

Abstract
Illuminating the primary sequence encryption of enhancers is central to understanding the regulatory architecture of genomes. We have developed a machine learning approach to decipher motif patterns of hindbrain enhancers and identify 40,000 sequences in the human genome that we predict display regulatory control that includes the hindbrain. Consistent with their roles in hindbrain patterning, MEIS1, NKX6-1, as well as HOX and POU family binding motifs contributed strongly to this enhancer model. Predicted hindbrain enhancers are overrepresented at genes expressed in hindbrain and associated with nervous system development, and primarily reside in the areas of open chromatin. In addition, 77 (0.2%) of these predictions are identified as hindbrain enhancers on the VISTA Enhancer Browser, and 26,000 (60%) overlap enhancer marks (H3K4me1 or H3K27ac). To validate these putative hindbrain enhancers, we selected 55 elements distributed throughout our predictions and six low scoring controls for evaluation in a zebrafish transgenic assay. When assayed in mosaic transgenic embryos, 51/55 elements directed expression in the central nervous system. Furthermore, 30/34 (88%) predicted enhancers analyzed in stable zebrafish transgenic lines directed expression in the larval zebrafish hindbrain. Subsequent analysis of sequence fragments selected based upon motif clustering further confirmed the critical role of the motifs contributing to the classifier. Our results demonstrate the existence of a primary sequence code characteristic to hindbrain enhancers. This code can be accurately extracted using machine-learning approaches and applied successfully for de novo identification of hindbrain enhancers. This study represents a critical step toward the dissection of regulatory control in specific neuronal subtypes.
In metazoans, precise spatiotemporal patterns of gene expression are modulated by the exquisite contributions of transcriptional regulatory sequences. These include enhancers that activate transcription in a manner frequently observed to be independent of distance, position, and orientation with respect to the promoter of their target genes (Banerji et al. 1981). Empirically validated enhancers are typically a few hundred base pairs long and comprise binding sites for multiple transcription factors (TFs). In turn, TFs bound to these sequences also interact with common co-activators, communicating with the basal transcription machinery assembled at the promoter, and increasing the rate of transcription (Bulger and Groudine 2011). Identifying the combinatorial protein–DNA and protein–protein interactions that determine spatial and temporal enhancer function is crucial to understanding how distinct cellular and developmental programs are established.
The systematic discovery of enhancers has proven challenging, since they are often located at great genomic distances from the genes they regulate (Lettice et al. 2003). The classical approach to enhancer identification involves the use of sequence constraint in the proximity to genes with known biology or expression in a tissue of interest. However, this approach is limited in that comparative genomics offers no information regarding the specific regulatory role of the sequences (Noonan and McCallion 2010). Recent advances in sequencing technologies have enabled the identification of protein–DNA interactions and chromatin structural conformation at the whole-genome level (Barski and Zhao 2009; Visel et al. 2009; Ernst et al. 2011). For instance, the ENCODE project has annotated ∼15 histone variants and modifications, as well as binding events for ∼150 TFs and transcriptional co-factors in many human cell lines, identifying hundreds of thousands of sequence intervals harboring active chromatin (The ENCODE Project Consortium 2007). Despite the unprecedented scale of the ENCODE project, enhancers identified using the TFs, co-factors, and histone marks likely account for only a fraction of all tissue-specific enhancers utilized in any vertebrate (He et al. 2011). Identified sequences are tissue-specific and cannot be used to infer the gene regulatory activity in other tissues (Visel et al. 2009). The complete discovery and validation of enhancers in the human genome spanning all cell types and developmental stages will remain an elusive goal for years to come. Experimental efforts must be accompanied by large-scale computational predictions that are capable of deciphering the DNA sequence encoding tissue-specific regulatory elements and can be applied to annotate complete genomes. Accurate computational predictions not only permit whole-genome annotations of tissue-specific enhancers in a single species, but they can also be applied to annotation of related species in a straightforward manner (Lee et al. 2011). Computational approaches based on the analysis of sequence motifs shared among enhancers with the same or similar regulatory activities are not only capable of accurately predicting enhancers with specific biological functions de novo, but also contribute to our understanding of the combinatorial networks of TFs underlying particular spatio-temporal patterns of gene expression.
We previously proposed a novel computational strategy that combines comparative genomics, Gibbs sampling, and linear regression to systematically identify heart enhancers in the human genome (Narlikar et al. 2010). The reliability of our approach has been evaluated not only computationally, but also in vivo, using transgenic reporter assays in zebrafish and mouse, with a validation rate of 62% for our heart enhancer predictions. High-throughput experimental approaches, such as genome-wide chromatin immunoprecipitation coupled with massively parallel sequencing (ChIP-seq) against EP300, a histone acetyltransferase and transcriptional co-activator protein, predict the genomic location of heart developmental enhancers with comparable accuracy (Blow et al. 2010). These different strategies uncover only partially overlapping sets of putative heart enhancers. Thus, we observed only 17% of the sequences predicted by ChIP-seq experiments overlapping with our candidate heart enhancer sequences (Narlikar et al. 2010). Weak sequence conservation (Blow et al. 2010) alone does not explain this result, since ∼80% of predictions based on ChIP-seq are conserved between human and mouse, which is the only evolutionary constraint imposed by our method. Instead, as current evidence suggests, the small overlap is more likely to be attributable to the different nature of the enhancer signatures captured by each model (He et al. 2011).
In this study we asked whether regulatory signatures (vocabularies) could be uncovered from a more complex cellular substrate, the central nervous system (CNS). In particular, we set out to determine the sequence basis of regulatory control in the hindbrain (Hb). The Hb, or rhombencephalon, is the most primitive part of the human brain, and likely evolved from a homologous structure present in Urbilateria around 550 million years ago (Ghysen 2003). It includes the cerebellum, pons, and medulla oblongata, which are structures that control functions as fundamental and diverse as respiration, heart rate, reflex, and voluntary movements. Impaired Hb development and function are associated with many disorders such as autism, ADHD (attention deficit hyperactivity disorder), schizophrenia, cerebral palsy, and various sleep disorders (Berquin et al. 1998; Aston-Jones 2005; Andreasen and Pierson 2008). As with other complex diseases and phenotypes, most variants identified by genome-wide association and sequencing population studies are found in noncoding regions of the genome, and therefore suspected to play a role in regulatory control (Cooper and Shendure 2011). Understanding the gene regulatory landscape of the human genome in Hb development and structure is an important step toward uncovering the noncoding substrate of the genomic component of brain disorders.
We introduce a machine learning approach, based on the distribution of transcription factor binding sites (TFBSs) in enhancers, which are capable of accurately identifying enhancers whose regulatory control includes the nascent Hb. Our classifier performs very well in de novo discovery of Hb enhancers, with 88% (30/34) of computational predictions validated in vivo using transgenic zebrafish reporter assays. We also analyze the impact of small collections TFBSs on the Hb function of the host enhancers, and present a map of 40,000 Hb enhancers in the human genome. In summary, our data show how the application of effective computational methods for enhancer prediction can greatly improve our understanding of the gene regulatory networks controlling human development and disease.
Results
Building a training set of Hb enhancers
In order to construct a model for Hb enhancer activity, we first compiled a data set of 211 enhancers for which Hb activity has been validated in vivo with reporter assay systems in transgenic mice and zebrafish (Supplemental Table S1). Most of these sequences (n = 192) were obtained from the VISTA Enhancer Browser (Visel et al. 2007) and an additional 20 enhancers were identified in our laboratory in the context of ongoing in vivo transgenic enhancer screens in zebrafish. This data set of Hb enhancers bears genomic features consistent with other enhancer sets. The GC and repeat-content of the Hb enhancers are close to the genome averages (Supplemental Fig. S1). Thirty-nine percent of the Hb enhancers in this catalog are intronic and 61% are intergenic, displaying a genomic distribution close to the expected (for comparison, 44% of enhancers in the VISTA database are intronic). On the other hand, these Hb enhancers are especially well conserved among vertebrates—99% of the Hb enhancers are conserved between human and mouse genomes, and 82% are also conserved between human and chicken genomes. The average phastCons evolutionary conservation score (Siepel et al. 2005) of Hb enhancers is 1.6, significantly higher than the corresponding scores of the heart and limb enhancers (0.5 and 1.2, respectively; Wilcoxon rank-sum test P-value << 0.001).
Enhancers driving expression in the nervous system frequently direct expression in one or more additional tissues or developmental stages. Eighty percent of Hb VISTA enhancers also direct transcription in other tissues, such as midbrain (49%), forebrain (33%), neural tube (43%), and limb (8%), suggesting that the same elements may play pleiotropic roles in expression, and thus that regulatory lexicons may not always be discrete.
Designing an enhancer classifier
There is now broad interest in determining the extent to which computational power can be used to elucidate how transcriptional regulatory instructions are encrypted in primary DNA sequence. The increased volume of genomic sequence-based data sets far exceeds our present capacity to impute biological value to primary sequence and variation therein, particularly in noncoding sequence. We previously developed a linear regression approach that relies on sequence patterns to accurately predict sequences with similar regulatory activity in the human genome de novo beginning with a small catalog of known heart enhancers (Narlikar et al. 2010). Since then, a similar method based on support vector machines (SVMs) and primitive short sequence segments (k-mers) has also performed well in classifying enhancers from different expression domains, including forebrain- and midbrain-derived ChIP-seq data sets (Lee et al. 2011). However, the SVM method was unable to accurately distinguish between different brain enhancer data sets. This was likely complicated in part by the vastly increased cellular complexity of the sequence used in their training sets. Therefore, although both the SVM and the linear regression method exhibited similar performances (data not shown), we opted to combine the specificity of our original classifier with the advanced statistical model proposed by the latter approach (Lee et al. 2011). To this end, we constructed an SVM classifier operating on known TFBSs and overrepresented de novo identified motifs, which we dubbed EnhSVM (see Methods for details). We then used this strategy to determine if we could better discriminate among regulatory catalogs of CNS subdomains and extend this to define a classifier for the Hb, which currently has no ChIP-seq substrate available.
When applied to the collection of 11 tissue-specific experimentally validated sets of VISTA enhancers (forebrain, midbrain, hindbrain, neural tube, limb, heart, dorsal root ganglia, branchial arch, nose, cranial nerve, eye) our classifier was able to discriminate all enhancer sets from background genomic regions with accuracies exceeding 60% according to the area under the Receiver Operating Characteristic (ROC) curve (AUC) measurements in all cases (Supplemental Fig. S2). The vast majority of predictions produced by these models only overlapped predictions from related tissues, indicating that our method identifies cell type-specific enhancer signatures. CNS enhancer classifiers (forebrain, midbrain, hindbrain, neural tube) performed better than the rest (Supplemental Fig. S2), and the Hb classifier displayed the highest AUC accuracy at 91%.
Refinement of a hindbrain classifier
The embryonic Hb forms along the anterior–posterior axis and is initially segmented into a series of adjacent units called rhombomeres. The identity of these rhombomeres is correlated with domains of Hox gene expression and function, which in turn are determined by a gradient of retinoic acid along the anterior–posterior axis of the Hb (Schneider-Maunoury et al. 1998). Thus, the most anterior rhombomeres contribute to the metencephalon (pons and cerebellum), while the most posterior rhombomeres form the myelencephalon (medulla oblongata). In order to determine if we could further refine our classifier's predictive capacity, we separated the data set of Hb enhancers into 161 anterior and 153 posterior Hb enhancers based on expression patterns driven by the sequences in embryonic mice at developmental stage E11.5. The purpose of this step was twofold. First, although these two sets of enhancers are highly overlapping with ∼80% of the sequences driving reporter expression in both domains, we hypothesized that simple functional clustering should result in increasingly homogeneous data sets, more suitable for our method. Second, combinations of multiple classifiers, in this case trained on different Hb subsets, often outperform single classifiers (Kittler et al. 1998). Consequently, we trained and tested three independent Hb classifiers using a standard 10-fold cross-validation setup on five random partitions of the data, using three slightly different data sets: the complete Hb data set, the subset of Hb enhancers that are active in the anterior Hb, and the subset of enhancers which functions in the posterior Hb. However, no single classifier significantly outperforms the others. Indeed, all three Hb classifiers achieved average AUCs of ∼90%, with a true positive rate (TPR) of at least 47% at a false positive rate (FPR) of 5% (Fig. 1A).
Figure 1.

Hindbrain enhancers can be accurately predicted from DNA sequence. (A) Area under the ROC curve (AUC) for three Hb enhancer classifiers trained on three highly overlapping data sets (enhancers with activity in the anterior Hb, posterior Hb, and whole Hb). AUC values range from 0.5 (random discrimination) to a theoretical maximum of 1. We tested the performance of the classifiers in a cross-validation setting and obtained values of 0.89 (anterior Hb), 0.92 (posterior Hb), and 0.89 (combined Hb). (B) Overlap among the top-scoring 5% Hb enhancer predictions produced by all three Hb classifiers. (C) Fold-enrichment in 787 genes involved in Hb function in the neighborhood of positive predictions or putative Hb enhancers. Putative Hb enhancers were associated with the closest gene. P-values were computed using Fisher's exact test.
Hindbrain enhancers harbor putative binding sites for transcriptional regulators of cell identity
Our Hb classifiers rely on sequence motifs representing TFBSs that facilitate distinction of Hb enhancers from random genomic sequences (Methods). We analyzed the discriminatory power of individual motifs to reveal specific TFs likely to interact with Hb enhancers. All three Hb classifiers identified motifs that are known to bind the critical Hb TFs MEIS1, NKX6-1, HOX family members, and POU protein family members among the 100 most relevant sequence features for identifying Hb enhancers (Waskiewicz et al. 2001; Nelson et al. 2005; Kiyota et al. 2008). Similarly, binding motifs known to bind SOX2, a TF which is highly expressed in the Hb with roles in CNS development, were common to all three Hb classifiers (Supplemental Table S2; Kelberman et al. 2008). Many of these motifs are specific to Hb development and function, and their relevance differs for analogous classifiers trained on data sets of enhancers specific to other tissues (compared, for example, with motifs relevant to limb and heart gene expression regulation, Supplemental Table S2; Supplemental Fig. S3). As expected, distinct sets of Hb sequences, even if largely overlapping, showed slight differences in the contribution of each motif to the decision function of the corresponding classifier. For example, we observed differences in the relevance of the estrogen receptor ESR1 motif, which is particularly enriched among enhancers active in the posterior Hb. Thus, the motif for ESR1 is among the 100 most relevant sequence features for the Hb classifier focusing on posterior Hb, but not among the 100 most relevant sequence features for the other two Hb classifiers. Estrogen receptor-related proteins, which can bind ESR1-like motifs (Vanacker et al. 1999; Giguere 2002), have previously been implicated in anterior–posterior brain segmentation (Bardet et al. 2005). The ability of the Hb classifiers to recover motifs corresponding to known Hb TFs provides additional validation of our model. However, we must caution that it is likely that not all computationally predicted motifs are bound by a TF. Moreover, even if they are, assigning the identity of the TFs binding to these motifs is not straightforward, since the binding affinity catalog of TFs is not complete and many motifs are recognized by multiple TFs.
In order to determine the specificity of the motifs with high discriminatory power in the Hb classifiers, we compared them with those in forebrain, midbrain, and limb enhancer classifiers. These comparison classifiers were trained using EnhSVM on sequences identified using ChIP-seq with the enhancer-associated protein EP300 (Methods). While a negligible fraction (<5%) of EP300 peaks is shared among all data sets, overlap among EP300 peaks for closely related tissues, such as forebrain and midbrain, was higher (15%–20%), consistent with tissue-dependent EP300 binding specificity. Less than 10% of the motifs are shared among the 50 most relevant sequence features for the different classifiers. Additionally, <20% overlap with the motifs identified for Hb enhancers—an observation that highlights the ability of our Hb classifier to specifically capture the Hb enhancer code. The TFBSs shared by the Hb and other brain classifiers included binding sites for MEIS1, the NKX, SOX, and HOX homeobox factors, and ZHX2—developmental TFs that are characteristic of general brain regulatory pathways.
Genome-wide predictions identify novel hindbrain enhancers
Our training set is largely made up of conserved sequences and brain enhancers have been shown to frequently be deeply conserved (Visel et al. 2009), so to obtain a genome-wide map of putative human enhancers active in the Hb, we restricted our genome scan to sequences which are conserved among mammals, n = 337,000 (Siepel et al. 2005; Methods). We repeated the scans using the anterior Hb (aHb), the posterior Hb (pHb), and the Hb enhancer classifiers independently. Approximately 40% of the sequences scored positively for at least one classifier (Supplemental Fig. S4), but only 12% (40,000) scored positively for all three (we dubbed the overlap set HbEns, as it represents the most reliable prediction of Hb enhancers). Seventy-seven of the HbEns (0.2%) are known hindbrain enhancers from the VISTA Enhancer Browser (Supplemental Table S3; Visel et al. 2007), and 26,000 (60%) overlap enhancer marks (H3K4me1 or H3K27ac, Methods). Reflecting the similarity of the training data, we observed a large overlap among the highest scoring predictions obtained by each Hb classifier (Fig. 1B). The overlap correspondingly increases with an increase in score cutoff, suggesting that sequence signatures for general Hb activity, rather than anterior or posterior Hb, dominate the decision function of all classifiers.
The genomic distribution of the HbEns is similar to that observed for the training set. Approximately half of the candidate enhancers are intronic and half are intergenic (see Supplemental custom track 1 HbEns). Also, HbEns are fairly uniformly distributed with respect to the conserved sequences that served as the basis for the genome scans, with an average of four candidates per locus and a maximum of 102 in the case of PTPRD, a 2.3 Mb gene highly expressed in brain and recently associated with ADHD (Elia et al. 2010). Compared with all scanned conserved sequences, HbEns are enriched within the loci of genes that are known to play a role in Hb development (P-value = 2.8 × 10−9, hypergeometric test) (Supplemental Table S4). Moreover, higher scoring predictions are located significantly closer to genes associated with Hb development (Fig. 1C; Supplemental Table S3), indicating that our method identifies enhancers that are active in the Hb. Although all HbEns are, by definition, conserved among mammals, their level of evolutionary conservation is notably elevated. HbEns are significantly more conserved with respect to the conserved sequences that served as the basis for the genome scans (based on average phastCons scores [Siepel et al. 2005], P-value < 2.2 × 10−16, Wilcoxon rank-sum test). Additionally, 21% of HbEns are shared with chicken, 8% with frog, and 3% with zebrafish (Methods). Also, with respect to the conserved sequences that served as the basis for the genome scans, conservation in vertebrates is slightly, but significantly, enriched among HbEns (P-value is 2.3 × 10−11 for the overlap with regions that are also conserved in chicken, Fisher's exact test). Moreover, we found a statistical enrichment of DNase I hypersensitive sites (HSS) identified in genomic DNA isolated from human fetal brain among HbEns (1.2-fold enrichment as compared with low-scoring sequences, P-value < 2.2 × 10−16, Fisher's exact test), while we do not observe any enrichment for DNase I HSS in other fetal tissues, such as heart and lung. Although, the hindbrain is only a subset of the complex tissue analyzed in fetal brain, and may refer to a different developmental stage, the enrichment in brain DNase I HSS corroborates our predictions as tissue-specific enhancers.
Finally, to evaluate the ability of our method to accurately define tissue-specific sequence patterns, we compared the distribution of predicted Hb enhancers with forebrain, midbrain, and limb enhancer predictions obtained in the same manner. In particular, we sought to verify that our predictions are not generally shared between different tissues, which would suggest a failed attempt to define a tissue-specific classifier. After we trained additional classifiers on the corresponding EP300 ChIP-seq enhancer sets, we found that there is <20% overlap between the top 5% of high scoring predictions (16% forebrain, 13% midbrain, 9% limb). This overlap is further reduced to 12% when comparing the top 1% of high scoring predictions. This confirms our hypothesis that genome-wide predictions of classifiers trained on enhancers with different activities constitute largely disjoint sets, suggesting that the corresponding classifiers recognize sequence patterns linked to different biological functions.
The hindbrain classifier is a highly accurate predictor of hindbrain activity in zebrafish
In order to determine the accuracy of our method we set out to determine how frequently our predictions identify active Hb enhancers in vivo. In total we selected 55 sequences with a positive scaled summary Hb (see Methods) for functional evaluation in a zebrafish transgenic reporter assay (Supplemental Table S5). To avoid the introduction of biases based on genomic position, we included both intronic and intergenic sequences residing on 21 different human chromosomes (all except chr10 and Y) (Supplemental Table S5). In addition, six low scoring sequences with a scaled summary Hb score less than zero were selected as likely “negative” predictions. Predicted sequences may not identify all functional components within a complete enhancer, thus although our predictions were based on 100–200 bp sequence intervals, we designed primers to include ∼200 bp flanking each side of the original sequence. The average size of all assayed amplicons was 485 bp (Supplemental Table S6).
All sequences were tested for enhancer activity in the Hb using our established zebrafish transgenesis pipeline (Fisher et al. 2006; McGaughey et al. 2008). We define hindbrain expression as any expression in the CNS region that is posterior to the midbrain extending through the myelencephalon and delimited by the anterior portion of the spinal cord. Since the training set sequences directed expression in a number of non-Hb tissues, we do not require that expression is restricted to this region, and are therefore testing the sensitivity of the classifiers to Hb patterns rather than the specificity. The vast majority of constructs (51/55 putative Hb enhancers) directed reporter expression in some portion of the CNS in mosaic zebrafish embryos. Similarly, 6/6 low likelihood predicted sequences displayed mosaic signals in some part of the embryo, including portions overlapping the CNS. All embryos that displayed reporter expression in mosaics were raised to maturity and crossed with AB zebrafish to determine which sequences could direct EGFP expression in the Hb. In total we identified two or more founders for 34 putative Hb enhancers, of which 30 (88%) founder sets displayed concordant expression in the Hb (Supplemental Fig. S5), a predictive success rate that exceeds prior rates of enhancer validation using both computational predictions as well as EP300 ChIP-seq-based predictions ([Narlikar et al. 2010], 62% [Blow et al. 2010], 84%, Fig. 2). In contrast, none of the six low likelihood controls displayed consistent expression in the Hb when passed though the germline (Supplemental Fig. S5).
Figure 2.

Experimental validation of tissue-specific enhancer candidates in transgenic zebrafish and mouse assays. Our computational approach trained on small empirical data sets (red bars) resulted in validation rates comparable to those for ChIP-seq-derived data sets using an EP300 antibody (gray bars) for the heart. Similarly, the validation rates of computational Hb classifiers trained on small empirical data sets were also comparable to those obtained with EP300 ChIP-seq experiments in other brain tissues.
The patterns observed in stable lines displayed marked pleiotropy in their range of reporter expression both in Hb regions as well as in non-Hb regions, likely reflecting the heterogeneity of the training set. Figure 3 provides eight examples that illustrate the diverse patterns of expression observed in our validation set. Although the models trained on anterior and posterior sets of sequences did not appear to be particularly predictive of the relative position of Hb expression, we found that the resulting patterns could be grouped into categories displaying similar expression. HB41, HB34, and HB02 share an expression profile that includes the cerebellum, part of the anterior Hb, in addition to varying levels of expression along the length of the Hb (Fig. 3A–C). However, HB02 also directs non-neuronal expression in the lens of the eye and myotome (Fig. 3C), which may be a result of position effects based on the site of amplicon insertion in the zebrafish genome as it was not observed in all stable lines. Some sequences, like HB15 (Fig. 3D), show expression in the Hb and very little extraneous expression. In contrast, HB25 and 51 share a different expression profile displaying strong expression in the dorsal Hb as well as the tegmentum, a structure in the midbrain that is continuous with the medulla oblongata (Fig. 3E,F; Thisse and Thisse 2004; Thisse et al. 2004). HB10 shows distinct expression in the Hb, spinal cord, and dorsal diencephalon, as well as faint expression in the tegmentum and non-neuronal expression in the myotome and fin buds (Fig. 3G). In contrast to the distinct Hb expression seen in HB10, many domains within the CNS are faintly marked by reporter expression directed by HB50, including Hb neurons, cerebellum, tegmentum, dorsal diencephalon, and telencephalon (Fig. 3H). The varied patterns of expression observed within the Hb validation set are consistent with the diverse nature of the motifs comprising the classifier. This is expected given that the training set is comprised of sequences that displayed significant pleiotropy and included sequences that directed expression in an array of Hb subdomains, as well as in non-Hb tissues. Consequently, we expected that TFBSs contained within these amplicons, and contributing to their prediction, would be diverse. However, we also anticipated that they would include sites for factors whose endogenous expression overlap with domains of reporter expression.
Figure 3.

Predicted enhancers display pleiotropic expression patterns in the hindbrain. (A–H) GFP reporter expression from eight stable lines corresponding to Hb predictions showing expression across the Hb as well as in some non-Hb domains. Dorsal view images were taken at 3 dpf (for lateral images, see Supplemental Figures), anterior to the left. (A) HB41, (B) HB34, (C) HB02, (D) HB15, (E) HB25, (F) HB51, (G) Hb10, (H) HB50. (Cb) cerebellum; (OV) otic vesicle; (Hb) hindbrain; (L) lens; (My) myotome; (dDi) dorsal diencephalon; (Tm) tegmentum; (CG) cranial ganglia; (fb) fin bud.
In vivo validated Hb enhancer sequences are enriched for the 100 most relevant motifs for discriminating Hb enhancers compared with random sequences with similar GC content (Supplemental Table S7). TFBSs for proteins in the POU, NKX, or PAX families, as well as LHX3 are especially common in our validation set (Supplemental Table S8). Consistent with the in silico evaluations of TFBSs identified in HbEns collectively, factors in these families play critical roles in neuronal development. Furthermore, the observed reporter expression for each is largely consistent with previously published expression patterns for one or more of the corresponding TFs. POU domains are found in a large family of TFs and bind the consensus sequence ATGCAAAT (Verrijzer and Van der Vliet 1993). They are expressed mainly in the CNS, and act as regulators of neurogenesis in zebrafish (Spaniol et al. 1996). Consistent with these data, POU family TFBSs were the most commonly identified sites in our validation set and showed an enrichment of 2.6 over GC matched control sequences (P-value = 0.01, Fisher's exact test) (Supplemental Table S8) and many of our elements share expression domains with POU factors. NKX proteins are necessary for the proper development of motor neurons in the hindbrain (Pattyn et al. 2003) and consistent with this role we see a significant enrichment (2.4, P-value = 0.005, Fisher's exact test) for NKX family TFBSs in our validated set of Hb enhancers. Similarly, the PAX gene family similarly comprises a large group of highly conserved TFs required for neuronal development (Wang et al. 2010; Thompson and Ziman 2011). Furthermore, 10/30 validated predictions contained at least one PAX family motif (enrichment of 1.8 as compared with random genomic sequences with similar length and GC-content, P-value = 0.05, Fisher's exact test). Finally, a number of sequences share in common an LHX3 motif that binds a LIM domain TF with a role in neuronal specification (Cepeda-Nieto et al. 2005; Gadd et al. 2011), resulting in an enrichment of 4.6 (P-value = 0.0002, Fisher's exact test). HB25 and HB51 both contain an LHX3 TFBS and share many overlapping domains of reporter expression, including in the Hb and spinal column, which is consistent with endogenous lhx3 expression (Fig. 3E,F; Supplemental Fig. S5; Thisse and Thisse 2004; Thisse et al. 2004). In contrast to the HbEns sequences tested, only one of the low likelihood controls contained any of these motifs, supporting their high predictive power in our model. Taken collectively, these data provide compelling evidence that the validated sequences may play important roles in regulating transcription in the developing Hb.
Enhancer activity is due to the presence of specific transcription factor motifs
Our data suggest that TFBSs contributing to the classifier might independently or collectively explain aspects of the observed regulatory control of the sequences within which they reside. We selected two sequences with Hb regulatory control (HB01 and HB16) to examine more closely, surveying the distribution of TFBSs within each predicted sequence. We then identified smaller sequence fragments for analysis in zebrafish based on the clustering of TFBSs therein.
The full-length HB01 sequence directed distinct expression in the rhombomeres, as well as the midbrain Hb boundary, cranial ganglia, and dorsal diencephalon (Fig. 4B,E). We amplified two smaller fragments (HB01_I and HB01_II) from within this full-length sequence based on the pattern of TFBSs clusters. HB01_I is a 56-bp sequence containing motifs for PITX2, CDX, CEBPG, NKX3-1, and BCL6 (Fig. 4A). Upon passage through the germline, HB01_I displayed broad reporter expression in the CNS (Fig. 4C,F). This pattern encompassed the expression domains marked by the full-length HB01. The expanded expression domains marked by HB01_I could reflect the increased efficiency of TFBSs being placed closer to the minimal promoter (Nolis et al. 2009). It may also reflect the absence of other regulatory sequence motifs within or beyond the initial predicted interval which otherwise act in the full-length construct to moderate transcriptional activity (Gompel et al. 2005). Notably HB01_II, which is 93 bp and contains motifs for HNF3, POU2F1, NKX2-5, MYOG, SOX10, and HMGA1 (Fig. 4A), did not show any mosaic expression, and was determined to be insufficient for enhancer activity in the Hb in this assay (Fig. 4D,G).
Figure 4.

TF clustering reveals functional sequence domains. (A,H) UCSC Genome Browser custom track showing injected construct, classifier predicted HB sequence, and fragments tested for Hb expression (black bars, top to bottom). Colored bars mark TFBS for various factors. (B–G, I–P) GFP reporter expression observed with each sequence (lateral view, top; dorsal view, bottom). All images taken at 2 dpf, anterior to the left. (A) HB01 custom track with two subcloned fragments, (B) full-length HB01, lateral view, (C) HB01_I, lateral view, (D) HB01_II, no G0 GFP reporter expression observed, (E) full-length HB01, dorsal view, (F) HB01_I, dorsal view, (G) HB01_II, no G0 GFP reporter expression observed. (H) HB16 custom track with three subcloned fragments, (I) full-length HB16, lateral view, (J) HB16_I, lateral view, (K) HB16_II, lateral view, (L) HB16_III, lateral view, (M) full-length HB16, dorsal view, (N) HB16_I, dorsal view, (O) HB16_II, dorsal view, (P) HB16_III, dorsal view. (CG) cranial ganglia; (M-H) midbrain hindbrain boundary; (Hb) hindbrain; (Fb) forebrain; (Tm) tegmentum; (My) myotome; (L) lens.
Similarly, HB16 displays prominent expression in the dorsal Hb and fainter expression in the ventral Hb and lateral tegmentum (Fig. 4I,M). Once again we amplified three short fragments from within the initially predicted sequence based on TFBS clusters (Fig. 4H). HB16_I is a 29-bp fragment containing a GATA1 motif; HB16_II is 54 bp in length and contains MEOX2, NKX6-1, EN1, TAL1, NKX2 family, JUN, and PAX4 motifs; and HB16_III is a 23-bp fragment encompassing a HOXA4 motif (Fig. 4H). Upon passage through the germline, both HB16_I and HB16_II directed expression in the Hb (Fig. 4J,K,N,O). In contrast, HB16_III only drove expression in the myotome of stable lines (Fig. 4L,P).
Notably, the reporter expression in the Hb neurons and the lateral tegmentum directed by HB16_I are similar to those of the endogenous gata3 (Fig. 4J,N; Thisse and Thisse 2004; Thisse et al. 2004). This pattern is further consistent with expression directed by full-length HB16. Furthermore, HB16_II directs expression along the entire length of the ventral and medial Hb and spinal column (Fig. 4K,O). As such, it overlaps much of the Hb domain marked by HB16 and resembles the endogenous expression of nkx6 family, nkx2 family, and tal1 RNA (Thisse and Thisse 2004; Thisse et al. 2004; Binot et al. 2010). The observed tegmental reporter expression is also consistent with endogenous expression of tal1 (Thisse and Thisse 2004; Thisse et al. 2004). A potential role for the JUN TFBS identified in HB16 is not immediately obvious but these factors display much broader expression domains throughout the CNS and may in part account for expression domains extending dorsally. Although not conclusive, these data suggest that the expression of TFs corresponding to motifs contributing to our classifier are consistent with their predicted biological roles in modulating expression in the Hb and show that enhancers can be further broken down into their component TFBS fragments and continue to faithfully drive reporter expression in the predicted tissue.
Discussion
The exquisite orchestration of transcriptional control is essential for the normal development and homeostasis of multicellular organisms. Systematic identification of sequences responsible for these activities, however, has proven a significant challenge. Although the encryption of regulatory instructions in DNA sequence is well established, the absence of an established vocabulary has precluded the prediction of biological activities rendered by noncoding functional genome components based on inspection of the primary sequence. Two strategies commonly employed in the identification of transcriptional enhancers are evolutionary sequence constraint and ChIP-seq. Although sequence constraint has been used with significant success, it can impute little regarding the likely biological activity of any identified sequence. Similarly ChIP-seq profiling of TFs, histone modifications, and transcriptional co-activators such as EP300 has recently emerged as a powerful tool for the identification of enhancers active in various tissues; however, not all enhancers are captured by affinity-based methods, and not all cell types are amenable to these assays. Recent efforts to identify sequence motifs (active TFBS) have proven increasingly powerful, allowing the elucidation of early language structure for regulatory control in specific tissues (Narlikar et al. 2010; Lee et al. 2011).
We have integrated these computational strategies, employing machine learning to train a sequence-based classifier on a set of largely published in vivo validated enhancers in the Hb. The result is a highly accurate predictor of enhancer activity in the Hb. When applied to the human genome, 88% (30/34) of sequences demonstrate Hb regulatory control when assayed in vivo (stable zebrafish transgenesis). In contrast, even among sequences identified as being deeply conserved only ∼8% were observed to drive expression in the Hb (Pennacchio et al. 2006). The motifs identified by our classifier frequently represent TFBSs for factors with known roles in regulating transcription in the Hb and with endogenous expression patterns overlapping with that of reporter expression. Furthermore, we show that, consistent with our classifier, clusters of TFBS (∼30–100 bp) contributing to predicted Hb regulatory control can account for aspects of Hb regulatory expression observed in the original (∼500 bp) sequence from which they were derived.
Although the vocabulary described is an effective predictor of Hb activity, we observed pleiotropy among Hb domains marked by reporter expression as well as expression in domains outside the Hb, including non-neural tissues. These observations are consistent with the complexity of vertebrate enhancers known to display a broad expression pattern across multiple tissues (Visel et al. 2007). It is particularly important to keep in mind that the Hb enhancers in our training data set were not exclusively expressed in the Hb, but largely displayed multi-tissue expression patterns. From the sequence analysis perspective, our training set contained a large group of Hb enhancers and several smaller clusters of other expression subdomains. All non-Hb signatures in our training created a plethora of misleading signals confusing the classifier. However, the high Hb validation rate of HbEns reflects the ability of the classifier to sensitively extract the Hb sequence encryption from the noisy input data set. Knowing that Hb sequence encryption often resides within enhancers with broad expression patterns and does not represent a code of exclusive Hb expression, we were not surprised to observe that expression is not specific to Hb in experimentally validated HbEns.
As additional support for the utility of our model, we find that our predicted Hb enhancers are enriched for a particularly large number of CNS TFBSs compared with TFs known to be active in other tissues. Our experimental data also suggest that Hb enhancers can be divided into independent functional subunits, here tested as TFBS clusters, with similar activities but different sequence structures—an observation that highlights flexibility of the Hb sequence encryption with potential for adaptation to additional functions and the use of different activation mechanisms. The observed biological behaviors of these TFBS clusters were consistent with the known patterns of expression of TF family members predicted to bind them. This raises the possibility that retraining algorithms using subsets of training or predicted sequence sets may define the sequence grammar specific to individual Hb sub-domains and cell types.
Computational methods are becoming increasingly powerful tools for enhancer prediction. Experimental validation rates for computer learning algorithms are comparable to those achieved by experimental ChIP-seq predictions and can be similarly independently correlated with the presence of features known to be present in active enhancers such as known TFBS motifs, specific histone marks, and increased conservation. This study demonstrates that, in addition to the sequence substrate provided by genome-wide ChIP-based strategies, the published literature may serve as a valuable entry point for such analyses of regulatory elements. We demonstrate that even a relatively small curated experimental data set can provide significant insight into the regulatory lexicon of a highly complex anatomical structure like the Hb, and that this vocabulary can likely be dissected and improved in subsequent cycles of investigation and/or by the refinement of the substrate on which it is trained. Therefore, this study adds to the ongoing project of genome annotation by identifying sequences that have a functional role in the Hb. The development of regulatory language is a pivotal step in the prediction of functional variation by inspection of the primary sequence and as such this study makes a significant first step in the development of a Hb lexicon.
Methods
Tissue-specific enhancer models
We extracted 771 human sequences from the VISTA Enhancer Browser (Visel et al. 2007) with validated in vivo enhancer activity in 23 tissues. We were able to retrieve at least 29 sequences each for 11 of these tissues.
Hindbrain enhancer models for mouse, chicken, frog, and zebrafish
Orthologous regions of the human Hb enhancer training set were identified using the liftOver utility from the UCSC Genome Browser (Karolchik et al. 2008). We discarded mapped sequences longer than 5 kb. We successfully mapped 100%, 86%, 74%, and 47% of the 211 Hb enhancers onto the mouse (mm9), chicken (galGal3), frog (xenTro2), and zebrafish (danRer5) genomes respectively (See Supplemental custom tracks 2–5).
Forebrain, midbrain, and limb enhancers identified using ChIP-seq
Genomic regions enriched for EP300 binding in mouse forebrain, midbrain, and limb tissues were extracted from Supplemental Tables 2–4 of Blow et al. (2010). We identified orthologous regions of the mouse coordinates with the liftOver utility from the UCSC Genome Browser (Karolchik et al. 2008). Sequences longer than 1 kb were discarded, resulting in a total of 2199 forebrain sequences, 1909 midbrain sequences, and 3155 limb sequences.
Background genomic sequences
For each enhancer in the training set, 10 controls with similar length, GC, and repeat-content were randomly drawn from the noncoding portion of the corresponding genome.
TFBS mapping
Putative TFBSs were identified by searching the sequences with MAST (Bailey and Elkan 1994) for 775 motifs in TRANSFAC Release 2009.2 (Matys et al. 2006) and JASPAR (Bryne et al. 2008). MAST was run independently on each individual sequence with default setup and parameters.
TF binding to de novo motifs
The identity of the TFs binding to the de novo motifs was queried using STAMP (Mahony and Benos 2007) and JASPAR (Bryne et al. 2008).
Association between TFs and TFBSs
TF annotation for known TFBSs was obtained from TRANSFAC, JASPAR, and the Broad Institute MsigDB database (Subramanian et al. 2005).
TFBS enrichment
Overrepresented TFBSs were determined by comparing the occurrence of the motifs among query sequences and background genomic sequence, and applying Fisher's exact test. We used a P-value threshold of 0.05. When indicated, we adjusted the P-values for multiple testing using the procedure suggested by Benjamini and Hochberg (1995).
Enhancer models
Each enhancer model was trained to distinguish between enhancers specific for a given tissue and other noncoding sequences, randomly drawn from the noncoding human sequence, with length, GC, and repeat-content distributions similar as those observed for the enhancers. The decision of the corresponding classifier was based on the presence or absence of two different types of motifs: 775 corresponding to binding specificities of vertebrate TFs compiled in public databases (TRANSFAC and JASPAR [Matys et al. 2003; Bryne et al. 2008]), and 20 short sequence patterns enriched among the set of enhancers, identified with PRIORITY (Narlikar et al. 2007), which should account for the binding of unknown TFs or TFs with unknown binding specificities. Thus, each sequence was represented as a feature vector indicating the number of matches per base pair to each of these motifs, computed using MAST (Bailey and Gribskov 1998). We built the classifier using linear SVMs (implemented in libsvm [Chang and Lin 2011]), assuming no prior knowledge of TFs active in the different tissues, with the goal being to discover them using the feature weights learned by the classifier.
Extracting homogeneous Hb enhancer data sets
Hb enhancers tend to drive expression in multiple tissues, and even show heterogeneous patterns of expression within the Hb. As a result it is unlikely that we would be able to identify a unique set of sequence features representing all Hb enhancers. Thus, similarly to the approach taken in Narlikar et al. (2010), we selected a large subset of these sequences sharing homogeneous sequence features as an attempt to reduce the sequence heterogeneity among the 212 human Hb enhancers. For this purpose, we repeated the 10-fold cross-validation on five random partitions of the Hb enhancer data set as well as on that of the corresponding controls, and selected only those Hb enhancers that were classified as such in at least 50% of the times in which they were tested for the final training set. Therefore, the final human Hb enhancer data set contained 124 sequences.
Performance assessment of enhancer classifiers
The performance of the classifiers was evaluated in a 10-fold cross-validation, using the area under the ROC curve (AUC). AUC values range from 0.5 (random discrimination) to a theoretical maximum of 1.
Linear SVMs
Training a linear SVM classifier is equivalent to solving the following constrained optimization problem (Shawe-Taylor and Cristianini 2002):
Given the training samples 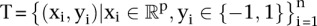 , find the values of
, find the values of 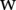 ,
, 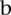 and
and 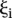 that minimize
that minimize
satisfying the constraints
and
The decision function of the classifier for an unknown sample 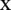 is given by
is given by
The dual form of this problem is:
Given the training samples 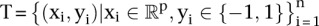 , find the values
, find the values 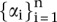 that maximize
that maximize
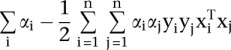 |
satisfying the constraints
and
Samples 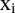 for which
for which 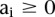 are called support vectors.
are called support vectors.
The vector  can be computed in terms of
can be computed in terms of 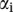 as
as
and, therefore, contains the weighted features of the support vectors.
SVM parameter selection
Linear SVMs have only one parameter, C, which controls the trade-off between errors on the training data and margin maximization. We found that the performance of the Hb enhancer classifier was relatively stable with respect to changes in C. We estimated C based on the training data as 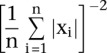 .
.
Additionally, because the training data are unbalanced (there are 10 controls for each enhancer sequence), misclassifications are penalized differently depending on the class of sequences (controls and enhancers), proportionally to the total number of sequences in each class.
Motif rankings
After obtaining a linear SVM model, the weight vector w can be used to decide the relevance of each feature (Guyon et al. 2002). The larger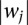 , the more important role of feature j in the decision function. We rank features—in our case, motifs—according to
, the more important role of feature j in the decision function. We rank features—in our case, motifs—according to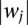 . We exclude de novo motifs from these ranks unless stated otherwise. It is important to note that this interpretation for
. We exclude de novo motifs from these ranks unless stated otherwise. It is important to note that this interpretation for 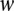 is only valid for linear SVMs.
is only valid for linear SVMs.
Hindbrain genes
We identified a set of 787 human genes likely to be involved in Hb function by retrieving genes with relevant phenotypes from the Online Mendelian Inheritance in Man (OMIM) database (Amberger et al. 2009) and the corresponding orthologs of genes with pertinent annotation in the Mammalian Phenotype (MP) Browser at the Mouse Genome Informatics website, The Jackson Laboratory, Bar Harbor, Maine (http://www.informatics.jax.org).
Genome scans
We applied three human Hb enhancer models trained on (1) the complete Hb data set, (2) the subset of Hb enhancers that are active in the anterior Hb, and (3) the subset of enhancers which functions in the posterior Hb to scan sequences highly conserved across mammals using the Most Conserved Elements database from the UCSC Table Browser (Siepel et al. 2005). Noncoding conserved sequences were determined based on annotation in UCSC Known and RefSeq (Hsu et al. 2006; Pruitt et al. 2009). Sequences within 100 bp of each other were clustered together and clusters <100 bp were excluded from the analysis. Using classifiers trained on the orthologous sequences of the complete data set of human Hb enhancers, we utilized an analogous procedure to predict Hb-specific enhancers in the mouse, chicken, frog, and zebrafish genomes.
Scaled summary Hb score
Each scanned sequence is given three scores, 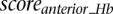 ,
, 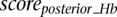 , and
, and 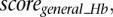 by the classifiers trained on the subset of Hb enhancers that are active in the anterior Hb, the subset of enhancers which functions in the posterior Hb, and the complete Hb data set, respectively. The scores are distributed in the range [−17,15], [−20,15], and [−22,15], respectively (see Supplemental Fig. S4). Scores >0 correspond to putative enhancers active in the anterior Hb, in the posterior Hb, and in the (general) Hb, respectively. Approximately 130,000 sequences scored >0 for at least one of the classifiers, while 40,000 sequences scored >0 for all three. Scores for all classifiers are subsequently linearly scaled according to
by the classifiers trained on the subset of Hb enhancers that are active in the anterior Hb, the subset of enhancers which functions in the posterior Hb, and the complete Hb data set, respectively. The scores are distributed in the range [−17,15], [−20,15], and [−22,15], respectively (see Supplemental Fig. S4). Scores >0 correspond to putative enhancers active in the anterior Hb, in the posterior Hb, and in the (general) Hb, respectively. Approximately 130,000 sequences scored >0 for at least one of the classifiers, while 40,000 sequences scored >0 for all three. Scores for all classifiers are subsequently linearly scaled according to
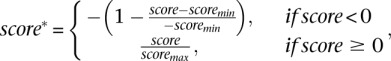 |
where 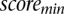 and
and 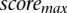 are the minimum and maximum scores obtained in the genome-wide scan, respectively.
are the minimum and maximum scores obtained in the genome-wide scan, respectively.
Finally, we define the scaled summary Hb score as the maximum between 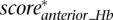 ,
, 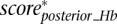 , and
, and 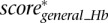 .
.
Association between enhancer predictions and loci
For defining gene loci in the human genome, we used the knownGene and RefSeq annotation tracks available at the UCSC Genome Browser (November 2011). Each locus was defined by one or more overlapping transcripts, prohibiting overlap among different loci. Putative Hb enhancers were associated with loci based on genomic proximity. Thus, each putative Hb enhancer is assumed to target the genes in the nearest locus.
DNase I hypersensitivity
We compared our putative Hb enhancers with human fetal brain, heart, and lung DNase I hypersensitive peaks from http://nihroadmap.nih.gov/epigenomics/.
H3K4me1 and H3K27ac
H3K4me1 and H3K27ac peaks were downloaded from http://genome.ucsc.edu/ENCODE/ (The ENCODE Project Consortium 2011) and correspond to multiple human cell lines (all available to date).
In vivo validation
Candidate Hb enhancers for validation were selected randomly from positively scoring sequences with rank less than or equal to ∼40,000. Controls were selected among sequences that scored among the bottom 1% (i.e., rank greater than or equal to ∼334,000) for all classifiers. Zebrafish were maintained as previously described (Kimmel et al. 1995; Westerfield 2000). Predicted enhancers were amplified by PCR from human genomic DNA and cloned using Gateway Technology (Invitrogen). PCR fragments were TA-cloned into the pCR8/GW/TOPO vector (Invitrogen) then TOPO-cloned using attL1 and attL2 sites into the pT2cfosGW vector for injection into zebrafish embryos. Short fragment sequences for HB01 and HB16 were synthesized as double-stranded oligos, A overhangs added, then cloned as predicted enhancers. At least 100 embryos were injected per construct at the two-cell stage with tol2 transposase as previously described (Fisher et al. 2006). Injected embryos were screened for GFP expression in the CNS at 24 and 48 hpf. Those showing CNS expression were raised to adulthood and crossed to AB zebrafish. G1 embryos were screened for Hb expression at 24, 48, and 72 hpf. GFP positive embryos were live-imaged at 72 hpf using a Carl Zeiss Lumar V12 Stereo microscope with AxioVision version 4.8 software. Embryos were fixed in 4% PFA (Sigma) overnight then post-fixed in 100% acetone (JT Baker) and washed in PBS with 0.5% Tween. Embryos were blocked in 10% goat serum and 1% BSA for two hours, then incubated with chicken anti-GFP (Invitrogen A10262, 1:1000) overnight. After washing, Alexa Fluor 488 goat anti-chicken IgG (Invitrogen A11039, 1:3000) was added and incubated overnight. After washing, embryos were stored in 80% glycerol at 4°C for future imaging.
Acknowledgments
This work was funded in part by the National Institute of Neurological Disease and Stroke (NS062972) to A.S.M., the Intramural Research Program of the NIH, National Library of Medicine to I.O., and a predoctoral training grant (GM07814) to X.R.
Author contributions: This study was conceived by A.S.M. and I.O. The zebrafish transgenic analyses were designed and executed by G.M.B., X.R., T.M., and A.S.M. The development of the Hb classifier and computational analyses were performed by L.T. and I.O. Z.E.S. computed the enrichment in DNaseI HS. All authors contributed to the interpretation of experimental data. The manuscript was written by A.S.M., L.T., I.O., X.R., G.M.B., and Z.E.S.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.139717.112.
Freely available online through the Genome Research Open Access option.
References
- Amberger J, Bocchini CA, Scott AF, Hamosh A 2009. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 37: D793–D796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, Pierson R 2008. The role of the cerebellum in schizophrenia. Biol Psychiatry 64: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G 2005. Brain structures and receptors involved in alertness. Sleep Med (Suppl 1) 6: S3–S7 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Bailey TL, Gribskov M 1998. Methods and statistics for combining motif match scores. J Comput Biol 5: 211–221 [DOI] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, Schaffner W 1981. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27: 299–308 [DOI] [PubMed] [Google Scholar]
- Bardet PL, Schubert M, Horard B, Holland LZ, Laudet V, Holland ND, Vanacker JM 2005. Expression of estrogen-receptor related receptors in amphioxus and zebrafish: Implications for the evolution of posterior brain segmentation at the invertebrate-to-vertebrate transition. Evol Dev 7: 223–233 [DOI] [PubMed] [Google Scholar]
- Barski A, Zhao K 2009. Genomic location analysis by ChIP-Seq. J Cell Biochem 107: 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Berquin PC, Giedd JN, Jacobsen LK, Hamburger SD, Krain AL, Rapoport JL, Castellanos FX 1998. Cerebellum in attention-deficit hyperactivity disorder: A morphometric MRI study. Neurology 50: 1087–1093 [DOI] [PubMed] [Google Scholar]
- Binot AC, Manfroid I, Flasse L, Winandy M, Motte P, Martial JA, Peers B, Voz ML 2010. Nkx6.1 and nkx6.2 regulate α- and β-cell formation in zebrafish by acting on pancreatic endocrine progenitor cells. Dev Biol 340: 397–407 [DOI] [PubMed] [Google Scholar]
- Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2010. ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryne JC, Valen E, Tang MH, Marstrand T, Winther O, da Piedade I, Krogh A, Lenhard B, Sandelin A 2008. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res 36: D102–D106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda-Nieto AC, Pfaff SL, Varela-Echavarria A 2005. Homeodomain transcription factors in the development of subsets of hindbrain reticulospinal neurons. Mol Cell Neurosci 28: 30–41 [DOI] [PubMed] [Google Scholar]
- Chang C-C, Lin C-J 2011. LIBSVM: A library for support vector machines. ACM Trans Intell Syst Technol 2: article 27 doi: 10.1145/1961189.1961199 [Google Scholar]
- Cooper GM, Shendure J 2011. Needles in stacks of needles: Finding disease-causal variants in a wealth of genomic data. Nat Rev Genet 12: 628–640 [DOI] [PubMed] [Google Scholar]
- Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, D'Arcy M, deBerardinis R, Frackelton E, Kim C, et al. 2010. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 15: 637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2011. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046 doi: 10.1371/journal.pbio.1001046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher S, Grice EA, Vinton RM, Bessling SL, McCallion AS 2006. Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science 312: 276–279 [DOI] [PubMed] [Google Scholar]
- Gadd MS, Bhati M, Jeffries CM, Langley DB, Trewhella J, Guss JM, Matthews JM 2011. Structural basis for partial redundancy in a class of transcription factors, the LIM homeodomain proteins, in neural cell type specification. J Biol Chem 286: 42971–42980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghysen A 2003. The origin and evolution of the nervous system. Int J Dev Biol 47: 555–562 [PubMed] [Google Scholar]
- Giguere V 2002. To ERR in the estrogen pathway. Trends Endocrinol Metab 13: 220–225 [DOI] [PubMed] [Google Scholar]
- Gompel N, Prud'homme B, Wittkopp PJ, Kassner VA, Carroll SB 2005. Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature 433: 481–487 [DOI] [PubMed] [Google Scholar]
- Guyon I, Weston J, Barnhill S, Vapnik V 2002. Gene selection for cancer classification using support vector machines. Mach Learn 46: 389–422 [Google Scholar]
- He A, Kong SW, Ma Q, Pu WT 2011. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci 108: 5632–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D 2006. The UCSC known genes. Bioinformatics 22: 1036–1046 [DOI] [PubMed] [Google Scholar]
- Karolchik D, Kuhn RM, Baertsch R, Barber GP, Clawson H, Diekhans M, Giardine B, Harte RA, Hinrichs AS, Hsu F, et al. 2008. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res 36: D773–D779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, de Castro SC, Huang S, Crolla JA, Palmer R, Gregory JW, Taylor D, Cavallo L, Faienza MF, Fischetto R, et al. 2008. SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. J Clin Endocrinol Metab 93: 1865–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF 1995. Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310 [DOI] [PubMed] [Google Scholar]
- Kittler J, Hatef M, Duin RPW, Matas J 1998. On combining classifiers. IEEE Trans Pattern Anal Mach Intell 20: 226–239 [Google Scholar]
- Kiyota T, Kato A, Altmann CR, Kato Y 2008. The POU homeobox protein Oct-1 regulates radial glia formation downstream of Notch signaling. Dev Biol 315: 579–592 [DOI] [PubMed] [Google Scholar]
- Lee D, Karchin R, Beer MA 2011. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res 21: 2167–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E 2003. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet 12: 1725–1735 [DOI] [PubMed] [Google Scholar]
- Mahony S, Benos PV 2007. STAMP: A web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 35: W253–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. 2003. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31: 374–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. 2006. TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34: D108–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA, McCallion AS 2008. Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res 18: 252–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar L, Gordan R, Hartemink AJ 2007. A nucleosome-guided map of transcription factor binding sites in yeast. PLoS Comput Biol 3: e215 doi: 10.1371/journal.pcbi.0030215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I 2010. Genome-wide discovery of human heart enhancers. Genome Res 20: 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB, Janiesch C, Sander M 2005. Expression of Nkx6 genes in the hindbrain and gut of the developing mouse. J Histochem Cytochem 53: 787–790 [DOI] [PubMed] [Google Scholar]
- Nolis IK, McKay DJ, Mantouvalou E, Lomvardas S, Merika M, Thanos D 2009. Transcription factors mediate long-range enhancer-promoter interactions. Proc Natl Acad Sci 106: 20222–20227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan JP, McCallion AS 2010. Genomics of long-range regulatory elements. Annu Rev Genomics Hum Genet 11: 1–23 [DOI] [PubMed] [Google Scholar]
- Pattyn A, Vallstedt A, Dias JM, Sander M, Ericson J 2003. Complementary roles for Nkx6 and Nkx2 class proteins in the establishment of motoneuron identity in the hindbrain. Development 130: 4149–4159 [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. 2006. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444: 499–502 [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR 2009. NCBI Reference Sequences: Current status, policy and new initiatives. Nucleic Acids Res 37: D32–D36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Maunoury S, Gilardi-Hebenstreit P, Charnay P 1998. How to build a vertebrate hindbrain. Lessons from genetics. C R Acad Sci III 321: 819–834 [DOI] [PubMed] [Google Scholar]
- Shawe-Taylor J, Cristianini N 2002. On the generalisation of soft margin algorithms. IEEE Trans Inf Theory 48: 2721–2735 [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaniol P, Bornmann C, Hauptmann G, Gerster T 1996. Class III POU genes of zebrafish are predominantly expressed in the central nervous system. Nucleic Acids Res 24: 4874–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B, Thisse C 2004. Fast release clones: A high throughput expression analysis. In ZFIN Direct Data Submission (http://zfin.org)
- Thisse B, Heyer V, Lux A, Alunni V, Degrave A, Seiliez I, Kirchner J, Parkhill JP, Thisse C 2004. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol 77: 505–519 [DOI] [PubMed] [Google Scholar]
- Thompson JA, Ziman M 2011. Pax genes during neural development and their potential role in neuroregeneration. Prog Neurobiol 95: 334–351 [DOI] [PubMed] [Google Scholar]
- Vanacker JM, Pettersson K, Gustafsson JA, Laudet V 1999. Transcriptional targets shared by estrogen receptor-related receptors (ERRs) and estrogen receptor (ER) α, but not by ERβ. EMBO J 18: 4270–4279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrijzer CP, Van der Vliet PC 1993. POU domain transcription factors. Biochim Biophys Acta 1173: 1–21 [DOI] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA 2007. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res 35: D88–D92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhong J, Wang YQ 2010. Comparative genomic analysis reveals the evolutionary conservation of Pax gene family. Genes Genet Syst 85: 193–206 [DOI] [PubMed] [Google Scholar]
- Waskiewicz AJ, Rikhof HA, Hernandez RE, Moens CB 2001. Zebrafish Meis functions to stabilize Pbx proteins and regulate hindbrain patterning. Development 128: 4139–4151 [DOI] [PubMed] [Google Scholar]
- Westerfield M. 2000 The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio), 4th ed. University of Oregon Press, Eugene, OR. [Google Scholar]