

Abstract
Eliminating the bacterial cloning step has been a major factor in the vastly improved efficiency of massively parallel sequencing approaches. However, this also has made it a technical challenge to produce the modern equivalent of the Fosmid- or BAC-end sequences that were crucial for assembling and analyzing complex genomes during the Sanger-based sequencing era. To close this technology gap, we developed Fosill, a method for converting Fosmids to Illumina-compatible jumping libraries. We constructed Fosmid libraries in vectors with Illumina primer sequences and specific nicking sites flanking the cloning site. Our family of pFosill vectors allows multiplex Fosmid cloning of end-tagged genomic fragments without physical size selection and is compatible with standard and multiplex paired-end Illumina sequencing. To excise the bulk of each cloned insert, we introduced two nicks in the vector, translated them into the inserts, and cleaved them. Recircularization of the vector via coligation of insert termini followed by inverse PCR generates a jumping library for paired-end sequencing with 101-base reads. The yield of unique Fosmid-sized jumps is sufficiently high, and the background of short, incorrectly spaced and chimeric artifacts sufficiently low, to enable applications such as mapping of structural variation and scaffolding of de novo assemblies. We demonstrate the power of Fosill to map genome rearrangements in a cancer cell line and identified three fusion genes that were corroborated by RNA-seq data. Our Fosill-powered assembly of the mouse genome has an N50 scaffold length of 17.0 Mb, rivaling the connectivity (16.9 Mb) of the Sanger-sequencing based draft assembly.
Paired-end sequencing of large DNA fragments cloned in Fosmid (Kim et al. 1992) or BAC (Shizuya et al. 1992) vectors were a mainstay of genome projects during the Sanger-based sequencing era. The large spans, particularly of BAC ends, helped resolve long repeats and segmental duplications and provided long-range connectivity in shotgun assemblies of complex genomes (Adams et al. 2000; Venter et al. 2001; Waterston et al. 2002). Fosmids are shorter than BACs but much easier to generate. Their consistent, narrow insert-size distribution centered around 35–40 kb enabled the scanning of individual human genomes with read pairs to detect structural variation such as insertions, deletions, and inversions (International Human Genome Sequencing Consortium 2004; Tuzun et al. 2005; Kidd et al. 2008).
Massively parallel genome-sequencing technologies no longer rely on cloning DNA fragments in a bacterial host. The platforms currently on the market (454, Illumina, SOLiD, Ion Torrent) replaced vectors with synthetic adapters and bacterial colonies with PCR-amplified “clones” of DNA fragments tethered to a bead (Margulies et al. 2005; McKernan et al. 2009) or with “colonies” of identical molecules grown by bridge PCR amplification on a solid surface (Bentley et al. 2008).
However, none of these platforms can handle DNA molecules much longer than 1 kb. Consequently, paired-end sequencing of DNA fragments >1 kb by these technologies requires “jumping” constructs (Collins and Weissman 1984; Poustka et al. 1987): the ends of size-selected genomic DNA fragments are brought together by circularization, the bulk of the intervening DNA is excised, and the coligated junction fragments are isolated and end-sequenced.
Suitable protocols exist for converting sheared and size-selected DNA samples to jumping libraries and for generating read pairs that span several kb of genomic distance which is generally sufficient to fashion accurate and highly contiguous de novo assemblies of microbial genomes from massively parallel short sequencing reads (MacCallum et al. 2009; Nelson et al. 2010; Nowrousian et al. 2010). However, early short-read assemblies of complex genomes, including human genomes, turned out fragmented—despite jumps up to ∼12 kb in length (Li et al. 2010a,b; Schuster et al. 2010; Yan et al. 2011). Without the equivalent of Fosmid or BAC end sequences, the N50 scaffold length (a measure of long-range connectivity) of these assemblies was <1.3 Mb. By comparison, largely owing to paired-end reads from large-insert clones, some of the best traditional Sanger-based mammalian draft assemblies had N50 scaffold lengths of >40 Mb (Lindblad-Toh et al. 2005; Mikkelsen et al. 2007).
Constructing a jumping library entails numerous physical and enzymatic DNA manipulations. Several steps, notably size selection and circularization of genomic DNA fragments in vitro, become increasingly difficult and inefficient as the desired jump length, and hence, fragment length, goes up. In contrast, Fosmid cloning employs a sophisticated biological machinery to carry out these critical steps: Large fragments are size-selected (and short competing fragments excluded) by packaging in bacteriophage λ; once inside the Escherichia coli host, cohesive ends mediate efficient circularization—aided by the cellular machinery and a powerful selection for circular amplicons.
To our knowledge, no jumping library constructed to date from uncloned DNA fragments has approached the average span (35–40 kb) and complexity (>105 independent clones per μg of input DNA) of a traditional Fosmid library. To close this technology gap, we and others have taken a hybrid approach wherein Fosmid libraries are constructed first and then converted to Fosmid-size jumps in vitro (Gnerre et al. 2011; Hampton et al. 2011).
Here, we present the experimental details of the “Fosill” concept (Gnerre et al. 2011) as well as extensive improvements of the original protocol. The term Fosill stands for paired-end sequencing of Fosmid libraries by Illumina, though we note that this approach should work for any massively parallel sequencing technology that can generate paired reads. We describe the methodology and novel cloning vectors that enable molecular barcoding of DNA inserts and multiplex Fosmid library construction without physical size selection of sheared genomic DNA. We demonstrate the power of Fosill to detect structural abnormalities in cancer genomes and to improve de novo assemblies of mammalian genomes from short reads.
Results
Modified Fosmid cloning vectors
To facilitate the conversion of Fosmids to Illumina-compatible Fosill jumping libraries, we modified the original Fosmid vector pFOS1 (Kim et al. 1992) such that the cloning site is flanked by Illumina forward and reverse sequencing primers and a pair of Nb.BbvCI nicking endonuclease sites (Fig. 1A). Our modified family of four pFosill vectors retains salient features of pFOS1 such as dual cos sites (Evans et al. 1989) and a pUC-derived pMB1 origin of replication which facilitates the preparation of large amounts of pFosill plasmid. In vitro packaging in bacteriophage λ removes the pUC portion of the plasmid, thereby rendering a single-copy amplicon under the control of the F-factor origin of replication oriS.
Figure 1.

pFosill cloning vectors. (A) General map of the pFosill family of modified pFOS1 Fosmid vectors. The cloning site for inserting the genomic DNA fragments is flanked by forward and reverse Illumina-primer sequences (ILMN-F and ILMN-R) and two Nb.BbvCI nicking endonuclease sites. Nicks (yellow triangles) are introduced on two different strands and are located 5′ of the cloning site. ILMN-F is the standard Illumina sequencing primer SBS-3. The reverse primer in pFosill-1 and pFosill-3 is the SBS-8 primer for standard paired-end sequencing. In pFosill-2 and pFosill-4, the reverse primer is SBS-12 for three-read multiplex paired-end sequencing. The pUC-derived portion between the two cos sites is not present in the final circularized Fosmids which replicate under the control of oriS and the F-factor functions repE and sopA-C that ensure proper partition of the Fosmid among the two daughter cells. Vectors are cut at the unique AatII site as well as two restriction sites at the cloning site and dephosphorylated. (B) Cloning site of pFosill-1 (SBS-8 version) and pFosill-2 (SBS-12). Sheared, end-repaired, and size-selected genomic insert fragments are inserted by blunt-end ligation between two dephosphorylated Eco72I sites 4 bp downstream from the ILMN sequencing primers. The SapI sites shown are not useful for cloning as pFosill-1 and -2 harbor three additional SapI sites. (C) pFosill-3 (SBS-8 version) and pFosill-4 (SBS-12) are digested with SapI which excises a single fragment that includes the 3′ ends of the sequencing primers. Sheared and end-repaired genomic insert fragments are ligated to an excess of adapters that provide an 8-bp barcode (orange), the 3′ end of the Illumina sequencing primers, and three non-self-complementary 5′ overhanging bases for sticky-end ligation to the SapI ends of the vector arms. Supplemental Table S1 summarizes the relevant features of all four pFosill vectors.
Genomic DNA fragments prepared by random shearing, end-repair, and size selection to ∼35–45 kb can be inserted by blunt-end ligation between two Eco72I sites of pFosill-1 and -2, four bases downstream from the sequencing primers (Fig. 1B). Alternatively, using pFosill-3 and -4, DNA fragments can be ligated first to an excess of SapI adapters and then inserted by sticky-end ligation (Fig. 1C). The adapters serve two purposes: First, capping the ends with non-self-complementary three-base overhangs helps prevent coligation artifacts and enables Fosmid cloning of sheared genomic DNA without physical size selection; second, genomic fragments can be tagged with unique barcodes in the adapters, thereby allowing pooled Fosmid cloning of multiple DNA samples at once. pFosill vectors are equipped with either SBS-8 (pFosill-1 and -3) or SBS-12 (pFosill-2 and -4) reverse Illumina primer sequences. Thus, the system is compatible with single- and multiplex paired-end sequencing chemistries.
Fosmid-to-Fosill conversion
The two Nb.BbvCI sites in the pFosill vectors are oriented such that digestion makes two symmetrical single-strand nicks, each located 5′ of the cloned fragment (Fig. 2A,B). Then, in the presence of dNTPs, DNA polymerase I translates the nicks in opposite directions into the insert (Fig. 2C). The insert is then fully cleaved at nicked sites using S1 nuclease which releases all but the ends of the cloned insert from the vector backbone (Fig. 2D). This is analogous to the nick-translation-directed cleavage protocol used to construct jumping libraries for SOLiD sequencing (McKernan et al. 2009). Of note, BbvCI sites present in the cloned genome fragment are being nicked as well and give rise to fragments that are no longer attached to the vector.
Figure 2.

Conversion of a Fosmid library to an Illumina-compatible Fosill jumping library. (A,B) The two Nb.BbvCI sites in the vector are nicked. (C) The nicks are translated in opposite directions into the cloned insert. (D) The insert is cleaved at the two translated nicks as well as at nicks originating at any BbvCI sites within the genomic DNA sequence. (E) Fragments are circularized by intramolecular ligation. (F) Recircularized vector molecules serve as templates for inverse PCR with full-length Illumina enrichment primers that include the sequences required for bridge-amplification and paired-end sequencing of the coligated termini of the original Fosmid insert on the Illumina flow cell.
After circularization by intramolecular ligation (Fig. 2E), the coligated insert termini are flanked by Illumina forward and reverse sequencing primer binding sites and can be PCR-amplified using full-length enrichment primers which append the sequences necessary for cluster amplification (Fig. 2F). The size range of PCR products is broad but can be controlled to some degree by the duration of the nick-translation (Supplemental Fig. S1). After a final size selection, the Fosill library is ready for paired-end sequencing by Illumina.
Testing and optimizing Fosill library construction
To test the Fosill strategy, we constructed a Fosmid library from 30 μg Schizosaccharomyces pombe genomic DNA. The library size, as estimated by plating small-scale test transductions on chloramphenicol plates, was 1.4 million colony-forming units (cfu). We performed a large-scale transduction with the remainder of the packaged library, amplified transductants in bulk by overnight liquid culture at 30°C, and prepared Fosmid DNA. We then converted 10 μg of Fosmid DNA to a Fosill jumping library, sequenced it in 2 × 101-base paired-end mode on a GAII instrument, and aligned the reads to the S. pombe reference genome sequence (Table 1, library S).
Table 1.
Summary statistics for four Fosill libraries
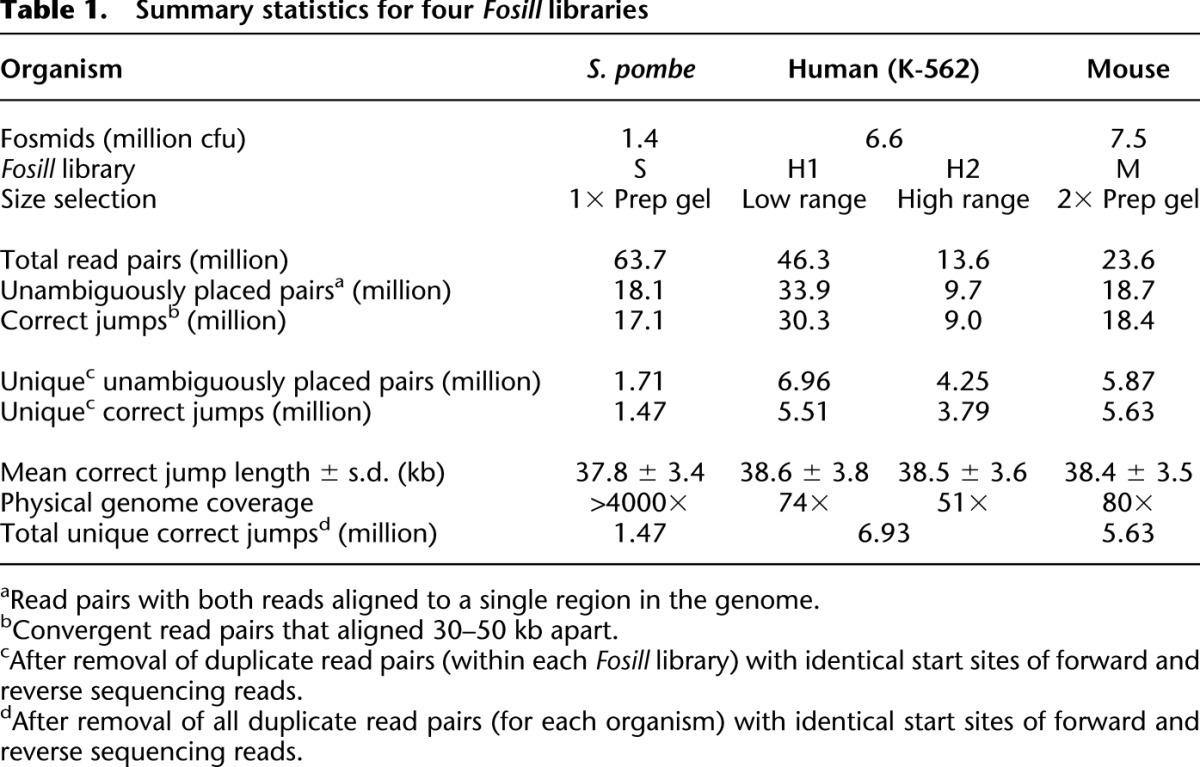
Of 18.1 million unambiguously placed read pairs, 17.1 million (94%) had the expected spacing (30–50 kb) and orientation (convergent). On average, these bona fide correct Fosmid jumps were 37.8 kb in length with a standard deviation of 3.4 kb. Less than 1% of the aligned read pairs were obvious chimeric artifacts that jumped wider than 100 kb or across chromosomes. The chimerism rate of the nonredundant set of unique read pairs (1.71 million) was higher (4.7%; Supplemental Table S1), presumably because singular artifacts and mapping errors have greater weight in this calculation. At this depth of sequencing (∼12×), we recover essentially all unique 30- to 50-kb jumps present in the Fosill library. Accordingly, the total number of unique Fosmid-size jumps was 1.47 million, approximately the same as the estimated size of the original Fosmid library (1.4 million cfu).
A fraction (6.3%) of nonredundant read pairs mapped <1 kb apart (Fig. 3A). Manual inspection suggested that a majority of these represented “nonjumps,” i.e., single small contiguous genome fragments and unequal jumps with one of the coligated end-fragments being too short to be aligned, possibly caused by uneven nick translation.
Figure 3.

Length distribution of genomic distance spanned by paired-end Fosill sequences. Shown are smoothed histograms of the spacing between unique read pairs in Fosill libraries from S. pombe 972h (A), human K-562 library H1 (gray) and H2 (black) (B), and mouse C57BL/6J (C) in their respective reference genomes. (y-axis) Percentage of all unique read pairs that fall in the 1-kb bin indicated on the x-axis. The percentages of unique read pairs spanning <1 kb and 30–50 kb are indicated.
Whatever the exact molecular mechanism, we reasoned that these undesired side products would tend to be shorter than average and enriched in the lower half of the broad smear of PCR-amplified Fosills (Supplemental Fig. S1). We tested this hypothesis with our second test Fosmid library, ∼6.7 million cfu generated from 60 μg of DNA from K-562, a well-studied human chronic myelogenous leukemia (CML) cell line (Lozzio and Lozzio 1975). We ran the PCR-amplified Fosills on a preparative gel, excised two size windows (450–700 bp and 700–900 bp) and sequenced them separately. As expected, the lower size cut contained a higher proportion of nonjumps (13.4% vs. 4.3%; Fig. 3B). The number of nonredundant 30- to 50-kb jumps within each fraction was 5.5 million and 3.8 million. Both size cuts combined had a complexity of 6.9 million correctly spaced unique read pairs (Table 1, libraries H1 and H2).
For our third test organism, mouse (library M), we sought to eliminate short nonjumps more thoroughly by repurifying the size-selected PCR product on a second preparative gel. Only ∼1% of nonredundant read pairs from these doubly size-selected Fosills aligned <1 kb apart (Fig. 3C); 96% (5.6 million distinct jumps) spanned 30–50 kb; 2.4% were classified as chimeric. The rate of chimerism for all unambiguously placed read pairs (18.7 million) was 1%. A detailed breakdown of the sequencing reads from all four test libraries is available in Supplemental Table S2.
To fine-tune the protocol, we used barcoded primers for PCR amplification, thereby tagging different aliquots of a human (NA12892) Fosill library. PCR products were size-selected separately and then combined and sequenced as a pool. Narrowing the size window by raising the lower size cut-off reduced the percentage of “nonjumps” from 1.8% to 0.5% while maintaining the total number of correct jumps (Supplemental Fig. S2A). We also found narrow automated size-fractionation on gel cartridges more effective than manual gel cuts (Supplemental Fig. S2B).
Fosmid library construction is a low-throughput process, and size selection of large DNA fragments on a pulsed-field gel is a particularly cumbersome step and often a source of sample loss. Multiplexing of samples prior to Fosmid library construction would increase throughput of the process and potentially reduce sample loss. To test multiplex Fosmid cloning of DNA fragments without prior size selection, we prepared four aliquots of sheared, end-repaired mouse DNA (starting with 2 μg of genomic DNA per aliquot) and ligated each separately to an excess of barcoded SapI adapters. We then combined the four tagged reactions, constructed a single, four-plex barcoded Fosmid library and converted it to Fosills.
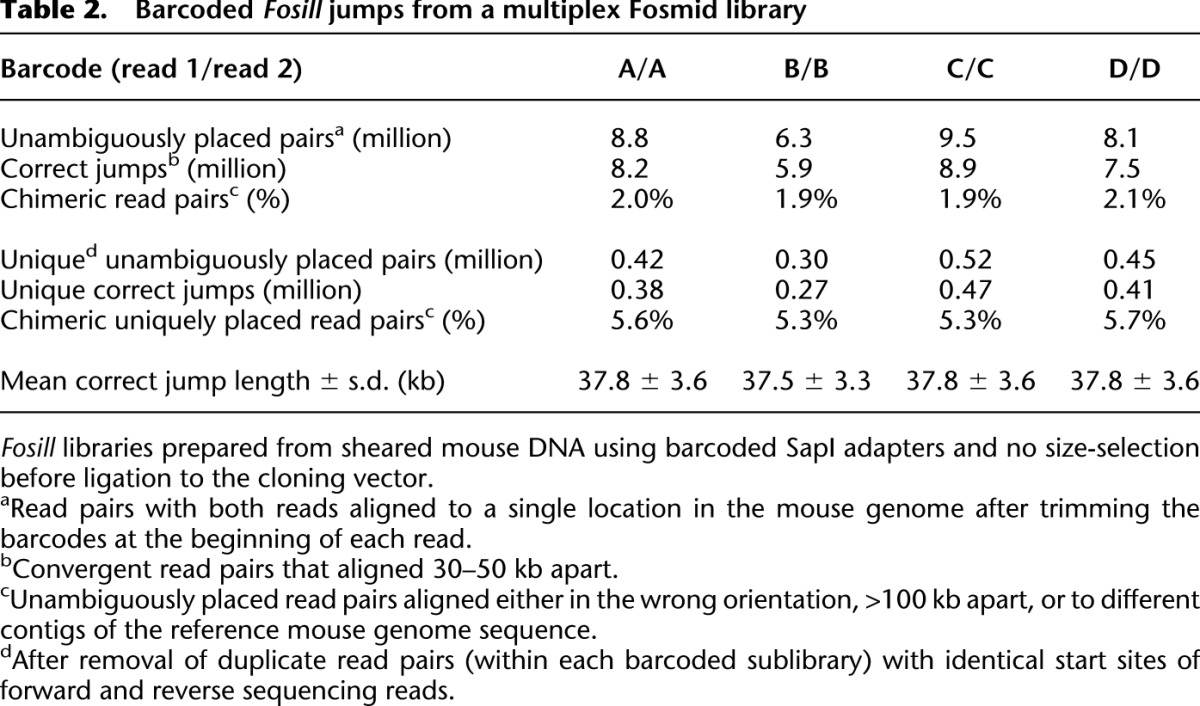
Only 0.6% of all read pairs had discordant barcodes at the beginning of forward and reverse sequencing reads (Supplemental Table S3), suggesting a low rate of “recombination” between Fosmids during the conversion process. It should, therefore, be possible to construct tagged libraries in multiplex format for several genomes at once.
The total number of concordantly tagged and unambiguously mapped read pairs ranged from 4.8 to 8.9 million and represented 0.30 to 0.52 million unique correct jumps per sublibrary (Table 2), i.e., 0.15 to 0.26 million per μg of input DNA. Mean spacings and standard deviations of correct jumps were similar to those from size-selected genomic DNA fragments (see Supplemental Fig. S3 for a side-by-side comparison of jump-size distributions). The rate of chimerism (∼2% for all mapped read pairs; 5%–6% for unique read pairs) was slightly higher than for the “traditional” mouse Fosill library (1% and 2.4%; Supplemental Table S2) but still acceptable. Based on these data, we conclude that multiplexed gel-free Fosmid cloning is a viable option for processing multiple DNA samples less hands-on and in a shorter amount of time.
Table 2.
Barcoded Fosill jumps from a multiplex Fosmid library
Detection of structural rearrangements
To assess the power of Fosills to detect gross structural rearrangements, we searched for loci in the K-562 genome spanned by jumps which were aberrantly spaced or oriented or interchromosomal in the human reference genome. We identified 21 distinct rearrangements with 10 or more independent supporting read pairs (Supplemental Table S4). A subset that includes the three most frequently observed events is listed in Table 3. While the K-562 is a well-studied cancer cell line, and many structural abnormalities in its genome are known, the data analysis was performed in a blind fashion without explicit a priori expectations of structural variants.
Table 3.
Examples of rearrangements in the K-562 genome identified by Fosill
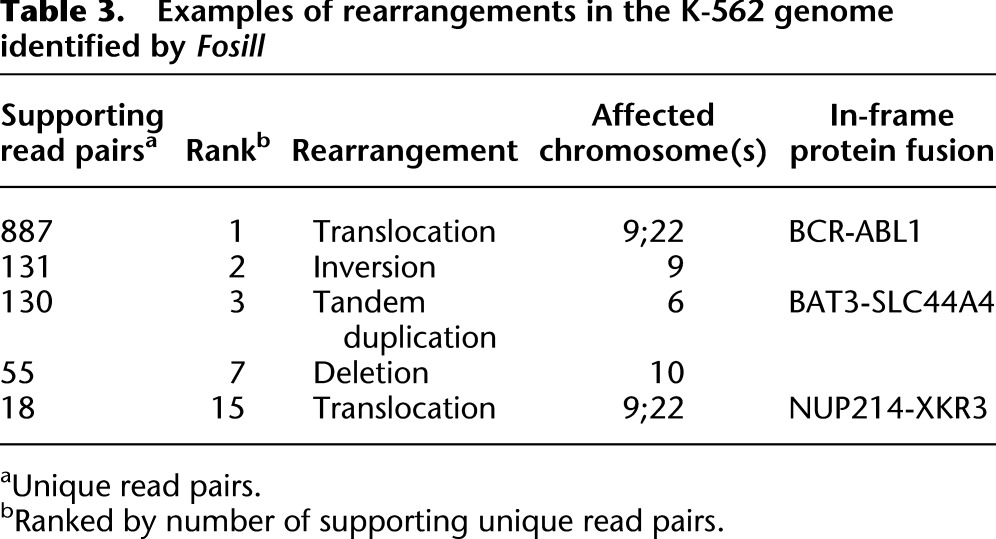
The t(9;22) translocation that gives rise to the BCR-ABL1 fusion protein was framed by a total of 887 unique read pairs that appear chimeric when aligned to the reference genome. The large number of BCR-ABL1 hits is consistent with extensive amplification of this locus (Ross et al. 2009). Given the complexity (6.9 million) and average spacing (38.5 kb) of Fosill jumps, one would expect ∼90-fold coverage for a single-copy locus.
We detected two more rearrangements, a tandem duplication on chromosome 6 and a second t(9;22) translocation, that could plausibly encode in-frame fusion proteins. Of note, chimeric transcripts supporting all three gene fusions (BCR-ABL1, BAT3-SKC44A4 and NUP214-XKR3) have been previously identified in the K-562 transcriptome by RNA-seq (Levin et al. 2009; Berger et al. 2010). We were able to pinpoint both the BCR-ABL1 and the BAT3-SKC44A4 junctions by 32 and seven “chimeric” single Fosill sequencing reads, respectively. The BCR-ABL1 junction matched the junction sequence reported in the literature (Chissoe et al. 1995; Shibata et al. 2010). Maps showing the location of breakpoints and read pairs implicating the three gene fusions and the other two rearrangements listed in Table 2 (an inversion and a deletion event) are available in Supplemental Figure S4.
Impact on de novo genome assemblies
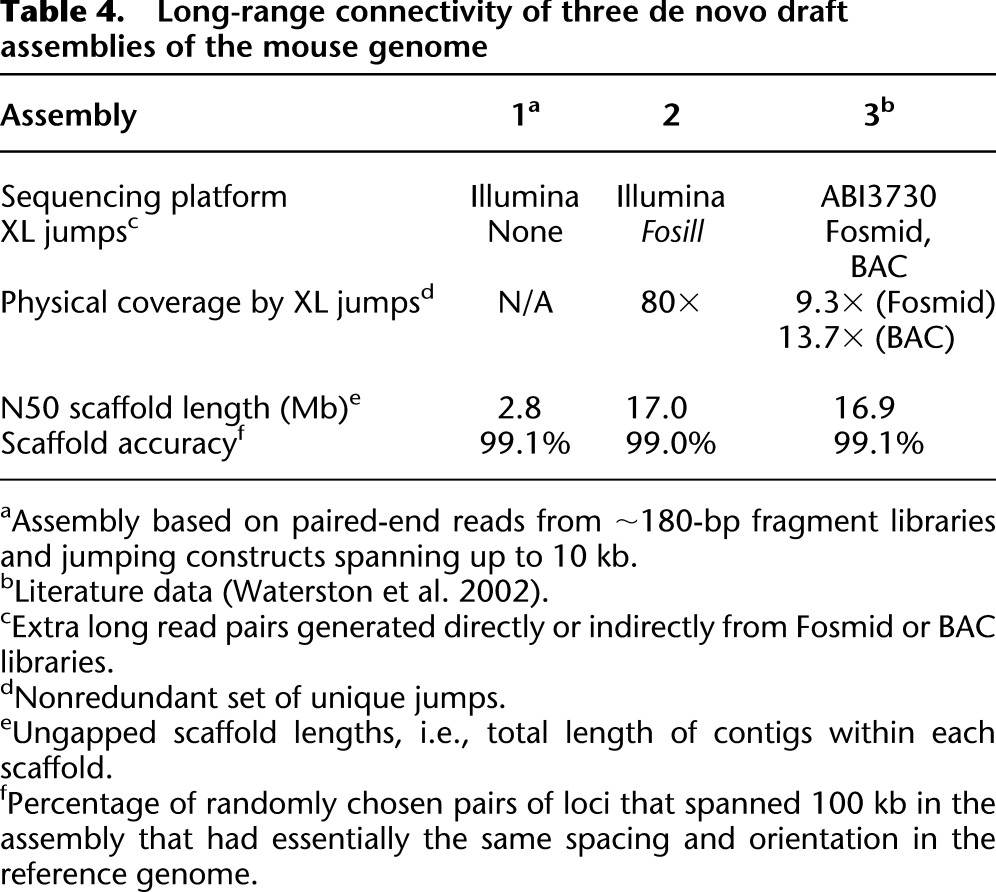
To demonstrate the effect of Fosill jumps on the long-range connectivity of de novo genome assemblies, we performed three Illumina-based ALLPATHS-LG draft assemblies (Gnerre et al. 2011) of the mouse genome (Table 4). Assembly 1, without Fosills, had an N50 scaffold length of 2.8 Mb. Adding data from the Fosill library (80-fold physical coverage) improved the N50 scaffold length to 17.0 Mb, rivaling the long-range connectivity (16.9 Mb) of the capillary-sequencing-based draft assembly 3 (Waterston et al. 2002). The scaffold accuracy, defined as the percentage of pairs of loci that were 100 kb apart in the assembly and had matching spacing and orientation in the reference genome as described previously (MacCallum et al. 2009; Gnerre et al. 2011), was essentially the same for all three assemblies.
Table 4.
Long-range connectivity of three de novo draft assemblies of the mouse genome
Discussion
Avoiding the cloning step in a microbial host has been a major factor in the efficiency and economy of massively parallel sequencing technologies. However, relying solely on enzymatic reactions in vitro to circularize and amplify genomic DNA fragments has made it a major technical challenge to produce the modern equivalent of the Fosmid- or BAC-end sequences that were crucial for assembling and analyzing complex genomes in the past. Fosill is a hybrid approach that employs packaging in bacteriophage λ and cloning in E. coli, followed by in vitro manipulations and PCR amplification to convert Fosmids to Fosmid-sized Illumina jumps. After library amplification by simple outgrowth in liquid culture, each Fosmid clone is represented multiple times. Hence, unavoidable DNA losses during the subsequent in vitro manipulations do not necessarily cause a concomitant drop in library complexity.
In this respect, Fosills are similar to Fosmid “diTags” (Hampton et al. 2011). The principal advantage of our method is that it allows much longer sequencing reads (up to 2 × 101 bases in the current study, but even longer reads are possible), whereas the EcoP15I digest strictly limits the diTags to 2 × 26 bases. Fosill reads have therefore more power to discriminate between different instances of repeat elements or segmental duplications, a significant benefit for mapping and assembly, particularly of mammalian and human genomes. We optimized Fosill using genomic DNA from three organisms (fission yeast, mouse, and human) and carried out pilot studies to test its suitability for two key applications: identification and mapping of chromosomal rearrangements and de novo assembly of complex genomes.
In the first pilot study, we found 21 gross abnormalities in the well-studied CML cell line (K-562), each event supported by at least 10 unique jumps. Three of them give rise to gene fusions that are corroborated by RNA-seq data. Scanning a genome with Fosill read pairs requires fewer read pairs than with short-range jumps or by direct paired-end sequencing of fragments. For example, 7.4 million unique Fosill jumps contained 887 read pairs implicating the hallmark t(9;22) translocation in the K-562 genome. This structural variant can be detected by sequencing a standard ∼300-bp fragment library. However, the detection sensitivity is ∼400-fold lower (175 implicating hits in 611 million unique read pairs; C-Z Zhang and M Meyerson, pers. comm.). If we count all aligned read pairs, including duplicates, (43.6 million Fosill jumps vs. 685 million standard read pairs), Fosill sequencing is 80 times more sensitive. Perhaps more importantly, considering the low cost of sequencing, long-distance jumps can span breakpoints that are flanked by long stretches of repetitive sequence on either side. They are also more suitable for mapping insertion events via read pairs that are closer in the reference genome than expected for bona fide Fosmid ends (International Human Genome Sequencing Consortium 2004).
The impact of Fosill on de novo assemblies of the mouse genome was profound. It boosted the N50 scaffold length from 2.8 Mb to 17.0 Mb without compromising the scaffolding accuracy. By this measure, to our knowledge, our Fosill-powered assembly has better long-range connectivity than any Fosill-free de novo short-read assembly of a mammalian genome reported to date.
For these studies, we sequenced deeply and generated redundant read pairs to maximize the yield of unique Fosill jumps. This adds relatively little cost to a genome project, for the total number of Fosill reads is small compared to the required number of short-insert reads. In almost all cases, the yield of distinct jumps of the expected length corresponded well to the estimated size of the initial Fosmid library, indicating little, if any, loss of complexity during the conversion process.
Our current set of modified Fosmid vectors enables barcoding and multiplexing at two stages. Barcodes can be introduced during the final PCR amplification of the Fosill library for standard three-read multiplex paired-end sequencing. “Inline” barcodes can be added by ligating excess amounts of barcoded SapI adapters to tag genomic DNA fragments before ligating them to the Fosmid cloning vector. These adapters also help prevent coligation of unrelated DNA fragments and thus the formation of packageable chimeric inserts. This is crucial for cloning inserts without running a preparative gel or sucrose gradient, i.e., relying exclusively on bacteriophage λ for size selection. Based on our experience so far, gel-free Fosmid cloning streamlines the process significantly, increases the yield of Fosmid clones per microgram of input DNA and produces Fosill libraries of acceptable size and quality.
We expect multiplex Fosmid-library construction from multiple DNA samples at once to be most useful for smaller genomes. There are also less obvious benefits of pooling multiple differently tagged aliquots of the same DNA sample, particularly for large genomes: First, a variety of “inline” barcodes at the beginning of each read improves the optical separation of adjacent clusters on the Illumina flowcell and thus allows higher read densities; second, filtering out read pairs with discordant barcodes may remove chimeric artifacts that arose downstream from Fosmid cloning.
Despite these improvements, Fosmid cloning remains low-throughput and sensitive to the quantity and quality of the input DNA. Fosmid libraries are also subject to cloning bias. While Fosill appears to work for the extremely GC-rich (69%) genome of Rhodobacter sphaeroides (not shown), we expect cloning problems for extremely AT-rich DNA from organisms such as Dictyostelium discoideum or Plasmodium falciparum that proved recalcitrant to cloning as Fosmids or BACs in the past (Gardner et al. 2002; Eichinger et al. 2005). Nonetheless, we note that numerous large-insert clone libraries have been made that were sufficiently deep, unbiased, and comprehensive to support successful and high-profile genome projects (Osoegawa et al. 2000, 2001; Wei et al. 2007) and that sequencing libraries constructed without cloning have biases of their own (Dohm et al. 2008; Kozarewa et al. 2009; Aird et al. 2011).
In principle, one can easily modify other cloning vectors, for example, plasmid or BAC vectors, and generate shorter “Plasill” or longer “BACill” jumps. The former may or may not be a practical alternative to standard 3–10 kb in vitro jumping libraries. The latter would extend the jump range up to ∼200 kb. In routine practice, considering the steep drop in cloning efficiency for DNA fragments >150 kb, BACill jumps averaging ∼100 kb may be a more realistic proposition. We note, however, that BACs have a much wider size distribution than Fosmids. Thus, in our view, Fosmid jumps currently occupy the sweet spot, the best balance of cloning efficiency and jump range. Not only do they help assemble genomes, their tight size distribution also allows genome-wide scans of individuals by consistently spaced read pairs to analyze structural polymorphisms in the human population and to map gross rearrangements that cause or contribute to human disease.
Methods
Construction and preparation of pFosill cloning vectors
pFosill-1 was constructed by inserting a cloned synthetic DNA fragment (Bio Basic) between the HindIII and BamHI sites of pFOS1 (New England Biolabs [NEB]). Replacing pFosill-1 sequences between the BamHI site and SfiI sites with Illumina SBS-12 primer sequences and an Nb.BbvCI nicking site resulted in pFosill-2. pFosill-3 is a derivative of pFosill-1 that has all SapI sites (and several other restriction sites) outside of the Illumina primer sequences removed. pFosill-4 is a derivative of pFosill-3 containing Illumina SBS-12 instead of SBS-8 primer sequences.
pFosill plamids were propagated in E. coli Stbl2 cells (Invitrogen) grown at 30°C in LB or TB broth containing 100 μg/mL carbenicillin and 15 μg/mL chloramphenicol. A 200-μg aliquot of a Qiafilter plasmid mega preparation (Qiagen) was incubated for 30 min at 37°C in 500 μl containing 300 units “plasmid-safe” ATP-dependent DNase (Epicentre) to remove contaminating linear E. coli chromosomal DNA fragments. After heat inactivation (30 min at 70°C), the reaction was cleaned up with a 1.8-fold volume of AMPure XP beads (Beckman Coulter Genomics). The beads were washed according to the manufacturer's protocol and the plasmid DNA eluted in 200 μl T10E0.1 buffer.
Vectors were prepared for cloning as follows, using restriction endonucleases from Fermentas. pFosill plasmid (50 μg) was digested with 200 units AatII and either 200 units Eco72I (pFosill-1 and pFosill-2) or 200 units LguI, a SapI isoschizomer (pFosill-3 and pFosill-4). After 1 h at 37°C and heat inactivation for 20 min at 65°C, the reaction was cleaned up with a 1.8-fold volume of AMPure XP beads. The restriction fragments were eluted in 200 μl T10E0.1 and dephosphorylated by a two-step incubation (1 h at 37°C and 1 h at 55°C) with 2 × 25 units calf intestine alkaline phosphatase (NEB). The vector arms were cleaned up by two successive extractions with phenol/chloroform/isoamylalcohol, precipitated with ethanol, and resuspended in T10E0.1 to a final concentration (f.c.) of 0.5 μg/ul.
Preparation of genomic DNA fragments
S. pombe strain 972h was a kind gift of Nick Rhind (U. Mass. Medical School). K-562 cells were kindly provided by Robyn Issner (Epigenomics Program, Broad Institute). DNA from Mus musculus strain C57BL/6J was from the Jackson Laboratory. DNA from a normal human lymphoblastoid cell line (NA12892) was from the Coriell Institute. The preparation of genomic DNA fragments for Fosmid cloning was a modification of established protocols. Briefly, genomic DNA (typically two 15-μg aliquots in 125 μl T10E0.1) was HydroSheared (Digilab) by 60 passages at speed code 15 through a 0.006″ shearing assembly (Bird Precision). The fragments were end-repaired for 30 min at 20°C in 175 μl containing 1× T4 DNA ligase buffer, 0.25 mM dNTPs, 15 units T4 DNA polymerase, 50 units T4 polynucleotide kinase, and 5 units Klenow fragment (all from NEB). The reaction was stopped by adding EDTA to a f.c. of 50 mM and heat inactivation for 10 min at 70°C.
For Fosmid cloning of size-selected DNA, 7.5 μg end-repaired fragments were loaded into a 42-mm-wide well on a 1% SeaPlaque GTG (Lonza) 0.5× TBE agarose gel and run at 14°C on a CHEF-DRIII system (BioRad) set to 6 V/cm, 120°, and a switching time ramped from 1.2 to 6 sec over 19 h, along with 8.3 kb to 48.5 kb DNA size markers (BioRad). Marker lanes were cut from the gel and stained with SYBR green I (Invitrogen). The gel was reassembled on a Dark Reader Transilluminator (Clare Chemicals) and a gel slice between the positions of the 33.5 kb and 48.5 kb size markers excised from the unstained preparative portion of the gel. The gel slice was equilibrated twice against two volumes of 1× β-agarase buffer (NEB) supplemented with 40 mM NaCl for 30 min each on ice, the buffer removed, and the agarose melted at 70°C for 10 min. After cooling to 42°C, the agarose was digested at 42°C by two successive 2-h incubations using 1/100th gel volume of 1 unit/μl β-agarase (NEB) each. After heat inactivation at 70°C for 10 min, the tube was chilled on ice for 5 min and centrifuged at 4°C for 20 min at 10,000 rpm. The volume of the supernatant (∼2 mL) was reduced to ∼350 μl by centrifugal ultrafiltration at 2000g using an Amicon Ultra, 0.5-mL 100K concentrator (Millipore). The size-selected genomic DNA was cleaned up by two successive extractions with phenol/chloroform/isoamylalcohol, ethanol precipitated, and resuspended overnight in 20 μl T10E0.1.
For multiplex Fosmid cloning without size selection, barcoded SapI adapters were annealed from oligonucleotide pairs 5′-[PHOS]GATCTXXXXXXXX and 5′-[PHOS]XXXXXXXXAG, where X8 denotes the eight-base barcode. Aliquots (500 ng) of end-repaired fragments were heated at 70°C for 10 min, spun on Amicon Ultra 0.5-mL 100K centrifugal concentrators, and cleaned up by three washes with 500 μl T10E0.1 (∼10 min at 2000g for each step). Concentrated fragments (∼30 μl) were ligated to 115 ng of pre-annealed barcoded SapI adapter at 16°C for 2 h in 100 μl containing 1× T4 DNA ligase buffer (NEB) and 1000 units T4 DNA ligase (NEB). The ligations were inactivated at 70°C for 10 min, pooled, cleaned up, and concentrated to ∼30 μl on Amicon Ultra 0.5-mL 100K centrifugal concentrators spun at 2000g as described above.
Fosmid library construction
All Fosmid libraries were made by a unified protocol using pFosill-1 and pFosill-2 for cloning size-selected DNA fragments and pFosill-3 and pFosill-4 for cloning non-size-selected, SapI-adapter-ligated DNA fragments. The number of ligation reactions depended on the total amount of insert DNA fragments. Each 10-μl ligation contained 250 ng inserts, 500 ng cut and dephosphorylated pFosill vector, 1× T4 DNA ligase buffer, and 2000 units T4 DNA ligase (NEB), and was incubated overnight at 25°C. The ligations were heat-inactivated at 70°C for 10 min, split into 2 × 5 μl, and packaged using MaxPlax λ packaging extracts (Epicentre). Each 5-μl aliquot was packaged by two successive additions of 25 μl packaging extract and 90-min incubations at 30°C. After adding 940 μl Phage dilution buffer (SM buffer with 0.01% gelatin) and 70 μl DMSO (Sigma-Aldrich), packaged Fosmid libraries were titered and stored at −80°C.
Batches of λ-competent T1-resistant E. coli GC10T1 cells, frozen and stored at OD600 ∼0.1, were prepared by standard protocols (Garnes et al. 2003). Thawed cells (1 mL) were mixed with 9 mL 10 mM MgSO4 and 1 mL packaged Fosmid library. After 20 min at room temperature, 40 mL of prewarmed (37°C) LB broth was added and the incubation continued for 45 min. at 37°C with shaking at 225 rpm. The number of Fosmid clones was estimated by plating 40 μl of the 51-mL culture onto LB agar plates supplemented with 15 μg/mL chloramphenicol. Three 51-mL cultures were combined into a 2-L flask containing 600 mL 2× YT media supplemented with 15 μg/mL chloramphenicol and grown overnight at 30°C with shaking. Fosmid DNA was isolated from the 750-mL culture using Qiagen's QIAfilter Plasmid Mega Purification kit.
Conversion of Fosmids into Fosills
Ten μg of Fosmid DNA were nicked for 1 h at 37°C in 250 μl containing 1× NEB Buffer 2 (NEB), 1× BSA (NEB), and 50 units Nb.BbvCI (NEB). Products were cleaned up using 1.8-fold volume AMPure XP beads (Beckman Coulter). Nicked Fosmids (800 ng) were incubated on ice for 45–55 min in 200 μl containing 1× NEB2 (NEB), 0.25 mM dNTP, and 50 units DNA polymerase I (NEB). The nick translation was stopped by adding EDTA to a f.c. of 50 mM. Products were cleaned up using 1.8-fold volume AMPure XP beads. Nicks were cleaved at 37°C for 15 min in 50 μl containing 300 mM NaCl, 1× S1 buffer, and 200 units nuclease S1 (Invitrogen). The reaction was stopped by EDTA (50 mM f.c.) and cleaned up using 1.8-fold volume AMPure XP beads. Ends were repaired at 20°C for 30 min in 100 μl containing 1× T4 Ligase Buffer, 0.25 mM dNTP, 9 units T4 DNA polymerase, 30 units T4 PNK, and 5 units Klenow DNA polymerase (all from NEB). After adding EDTA to 50 mM f.c., DNA was cleaned up using 1.0× AMPure beads and recircularized overnight at 16°C in 500 μl containing 1× T4 Ligase buffer and 4000 units T4 ligase (NEB). Products were purified using a Qiagen PCR clean-up kit and eluted in 50 μl T10E0.1. To determine the optimal number of PCR cycles for Fosill-library amplification, 50-μl test PCR reactions were set up containing 1× Phusion HF Mastermix (NEB), 2 μl of circularized DNA, and 0.5 μM PCR primers: PE1.0 and PE2.0 primers (Illumina) for single-plex Illumina paired-end sequencing runs (i.e., Fosills derived from pFosill-1 and pFosill-3); 5′- AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGC and 5′- CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGC for three-read multiplex paired-end sequencing runs with X8 denoting the eight-base barcode (i.e., Fosills derived from pFosill-2 and pFosill-4). The 50-μl PCR reactions were split into four 10-μl aliquots and run on GeneAmp 9700 thermocyclers (Applied Biosystems) as follows: 98°C for 30 sec; 12, 15, 18, or 21 cycles of 98°C for 10 sec, 65°C for 30 sec, and 72°C for 30 sec; and a final extension for 7 min at 72°C. PCR products were run on Criterion 5% 1× TBE polyacrylamide gels (Bio-Rad) and stained with SYBR Green I. The optimal number N of PCR cycles determined from this gel was used for amplification of the remaining library in a 600-μl PCR reaction containing 48 μl circularized DNA, 1× Phusion HF Mastermix (NEB), and 0.25 μM of appropriate PCR primers. Thermocycling in 50-μl aliquots was as follows: 98°C for 3 min; N cycles of 98°C for 120 sec, 65°C for 30 sec, and 72°C for 30 sec; and a final extension at 72°C for 7 min. The PCR product was purified using 1.8-fold volume AMPure XP beads and eluted in 30 μl T10E0.1. The PCR product was size-selected on a standard preparative 1% agarose 1× TAE gel or on an automated Pippin 1.5% agarose DNA gel (Sage Science) with size-selection settings of 550–900 bp. The latter resulted in a 550- to 800-bp Fosill PCR product. PCR products size-selected by Pippin were cleaned up using 1.8-fold volume AMPure XP beads and eluted in 25 ul T10E0.1. Fosill libraries were sequenced by paired-end sequencing chemistry on Illumina GAII or HiSeq instruments.
Sequence analysis
Paired 76-base or 101-base Illumina reads were aligned to the S. pombe 972h (NC_003424.2, NC_003423.2, NC_003421.2), Mus musculus C57BL/6J (NCBI37/mm9), or Homo sapiens (GRCh37/hg19) reference genome sequences by BWA v0.5.9 (Li and Durbin 2009). Each read was aligned independently with bwa aln (-q 5 -l 32 -k 2 -t 4 -o 1), and then the paired alignments were combined using bwa sampe (-a 100000). MergeBamAlignments, from the picard package (http://picard.sourceforge.net/) v1.59, was used to return the unmapped reads to the aligned BAM file. A custom picard module was used to classify the reads based on the following definitions: (1) unambiguously mapped read pairs: pairs with both reads aligning with a mapping quality score >0 as assigned by BWA; (2) duplicate read pairs: pairs where both reads have identical start sites of forward and reverse sequencing reads; (3) correct jumps: read pairs where the reads face each other and are aligned 30–50 kb apart; (4) chimeric jumps: (a) pairs with unexpected orientation (inverted read pairs facing away from each other, and tandem reads aligning to the same strand in the same orientation), and (b) pairs with unexpected spacing (>100 kb or aligning to different contigs in the reference genome sequence, usually different chromosomes). “Inline” barcodes in Fosill read pairs derived from SapI cloning adapters were identified and quantified by comparing to the first eight bases of each read and requiring a perfect match to the expected barcode sequence. Chromosomal rearrangements were identified by dRanger (MS Lawrence, Y Drier, C Stewart, SB Gabriel, ES Lander, and G Getz, in prep.) as read pairs that map to different chromosomes or with unexpected spacing (>50 kb) or orientation. Single reads aligning to either side of a dRanger breakpoint were identified by BreakPointer (Y Drier, MS Lawrence, SL Carter, C Stewart, SB Gabriel, ES Lander, M Meyerson, R Beroukhim, and G Getz, in prep.). A high-level description of these tools can be found in Berger et al. (2011).
Data access
Cloning vectors described in this work are available to academic researchers upon request. Vector sequences have been deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession numbers JX069761 (pFosill-1), JX069762 (pFosill-2), JX069763 (pFosill-3), and JX069764 (pFosill-4). Illumina sequencing reads have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under BioProject ID 40079, accession number SRX116276 for library S, ID 51977 (SRX029163, SRX029164) for library M, ID 82383 (SRX118400 and SRX118399) for libraries H1 and H2, respectively, ID 52009 for barcoded human NA12892 libraries (SRX118354, SRX118355, and SRX118352 for 450, 500, and 550 lower size cut-offs; SRX116629, SRX116621, and SRX116628 for 2× gel, 1× gel, and Pippin), and ID 51977 (SRX115463) for inline-barcoded libraries from mouse. ALLPATHS-LG mouse genome assembly 2 has been deposited in GenBank under accession number AEKQ02000000 and is available at ftp://ftp.broadinstitute.org/pub/papers/assembly/Williams2012/.
Acknowledgments
We thank the staff of the Broad Institute Sequencing Platform for generating sequencing data, Jim Bochicchio and Carsten Russ for catalyzing project and data submissions, Nick Rhind for S. pombe 972h, Robyn Issner for K-562 cells, and Oleg Iartchuk who suggested converting Fosmid libraries to Illumina jumping libraries more than five years ago. This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200900018C, and with funds from the National Human Genome Research Institute (HG003067-05 through HG003067-10).
Author contributions: L.J.S.W. and A.G. conceived and designed experiments. L.J.S.W., D.T., and N.L. performed laboratory experiments. L.J.S.W., A.M.B., T.P.S., and S.K.Y. analyzed sequence data and calculated library metrics, I.M. and D.B.J. assembled genomes, M.S.L., Y.D., and G.G. mapped chromosomal rearrangements, C.N. and A.G. coordinated and directed the project, and L.J.S.W. and A.G. wrote the paper. L.J.S.W. and A.G. are listed as inventors on a related patent application filed by the Broad Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.138925.112.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195 [DOI] [PubMed] [Google Scholar]
- Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A 2011. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 12: R18 doi: 10.1186/gb-2011-12-2-r18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Levin JZ, Vijayendran K, Sivachenko A, Adiconis X, Maguire J, Johnson LA, Robinson J, Verhaak RG, Sougnez C, et al. 2010. Integrative analysis of the melanoma transcriptome. Genome Res 20: 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. 2011. The genomic complexity of primary human prostate cancer. Nature 470: 214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chissoe SL, Bodenteich A, Wang YF, Wang YP, Burian D, Clifton SW, Crabtree J, Freeman A, Iyer K, Jian L, et al. 1995. Sequence and analysis of the human ABL gene, the BCR gene, and regions involved in the Philadelphia chromosomal translocation. Genomics 27: 67–82 [DOI] [PubMed] [Google Scholar]
- Collins FS, Weissman SM 1984. Directional cloning of DNA fragments at a large distance from an initial probe: A circularization method. Proc Natl Acad Sci 81: 6812–6816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm JC, Lottaz C, Borodina T, Himmelbauer H 2008. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res 36: e105 doi: 10.1093/nar/gkn425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, Berriman M, Song J, Olsen R, Szafranski K, Xu Q, et al. 2005. The genome of the social amoeba Dictyostelium discoideum. Nature 435: 43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GA, Lewis K, Rothenberg BE 1989. High efficiency vectors for cosmid microcloning and genomic analysis. Gene 79: 9–20 [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, et al. 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419: 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnes J, Ciancio M, Gnirke A 2003. Construction of large-insert bacterial clone libraries. In Genome mapping and sequencing (ed. I Dunham), pp. 53–92. Horizon Scientific Press, Wymondham, Norfolk, UK [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton OA, Miller CA, Koriabine M, Li J, Den Hollander P, Carbone L, Nefedov M, Ten Hallers BF, Lee AV, De Jong PJ, et al. 2011. Long-range massively parallel mate pair sequencing detects distinct mutations and similar patterns of structural mutability in two breast cancer cell lines. Cancer Genet 204: 447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, Hansen N, Teague B, Alkan C, Antonacci F, et al. 2008. Mapping and sequencing of structural variation from eight human genomes. Nature 453: 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim UJ, Shizuya H, de Jong PJ, Birren B, Simon MI 1992. Stable propagation of cosmid sized human DNA inserts in an F factor based vector. Nucleic Acids Res 20: 1083–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ 2009. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods 6: 291–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Berger MF, Adiconis X, Rogov P, Melnikov A, Fennell T, Nusbaum C, Garraway LA, Gnirke A 2009. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol 10: R115 doi: 10.1186/gb-2009-10-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, et al. 2010a. The sequence and de novo assembly of the giant panda genome. Nature 463: 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010b. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ III, Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438: 803–819 [DOI] [PubMed] [Google Scholar]
- Lozzio CB, Lozzio BB 1975. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45: 321–334 [PubMed] [Google Scholar]
- MacCallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, Malek J, McKernan K, Ranade S, Shea TP, et al. 2009. ALLPATHS 2: Small genomes assembled accurately and with high continuity from short paired reads. Genome Biol 10: R103 doi: 10.1186/gb-2009-10-10-r103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, et al. 2009. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res 19: 1527–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, et al. 2007. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447: 167–177 [DOI] [PubMed] [Google Scholar]
- Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, et al. 2010. A catalog of reference genomes from the human microbiome. Science 328: 994–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrousian M, Stajich JE, Chu M, Engh I, Espagne E, Halliday K, Kamerewerd J, Kempken F, Knab B, Kuo HC, et al. 2010. De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet 6: e1000891 doi: 10.1371/journal.pgen.1000891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, Tateno M, Woon PY, Frengen E, Mammoser AG, Catanese JJ, Hayashizaki Y, de Jong PJ 2000. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res 10: 116–128 [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, Mammoser AG, Wu C, Frengen E, Zeng C, Catanese JJ, de Jong PJ 2001. A bacterial artificial chromosome library for sequencing the complete human genome. Genome Res 11: 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poustka A, Pohl TM, Barlow DP, Frischauf AM, Lehrach H 1987. Construction and use of human chromosome jumping libraries from NotI-digested DNA. Nature 325: 353–355 [DOI] [PubMed] [Google Scholar]
- Ross DM, Schafranek L, Hughes TP, Nicola M, Branford S, Score J 2009. Genomic translocation breakpoint sequences are conserved in BCR-ABL1 cell lines despite the presence of amplification. Cancer Genet Cytogenet 189: 138–139 [DOI] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. 2010. Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Malhotra A, Dutta A 2010. Detection of DNA fusion junctions for BCR-ABL translocations by Anchored ChromPET. Genome Med 2: 70 doi: 10.1186/gm191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M 1992. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci 89: 8794–8797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. 2005. Fine-scale structural variation of the human genome. Nat Genet 37: 727–732 [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. 2001. The sequence of the human genome. Science 291: 1304–1351 [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Wei F, Coe E, Nelson W, Bharti AK, Engler F, Butler E, Kim H, Goicoechea JL, Chen M, Lee S, et al. 2007. Physical and genetic structure of the maize genome reflects its complex evolutionary history. PLoS Genet 3: e123 doi: 10.1371/journal.pgen.0030123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, et al. 2011. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol 29: 1019–1023 [DOI] [PubMed] [Google Scholar]