Scholar]
- Fernández-Miragall O, Ramos R, Ramajo J, Martínez-Salas E 2006. Evidence of reciprocal tertiary interactions between conserved motifs involved in organizing RNA structure essential for internal initiation of translation. RNA 12: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno M, Jiménez A, Vázquez D 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem 72: 323–330 [DOI] [PubMed] [Google Scholar]
- Garber M, Guttman M, Clamp M, Zody MC, Friedman N, Xie X 2009. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpfert U, Kullmann M, Hengst L 2003. Cell cycle-dependent translation of p27 involves a responsive element in its 5′-UTR that overlaps with a uORF. Hum Mol Genet 12: 1767–1779 [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann SR, King MW, Bentley DL, Anderson CW, Eisenman RN 1988. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt's lymphomas. Cell 52: 185–195 [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Harigai M, Miyashita T, Hanada M, Reed JC 1996. A cis-acting element in the BCL-2 gene controls expression through translational mechanisms. Oncogene 12: 1369–1374 [PubMed] [Google Scholar]
- Helsens K, Van Damme P, Degroeve S, Martens L, Arnesen T, Vandekerckhove J, Gevaert K 2011. Bioinformatics analysis of a Saccharomyces cerevisiae N-terminal proteome provides evidence of alternative translation initiation and post-translational N-terminal acetylation. J Proteome Res 10: 3578–3589 [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG 2011. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol Mol Biol Rev 75: 434–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann K, Ambrosch I, Elbling L, Micksche M, Berger W 2001. A small upstream open reading frame causes inhibition of human major vault protein expression from a ubiquitous mRNA splice variant. FEBS Lett 494: 99–104 [DOI] [PubMed] [Google Scholar]
- Hurtaud C, Gelly C, Bouillaud F, Lévi-Meyrueis C 2006. Translation control of UCP2 synthesis by the upstream open reading frame. Cell Mol Life Sci 63: 1780–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono M, Mignone F, Pesole G 2005. uAUG and uORFs in human and rodent 5′ untranslated mRNAs. Gene 349: 97–105 [DOI] [PubMed] [Google Scholar]
- Imataka H, Olsen HS, Sonenberg N 1997. A new translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J 16: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Loughran G, Atkins JF 2008. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc Natl Acad Sci 105: 10079–10084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Firth AE, Michel AM, Atkins JF, Baranov PV 2011. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res 39: 4220–4234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Turcott E, Englehardt S, Mize Gregory J, Morris David R 2003. The two upstream open reading frames of oncogene mdm2 have different translational regulatory properties. J Biol Chem 278: 25716–25721 [DOI] [PubMed] [Google Scholar]
- Jousse C, Bruhat A, Carraro V, Urano F, Ferrara M, Ron D, Fafournoux P 2001. Inhibition of CHOP translation by a peptide encoded by an open reading frame localized in the chop 5′UTR. Nucleic Acids Res 29: 4341–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochetov AV 2008. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays 30: 683–691 [DOI] [PubMed] [Google Scholar]
- Komar AA, Lesnik T, Reiss C 1999. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett 462: 387–391 [DOI] [PubMed] [Google Scholar]
- Kozak M 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 361: 13–37 [DOI] [PubMed] [Google Scholar]
- Kwon HS, Lee DK, Lee JJ, Edenberg HJ, Ahn YH, Hur MW 2001. Posttranscriptional regulation of human ADH5/FDH and Myf6 gene expression by upstream AUG codons. Arch Biochem Biophys 386: 163–171 [DOI] [PubMed] [Google Scholar]
- Lee J, Park EH, Couture G, Harvey I, Garneau P, Pelletier J 2002. An upstream open reading frame impedes translation of the huntingtin gene. Nucleic Acids Res 30: 5110–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-Y, Cevallos RC, Jan E 2009. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2α phosphorylation. J Biol Chem 284: 6661–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln AJ, Monczak Y, Williams SC, Johnson PF 1998. Inhibition of CCAAT/enhancer-binding protein α and β translation by upstream open reading frames. J Biol Chem 273: 9552–9560 [DOI] [PubMed] [Google Scholar]
- Lock P, Ralph S, Stanley E, Boulet I, Ramsay R, Dunn AR 1991. Two isoforms of murine hck, generated by utilization of alternative translational initiation codons, exhibit different patterns of subcellular localization. Mol Cell Biol 11: 4363–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsch JR, Dever TE 2010. Molecular view of 43 S complex formation and start site selection in eukaryotic translation initiation. J Biol Chem 285: 21203–21207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Yachie N, Okada Y, Saito R, Tomita M 2007. Bioinformatic analysis of post-transcriptional regulation by uORF in human and mouse. FEBS Lett 581: 4184–4188 [DOI] [PubMed] [Google Scholar]
- Miyasaka H, Kanai S, Tanaka S, Akiyama H, Hirano M 2002. Statistical analysis of the relationship between translation initiation AUG context and gene expression level in humans. Biosci Biotechnol Biochem 66: 667–669 [DOI] [PubMed] [Google Scholar]
- Mize GJ, Ruan H, Low JJ, Morris DR 1998. The inhibitory upstream open reading frame from mammalian S-adenosylmethionine decarboxylase mRNA has a strict sequence specificity in critical positions. J Biol Chem 273: 32500–32505 [DOI] [PubMed] [Google Scholar]
- Muller EA, Danner DJ 2004. Tissue-specific translation of murine branched-chain α-ketoacid dehydrogenase kinase mRNA is dependent upon an upstream open reading frame in the 5′-untranslated region. J Biol Chem 279: 44645–44655 [DOI] [PubMed] [Google Scholar]
- Nabeshima Y, Fujii-Kuriyama Y, Muramatsu M, Ogata K 1984. Alternative transcription and two modes of splicing results in two myosin light chains from one gene. Nature 308: 333–338 [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Niimura Y, Gojobori T, Hiroshi T, Miura K 2008. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res 36: 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans D 1964. Puromycin inhibition of protein synthesis: Incorporation of puromycin into peptide chains. Proc Natl Acad Sci 51: 585–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Galagan JE 2007. Dual modes of natural selection on upstream open reading frames. Mol Cell Biol 24: 1744–1751 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packham G, Brimmell M, Cleveland JL 1997. Mammalian cells express two differently localized Bag-1 isoforms generated by alternative translation initiation. Biochem J 328: 807–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B 2001. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 276: 8705–8712 [DOI] [PubMed] [Google Scholar]
- Pegg AE 2006. Regulation of ornithine decarboxylase. J Biol Chem 281: 14529–14532 [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2011 R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Reigadas S, Pacheco A, Ramajo J, López de Quinto S, Martinez-Salas E 2005. Specific interference between two unrelated internal ribosome entry site elements impairs translation efficiency. FEBS Lett 579: 6803–6808 [DOI] [PubMed] [Google Scholar]
- Robert F, Carrier M, Rawe S, Chen S, Lowe S, Pelletier J 2009. Altering chemosensitivity by modulating translation elongation. PLoS ONE 4: e5428 doi: 10.1371/journal.pone.0005428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwedel J, Kügler S, Engebrecht T, Purschke W, Müller PK, Kruse C 2003. Evidence for posttranscriptional regulation of the multi K homology domain protein vigilin by a small peptide encoded in the 5′ leader sequence. Cell Mol Life Sci 60: 1705–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapetschnig A, Koch F, Rischitor G, Mennenga T, Suske G 2004. Complexity of translationally controlled transcription factor Sp3 isoform expression. J Biol Chem 279: 42095–42105 [DOI] [PubMed] [Google Scholar]
- Sarrazin S, Starck J, Gonnet C, Doubeikovski A, Melet F, Morle F 2000. Negative and translation termination-dependent positive control of FLI-1 protein synthesis by conserved overlapping 5′ upstream open reading frames in Fli-1 mRNA. Mol Cell Biol 20: 2959–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG 2009. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 136: 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AF, Richard CW, Suzow J, Stephan D, Weremowicz S, Morton CC, Adra CN 1996. Cloning of human RTEF-1, a transcriptional enhancer factor-1-related gene preferentially expressed in skeletal muscle: Evidence for an ancient multigene family. Genomics 37: 68–76 [DOI] [PubMed] [Google Scholar]
- Sugihara H, Andrisani V, Salvaterra PM 1990. Drosophila choline acetyltransferase uses a non-AUG initiation codon and full length RNA is inefficiently translated. J Biol Chem 265: 21714–21719 [PubMed] [Google Scholar]
- Tanaka H, Yoshida T, Miyamoto N, Motoike T, Kurosu H, Shibata K, Yamanaka A, Williams SC, Richardson JA, Tsujino N, et al. 2003. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc Natl Acad Sci 100: 6251–6256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikole S, Sankararamakrishnan R 2006. A survey of mRNA sequences with a non-AUG start codon in RefSeq database. J Biomol Struct Dyn 24: 33–42 [DOI] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388: 539–547 [DOI] [PubMed] [Google Scholar]
- Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats A-C, Vagner S 2003. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol Cell 95: 169–178 [DOI] [PubMed] [Google Scholar]
- Uhlmann-Schiffler H, Rössler OG, Stahl H 2002. The mRNA of DEAD box protein p72 is alternatively translated into an 82-kDa RNA helicase. J Biol Chem 277: 1066–1075 [DOI] [PubMed] [Google Scholar]
- van den Beucken T, Magagnin MG, Savelkouls K, Lambin P, Koritzinsky M, Wouters BG 2007. Regulation of Cited2 expression provides a functional link between translational and transcriptional responses during hypoxia. Radiother Oncol 83: 346–352 [DOI] [PubMed] [Google Scholar]
- Volkova OA, Kochetov AV 2010. Interrelations between the nucleotide context of human start AUG codon, N-end amino acids of the encoded protein and initiation of translation. J Biomol Struct Dyn 27: 611–618 [DOI] [PubMed] [Google Scholar]
- Watatani Y, Ichikawa K, Nakanishi N, Fujimoto M, Takeda H, Kimura N, Hirose H, Takahashi S, Takahashi Y 2008. Stress-induced translation of ATF5 mRNA is regulated by the 5′-untranslated region. J Biol Chem 283: 2543–2553 [DOI] [PubMed] [Google Scholar]
- Zavolan M, Kondo S, Schonbach C, Adachi J, Hume DA, Hayashizaki Y, Gaasterland T 2003. Impact of alternative initiation, splicing, and termination on the diversity of the mRNA transcripts encoded by the mouse transcriptome. Genome Res 13: 1290–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Reynolds K 1994. Tissue specific expression of the retinoic acid receptor-β2: Regulation by short open reading frames in the 5′-noncoding region. J Cell Biol 127: 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]