

Abstract
SecA is a central component of the general secretion system that is essential for bacterial growth and thus an ideal target for the development of antimicrobial agents. A series of fluorescein analogs were first screened against the ATPase activity using the truncated unregulated SecA catalytic domain. Rose Bengal (RB) and Erythrosin B (EB) were found to be potent inhibitors with IC50 values of 0.5 µM and 2 µM, respectively. RB and EB inhibit the catalytic SecA ATPase more than the F1F0-proton ATPase. We used three assays to test the effect of these compounds on full length SecA ATPase: in solution (intrinsic ATPase), in membrane preparation, and translocation ATPase. RB and EB show the following trend in terms of IC50 values: translocation ATPase < membrane ATPase < intrinsic ATPase. Very importantly, the potency of these fluorescein analogs in inhibiting the truncated SecA ATPase correlates with their ability to inhibit the biologically relevant protein translocation activity of SecA. The in vitro translocation of proOmpA precursors into membrane vesicles is strongly inhibited by RB with IC50 of about 0.25 µM, making RB the most potent inhibitor of SecA ATPases and SecA-dependent protein translocation thus far. The ability of these compounds to inhibit SecA directly translates into antibacterial effects as well. Our findings show the value of fluorescein analogs as probes for mechanistic studies of SecA functions, and for the potential development of new antimicrobial agents with SecA as the target.
Keywords: SecA, ATPase inhibitor, protein translocation, antimicrobial, membrane protein
Introduction
In all growing cells, the translocation of proteins into various cellular and extra-cellular compartments is essential for maintaining normal physiological functions. In bacteria, more than 30% of the proteins are located in or outside the cellular cytoplasmic membrane. The general secretion (Sec)-pathway mediates the transport of most unfolded proteins across or inserted into the cytoplasmic membrane before these proteins reach their final destinations in the cytoplasmic membrane, periplasmic space or outer membrane.[1] The bacterial Sec-pathway consists of a series of membrane proteins including SecY, SecE, and SecG that constitute an oligomeric complex.[2] SecA is the major component of the bacterial Sec-system that is found both in the cytoplasm and the membrane[3] and functions as an ATPase that provides the energy for the Sec-dependent protein translocation. The intrinsic ATPase activity of the native SecA is relatively low because of the existence of the inhibitory C-terminal domain, while the membrane ATPase could be stimulated by anionic phospholipids. Binding to SecYEG complex and precursor proteins further fully activates the ATPase activity referred to as SecA translocation ATPase.[4] It has been suggested that the ATPase activities are stimulated by anionic phospholipids through conformational changes and the formation of the complex, which apparently negate the inhibitory effect of the C-terminal domain.
Inhibitors of SecA can be potential antimicrobial agents and useful tools for probing the roles of SecA in the secretion and membrane integration of various proteins. They can also be used in studying the conformational changes of SecA during protein translocation. Sodium azide is a well-known SecA inhibitor.[5] However, azide inhibits only the translocation ATPase but not the intrinsic/native and membrane ATPases of SecA.[5a, 6] It has been reported that a natural fungal fermentation product CJ21058 also inhibits the translocation ATPase, though the effect on the intrinsic or membrane ATPase of SecA has not been reported.[7] More recently, a secondary metabolite (pannomycin) from fungi was isolated by antisense-based screening against SecA.[8] This compound only shows weak antibacterial activity with MIC in the mM range, and the inhibitory effect against SecA function was not reported. By virtual screening and optimization, we recently found several thiouracil-based compounds capable of inhibiting the intrinsic SecA ATPase (IC50 20–60 µM).[9] Further optimization of these analogs led to low µM inhibitors.[10] Later, a report by others described a series of thiazolo[4,5-d]pyrimidine derivatives with the most potent compound having an IC50 value of 135 µM against SecA intrinsic ATPase, but showed minor effects on the translocation ATPase.[11]
It has been reported that some halogenated fluorescein analogs could influence the activity of phosphatase as non-hydrolyzable nucleotide analogs.[12] In this work, the effects of several fluorescein analogs were tested against Escherichia coli SecA (EcSecA) and Bacillus subtilis SecA (BsSecA) ATPase activity using truncated SecA and full length SecA under three different settings including in solution (intrinsic ATPase), in membrane preparation, and translocation ATPase in the presence of a precursor protein. Rose Bengal (RB) and Erythrosine B (EB) show strong inhibitory effects on the three forms of SecA ATPase activity and SecA-mediated in vitro protein translocation, as well as antimicrobial effects.
Results and Discussion
Before the discussion of the results, it is important to first describe the various types of experiments and their relevance. As discussed earlier, SecA exerts it transporter functions while integrated into membranes in a bound form with the SecYEG complex. However, SecA’s ATPase is functional in solution alone or in membranes. In addition, SecA itself has a C-terminal regulatory domain, which becomes disengaged upon integration into the membrane. Thus, there are several ways to test inhibitory effects against SecA. The ATPase activity can be examined using the truncated SecA without the C-terminal regulatory domain in solution (e.g., EcN68, unregulated ATPase), SecA alone in solution (regulated intrinsic ATPase), SecA in membrane (membrane ATPase) and SecA in the presence of membrane and precursor proteins (translocation ATPase). For functional assays, we can examine the translocation of proOmpA into membrane vesicles. Of course, the ultimate test is the antimicrobial activity. For practical reasons, the initial screening can be done with the truncated form (unregulated ATPase) because of its ease of experiments and sensitivity. The regulated ATPase actually does not reflect the function of SecA in membranes because of the engagement of the regulatory domain, which is not the case in membranes. In addition, integration into the membrane is known to cause conformational changes, which could perturb the binding domain. Therefore, for reliable results, it is important to test the inhibitory activities using more than one method. Among these methods, the truncated EcN68 ATPase, membrane ATPase, translocation ATPase, and SecA-dependent protein translocation yield the intrinsic ability for the compounds to bind and inhibit the most relevant forms of the transporter/ATPase. On the other hand, experiments using the regulated intrinsic ATPase suggest the ability for the compound to affect the closed state of SecA. It is important to emphasize that because of the membrane nature and the need for complex formation before SecA can function as a transporter, SecA inhibition studies do need to involve all different methods in order to achieve a thorough understanding of the ability of these inhibitors to inhibit SecA ATPase.
A series of commercially available fluorescein analogs (Figure 1) were first screened against E. coli SecA ATPase (EcSecA) using the intrinsic ATPase of the truncated N-terminal catalytic domain (unregulated, EcN68).[13] The IC50 values of fluorescein analogs with significant inhibitory effects are summarized in Table 1. Among the screened compounds, RB and EB are the most effective, with IC50 of 0.5 µM and 2 µM, respectively (Figure 2). Since RB and EB are known to inhibit some ATPases from animal tissues,[14] we tested whether these compounds inhibit other E. coli ATPases such as the F1F0-ATPase. The IC50 values of RB and EB for F1F0-ATPase are about 10 µM and 30 µM, respectively (Figure 2). The data indicate that RB and EB may be general ATPase inhibitors. However, they are more effective on the catalytic SecA ATPase. It has been previously reported that some ATPases from animal tissues can be inhibited by RB and EB through photo-oxidation and subsequent reactions.[15] In our studies, all assays have been performed under the condition of normal room illumination without special light excitation. Moreover, reactions with or without light showed no difference in IC50 (data not shown). Thus the inhibitory effects against SecA ATPase activity are not likely caused by photo-oxidation.
Figure 1.
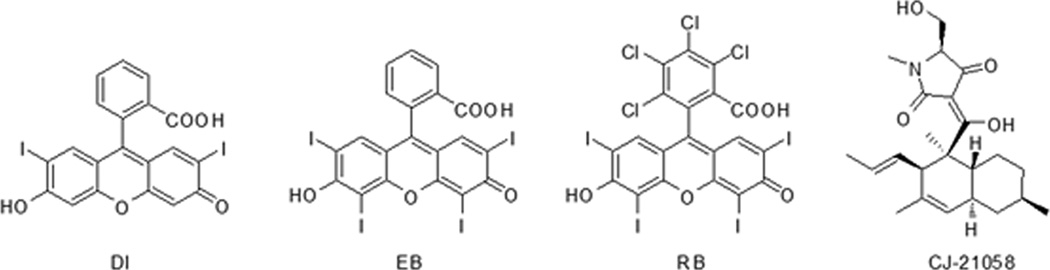
The chemical structures of DI, RB, EB, and CJ-21058.
Table 1.
Screening of fluorescein analogs using EcN68 SecA ATPase
| Chemical* | IC50 |
|---|---|
| Rose Bengal (RB) | 0.5 µM |
| Erythrosin B (EB) | 2 µM |
| Diiodofluorescein (DI) | 30 µM |
| Eosin Y (EY) | 25 µM |
| Dinitrofluorescein (DN) | 50 µM |
| Sodium azide | >10 mM |
Fluorescein analogs were applied to the intrinsic ATPase assay of EcN68 as described in Materials and Methods.
Figure 2. The inhibitory effect of RB and EB against different ATPases.

ATPase activities of the catalytic domain of SecA (EcN68) and the F1F0-proton ATPase were assayed with different concentrations of RB and EB. The inhibitory effects were illustrated by the percentage (%) of remaining ATPase activity as compared to the controls in the absence of inhibitors.
In order to fully understand the ability of these fluorescein analogs to inhibit the biological relevant SecA ATPase, we studied the effect of these compounds on all three forms of the SecA ATPase. Specifically, we next examined the inhibitory effects on the ATPase activity of full-length SecA alone (regulated intrinsic ATPase). As expected, the IC50 values (about 20–30 µM) for RB and EB are higher than with the truncated unregulated SecA ATPase. We further investigated the inhibitory effects of RB and EB on the membrane and translocation ATPases of EcSecA (Table 2). It is interesting to note that both RB and EB show the following trends in terms of their ability/affinity for different forms of SecA ATPase: EcN68 ATPase, translocation ATPase, membrane ATPase, and intrinsic ATPase. With RB, these numbers are 0.5, 0.9, 5, and 25 µM. In the presence of the C-terminal domain in the native regulated form, the IC50 is higher at 25 µM. EB shows similar trends in inhibiting the different forms of SecA, i.e., higher potency against truncated SecA, translocation and membrane ATPase than the regulated intrinsic SecA ATPase. However, the potency of EB is lower than that of RB with IC50 values of about 10–20 µM (Table 2). The significant differences of the sensitivities of three forms of ATPase of EcSecA also indicate that conformational changes of SecA induced by interaction with membranes and precursors may influence the accessibility of the enzyme to inhibitors. We also determined the inhibition profile of RB and EB on SecA from Gram-positive B. subtilis (BsSecA), which has high homology (51% identity) to EcSecA and has much higher intrinsic ATPase activity. As expected, both RB and EB show inhibitory effects on activity of BsSecA intrinsic ATPase, with RB as a strong inhibitor (Table 2).
Table 2.
IC50 of RB and EB* against different forms of ATPases of SecA
| Inhibitor |
RB (µM) |
EB (µM) |
|
|---|---|---|---|
| ATPase | |||
| Intrinsic | 25 | 21 | |
| EcSecA | Membrane | 5 | 12 |
| Translocation | 0.9 | 10 | |
| BsSecA | Intrinsic | 7 | 70 |
RB and EB were applied to the intrinsic, membrane, and translocation ATPase assays as described in the Material and Methods.
As discussed earlier, the ATPase activity is only relevant if its inhibition results in the same effect on protein translocation.[6, 16] Therefore, we further investigated the effects of RB and EB on the SecA-dependent protein translocation in vitro. We found that the in vitro translocation of precursor proOmpA into membrane vesicles is severely inhibited by RB and EB (Figure 3). Interestingly, the protein translocation is about three to four times more sensitive to RB and EB than the translocation ATPase. Consistent with the result of translocation ATPase, RB shows stronger inhibitory effect (with IC50 of 0.25 µM) than EB (with IC50 of 4 µM). Sodium azide is the most well-known SecA ATPase inhibitor; however, the intrinsic ATPase of SecA is not inhibited by sodium azide at concentrations as high as 10 mM. The inhibitory effect of azide against translocation ATPase of SecA and in vitro protein translocation are moderate, with IC50 of 5 mM and 0.6 mM, respectively.[6] On the other hand, RB inhibits the translocation ATPase and protein translocation very efficiently, with IC50 of 0.9 µM and 0.25 µM, respectively, which are about several thousand times more effective than azide.
Figure 3. The inhibitory effects of RB and EB against the SecA-dependent in vitro translocation of proOmpA.

The translocation of proOmpAprecursors into membrane vesicles was assayed in the presence of RB and EB. The insert is the expanded presentation for RB. The inhibitory effects were illustrated by the percentage (%) of translocated proteins as compared to the controls in the absence of inhibitors. The results were presented as line graphs with standard error of the mean.
The SecA-dependent protein translocation is essential for maintaining the normal physiology of bacteria. Therefore, it is important to study the effect of these inhibitors on bacterial growth. These fluorescein analogs inhibit bacterial growth in plate assays (Table 3). Gram-negative bacteria E. coli MC4100 (wild-type) is very resistant to the fluorescein analogs, while its permeable leaky mutant NR698[17] shows high sensitivity. Such results suggest that the outer-membrane barrier is the reason for the observed difference in activity. Among the tested fluorescein analogs, diiodofluorescein (DI), Eosin Y (EY), and dinitrofluorescein (DN) show the minimal inhibitory concentration (MIC) at the mM level, while RB and EB have stronger inhibition with MIC at the µM level. RB also completely inhibits the growth of E. coli NR698 in liquid culture at low concentration levels (50 µM, data not shown). RB demonstrates the same potency of bacteriostatic activity with or without 0.2% glucose in the media (data not shown), suggesting that F1F0-proton ATPase is not the primary target of the inhibition. The observed inhibition effect against bacterial growth validates the idea that SecA inhibitors can be used as antimicrobial agents. The inhibition potency for RB is in the single digit micromolar range, which is similar to the IC50 values obtained using truncated SecA and SecA in the presence of membrane and precursor proteins. In the case of EB, the MIC is much higher than the IC50 values obtained in the enzyme inhibition assays. Many reasons could contribute to such results. A key consideration is permeability. As seen with the results obtained using the wild-type strain of E. coli, minimal inhibition is observed. However, when a leaky mutant (NR698) was used, inhibitory potency increases substantially. It is interesting to note that azide has been reported to inhibit the translocation ATPase of SecA and the transport of precursor proteins across the inner membrane vesicles in vitro.[6] SecA mutants that lack the stimulated translocation ATPase show defects of preprotein translocation in vitro.[16] The in vitro translocation of precursor protein proOmpA into membrane vesicles is also inhibited by RB and EB. The in vitro translocation is even more sensitive to RB and EB than the translocation ATPase of EcSecA. Similar differences are also reported for azide, but the in vitro protein translocation and the cell growth show similar sensitivities.[6] In the case of RB and EB, in vivo growth is significantly less sensitive than in vitro protein translocation. This again may be due to the different membrane permeability of inhibitors. Azide is a small inorganic molecule, while RB and EB are much bigger organic molecules presumably with lower permeability across bacterial membranes. Since the permeability is important for the antibacterial effect of RB and EB, Gram-positive bacteria B. subtilis without the barrier of the outer-membrane was also examined. B. subtilis shows high sensitivities to these fluorescein analogs similar to E. coli NR698 (Table 3). Indeed, RB and EB are very effective against Gram-positive bacteria where permeability is not a major problem.
Table 3.
MIC† of fluorescein analogs on growth of microbes in plate assay
| Bacteria |
E. coli MC4100 | E. coli NR698 | B. subtilis 168 |
|---|---|---|---|
| Chemical* | |||
| Rose Bengal (RB) | >1 mM | 3.1 µM | 3.1 µM |
| Erythrosin B (EB) | >10 mM | 250–500 µM | 250–500 µM |
| Diiodofluorescein (DI) | >3 mM | 200–500 µM | 1 mM |
| Ecosin Y (EY) | NA | 1–2.5 mM | 2.5 mM |
| Dinitrofluorescein (DN) | NA | 10–20 mM | 10 mM |
Minimal inhibition concentrations (MIC) were determined by the concentration of inhibitors showing a clear zone after overnight incubation as compared to the surrounding area.
Fluorescein analogs were applied to the bacterial cells as described in Material and Methods.
In addition to the bacteriostatic studies, we are also interested in seeing whether these inhibitors have bactericidal effect. After one-hour treatment on exponential-phase cells, the CFU was determined after overnight incubation. RB shows strong bactericidal effects in a concentration dependent manner. With 100 µM of RB, cell survival decreases about 10 log units in leaky mutant E. coli NR698 (Figure 4a) and 8 log units in B. subtilis (Figure 4b). Cell density does not drop in the presence of 100 µM RB up to 90 min (data not shown), indicating that the bactericidal effects of RB on both bacteria are not caused by cell lysis. It has been reported that RB can inhibit the growth and kill Staphylococcus aureus in dark with unknown mechanisms, while some halogenated fluoresceins work as the photosensitizer in antimicrobial actions to kill various other bacteria, mainly through photo-oxidation.[18] As discussed earlier, under the experimental condition in this study, photo-oxidation is not likely the primary mechanism of the bacteriostatic and bactericidal effects. Taken together, the results suggest that SecA may be the target of fluorescein analogs, and the inhibition of ATPase and SecA-dependent protein translocation may contribute to the antibacterial effects.
Figure 4. The bactericidal effect of RB on Gram-positive and Gram-negative bacteria.

The bactericidal activities are illustrated by the number of surviving cells as CFU after one hour treatment with various concentrations of inhibitors (gray bar) as compared to the controls (white bar) in the absence of inhibitors.(A)Permeability leaky mutant E. coli NR698; (B) B. subtilis 168.
Because of the literature reports of other fluorescein analogs binding to enzymes containing nucleotide-binding sites,[14a, 19] we conducted in silico modeling using approaches described previously.[20] It needs to be noted that we have results from kinetic experiments indicating the RB and EB are competitive inhibitors against ATP at low ATP concentrations (data not shown). Such results indicate that these compounds bind to the high affinity ATP binding site. Thus the structures of RB, EB, and DI were docked into the high affinity ATP site of EcSecA. RB and EB show very similar binding profiles, while DI shows a different conformation because of the lack of the diiodo-moiety (Figure 5). For comparison, the binding mode of CJ-21058 (Figure 1),[7] a natural product SecA inhibitor, was also examined. RB and CJ-21058 seem to have the same position and with the same orientations.
Figure 5. The docking conformations of DI, EB, RB and CJ-21058 at the EcSecA ATP-site.

Protein secretion system is essential for bacterial viability and virulence; therefore, it has been considered as an ideal target for pharmaceutical interests.[21] The majority of secreted and membrane proteins are mediated by the Sec pathway, making the components of the Sec system potentially valuable for antibiotic targeting.[22] SecA is the central element of the Sec pathway and is highly conserved in bacteria. More importantly, SecA has no human counterpart, making it a good target for the development of new antibiotics.[21] Thus SecA inhibitors have the potential to be new antibacterial agents. In addition to sodium azide, some other SecA inhibitors are reported but not well defined.[7–11, 23] It is worthwhile to mention here that RB and EB are the first inhibitors against all three forms of SecA ATPase with low µM concentration levels. The inhibitory effects of RB and EB against the ATPase and in vitro protein translocation activities of SecA and bacterial growth may lead to some alternative antimicrobial strategies. The fluorescein analogs used in this study are hydroxyxanthenes. Xanthene derivatives are well known and have been used as food additives for some time. Although some xanthenes dyes have safety concerns, ten of those dyes are certifiable as regulated by FDA for food, drug, or cosmetic use.[18d] Rose Bengal is reportedly in phase II trials in a study for the treatment of metastatic melanoma.[24] Erythrosine B (FD&C Red No. 3) is at present the only xanthenes derivative with approval for food use.[25] These fluorescein analogs have several advantages as SecA inhibitors: the convenience of commercial availability, high solubility in water, known chemical structure for further modification, and relatively low or no toxicity for food and drug use.
Conclusion
SecA is essential for bacterial survival and is thus an attractive target for the development of antimicrobial agents. However, activities in this field have been lacking with a few mid- to high-micromolar inhibitors reported. We screened a series of fluorescein analogs and found the first sub-micromolar inhibitors RB, which can serve as a lead structural for the design of new antibacterial agents. It is also important to note that the screening results using the truncated SecA, not the native/regulated form, yielded potency similar to the more biologically relevant test, such as membrane SecA ATPase, translocation SecA ATPase, in vitro protein translocation, and antibacterial experiments.
Materials and Methods
Bacterial strains, medium, and chemicals
E. coli K-12 strain MC4100,[26] NR698 (MC4100 imp4213), a leaky mutant with increased outer membrane permeability[17] from T. Slhavy, BA13 (MC4100 secA13(am) supF(ts))[27] from D. Oliver, and B. subtilis strain 168 (lab stock) were used in this study. Luria-Bertani (LB) liquid and solid (1.5% agar) media with 0.2% glucose were used for bacterial growth. Fluorescein analogs were purchased from Sigma-Aldrich Corp (St. Louis, MO) and were dissolved in water (for Rose Bengal, Erythrosin B, and fluorescein) or 100% DMSO (for diiodofluorescein, Eosin Y, and dinitrofluorescein).
Determination of bacteriostatic and bactericidal concentrations of fluorescein analogs
Plate assay: 0.5 mL culture of bacterial cells (exponential phase, OD600=0.5) was mixed with 4 mL of LB with 0.2% of glucose and 0.75% soft agar and then was poured into petri dishes. After the soft agar solidified, 1 µL of the potential inhibitors was spotted on the surface of the culture. Bacteriostatic effect was judged by the appearance of a clear zone of growth inhibition after overnight incubation at 37°C. Liquid culture assay: Bacterial cells of exponential phase (OD600 about 0.5–0.8) were diluted to OD600=0.05 with LB with 0.2% of glucose. 90 µL of diluted culture was incubated at 37°C with shaking at 1,000 rpm (Eppendorf Thermomixer R, Brinkmann Instruments, Inc.) in the presence of 10 µL of inhibitors or equal volume of water as control. After 14 hours of incubation, cell growth was determined by OD600. Inhibition of cell growth (or bacteriostatic effect) was illustrated by decreasing OD. Bactericidal effect assay: 40 µL inhibitors were added into 360 µL of bacteria cultures (exponential phase, OD600=0.5) while the same volume of water was used as control. After one hour treatment at 37°C, cultures were spread on LB plate after serial dilutions, and the colony forming units (CFU) of surviving cells were enumerated after overnight incubation at 37°C. Inhibitory effect (or bactericidal effect) was illustrated by the decreasing of the log value of CFU. All assays were done at least in triplicate, and the results were presented as line or bar graphs with standard error of the means.
Preparations of various SecA proteins, F1F0-ATPase, proOmpA, and membrane vesicles
The N-terminal catalytic domain of SecA from E. coli (EcN68) was over-expressed from pIMBB28[13a] obtained from A. Economou. EcN68 was used for the early screening because it has higher intrinsic activity and is more sensitive to inhibitors. The full-length SecA from E. coli (EcSecA) and B. subtilis (BsSecA) were over-expressed from pT7-SecA[27] and pT7div[28] respectively, both obtained from D. Oliver. SecA proteins were purified as described.[29] F1F0-ATPase enriched membrane of E. coli strain KY7485[30] obtained from W. Brusilow was prepared as described.[31] F1F0-ATPase was partially purified by sucrose gradient fractionation and then reconstituted into liposomes by dialysis. Non-radiolabeled and 35S-labeled proOmpA were purified as described.[29b] SecA-depleted BA13 membrane vesicles were prepared as described,[32] and washed with 6M urea to reduce endogenous ATPase activity.
In vitro ATPase activity assay
ATPase activity assays were performed as described previously[4a] with minor modifications. For intrinsic and membrane ATPase assays, 50 µL reaction mixtures contained 1.8 µg EcN68, 1.5 µg EcSecA, or 1.5 µg BsSecA, 20 µg ovalbumin, 1.2 mM ATP, 50 mM Tris-HCl (pH7.6), 20 mM KCl, 20 mM NH4Cl, 2 mM Mg(OAc)2, 1 mM DTT, and (for membrane ATPase) 3 µg urea-washed E. coli BA13 membrane. For translocation ATPase assay, reaction mixtures contained 1 µg proOmpA in addition to membranes. For proton ATPase activity, reconstituted-liposomes containing partially purified F1F0-proton ATPase were assayed in the same condition as the intrinsic ATPase. All reactions were carried out at 40°C for an appropriate time in the linear ranges of the activity assay that was determined by the release of inorganic phosphate detected by the photometric method[33] with the absorption measured at 660 nm (SmartSpec Plus, Bio-Rad Laboratories, Inc.). The inhibitory effects were illustrated by the percentage (%) of remaining ATPase activity as compared to the controls in the absence of potential inhibitors. All assays were performed at least in triplicate, and the results were presented as line graphs with standard error of the mean.
In vitro protein translocation assay
Protein translocation assay was carried out as previously described using 35S-labeled proOmpA as a marker.[34] The protease-resistant translocated proteins were analyzed by SDS-PAGE, autoradiographed, and quantified by a densitometer (GS-800 Calibrated Densitometer, Bio-Rad, Hercules, CA).
Molecular simulation of docking complexes
For simulating the binding profiles of DI, EB, RB and CJ-21058, their structures were docked into the ATP site of EcSecA using the DOCK 6 program.[35] Residues within a radius of 6 Å around the center of ATP were defined as the active site to construct a grid. The active site included residues Gly80, Met81, Arg82, His83, Phe84, Gln87, Arg103, Thr104, Gly105, Glu106, Gly107, Lys108, Thr109, Leu110, Arg138, Asp209, Glu210, Arg509, and Gln578. The subsequent computational work was conducted as described previously.[36] Briefly, the docked complexes were solvated by using the TIP3P water model,[37] and then subjected to 500 steps of molecular mechanics minimization and molecular dynamics simulations at 300 K for 1.5 ns using the SANDER module in the AMBER 8 program.[38]
Acknowledgements
This work was supported in part by a NIH grant (GM 34766). We gratefully thank T. Silhavy, D. Oliver, A. Economou, and W. Brusilow for providing the bacterial strains and plasmids, and N. Ni for her help in editing the manuscript. Y. Huang has been funded by a fellowship from the Molecular Basis of Disease Program at Georgia State University.
References
- 1.Papanikou E, Karamanou S, Economou A. Nat Rev Microbiol. 2007;5(11):839–851. doi: 10.1038/nrmicro1771. [DOI] [PubMed] [Google Scholar]
- 2.a Driessen AJ, Nouwen N. Annu Rev Biochem. 2008;77:643–667. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]; b Mori H, Ito K. Trends Microbiol. 2001;9(10):494–500. doi: 10.1016/s0966-842x(01)02174-6. [DOI] [PubMed] [Google Scholar]
- 3.Cabelli RJ, Dolan KM, Qian LP, Oliver DB. J Biol Chem. 1991;266(36):24420–24427. [PubMed] [Google Scholar]
- 4.a Lill R, Dowhan W, Wickner W. Cell. 1990;60(2):271–280. doi: 10.1016/0092-8674(90)90742-w. [DOI] [PubMed] [Google Scholar]; b Wang L, Miller A, Kendall DA. J Biol Chem. 2000;275(14):10154–10159. doi: 10.1074/jbc.275.14.10154. [DOI] [PubMed] [Google Scholar]
- 5.a Nakane A, Takamatsu H, Oguro A, Sadaie Y, Nakamura K, Yamane K. Microbiology. 1995;141(Pt 1):113–121. doi: 10.1099/00221287-141-1-113. [DOI] [PubMed] [Google Scholar]; b Knott TG, Robinson C. J Biol Chem. 1994;269(11):7843–7846. [PubMed] [Google Scholar]
- 6.Oliver DB, Cabelli RJ, Dolan KM, Jarosik GP. Proc Natl Acad Sci U S A. 1990;87(21):8227–8231. doi: 10.1073/pnas.87.21.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugie Y, Inagaki S, Kato Y, Nishida H, Pang CH, Saito T, Sakemi S, Dib-Hajj F, Mueller JP, Sutcliffe J, Kojima Y. J Antibiot (Tokyo) 2002;55(1):25–29. doi: 10.7164/antibiotics.55.25. [DOI] [PubMed] [Google Scholar]
- 8.Parish CA, de la Cruz M, Smith SK, Zink D, Baxter J, Tucker-Samaras S, Collado J, Platas G, Bills G, Diez MT, Vicente F, Pelaez F, Wilson K. J Nat Prod. 2009;72(1):59–62. doi: 10.1021/np800528a. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Huang YJ, Tai PC, Wang B. Biochem Biophys Res Commun. 2008;368(4):839–845. doi: 10.1016/j.bbrc.2008.01.135. [DOI] [PubMed] [Google Scholar]
- 10.Chen W, Huang YJ, Gundala SR, Yang H, Li M, Tai PC, Wang B. Bioorg Med Chem. 2010;18(4):1617–1625. doi: 10.1016/j.bmc.2009.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang MY, De Jonghe S, Segers K, Anne J, Herdewijn P. Bioorg Med Chem. 2011;19(1):702–714. doi: 10.1016/j.bmc.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 12.Mignaco JA, Lupi OH, Santos FT, Barrabin H, Scofano HM. Biochemistry. 1996;35(13):3886–3891. doi: 10.1021/bi9518353. [DOI] [PubMed] [Google Scholar]
- 13.a Karamanou S, Vrontou E, Sianidis G, Baud C, Roos T, Kuhn A, Politou AS, Economou A. Mol Microbiol. 1999;34(5):1133–1145. doi: 10.1046/j.1365-2958.1999.01686.x. [DOI] [PubMed] [Google Scholar]; b Karamanou S, Sianidis G, Gouridis G, Pozidis C, Papanikolau Y, Papanikou E, Economou A. FEBS Lett. 2005;579(5):1267–1271. doi: 10.1016/j.febslet.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 14.a Morris SJ, Silbergeld EK, Brown RR, Haynes DH. Biochem Biophys Res Commun. 1982;104(4):1306–1311. doi: 10.1016/0006-291x(82)91392-4. [DOI] [PubMed] [Google Scholar]; b Fricke U. Br J Pharmacol. 1985;85(2):327–334. doi: 10.1111/j.1476-5381.1985.tb08865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Silbergeld EK, Anderson SM, Morris SJ. Life Sci. 1982;31(10):957–969. doi: 10.1016/0024-3205(82)90167-9. [DOI] [PubMed] [Google Scholar]
- 15.a Glaser E, Cadenas E, Andell S, Ernster L. Acta Chem Scand B. 1988;42(3):175–182. doi: 10.3891/acta.chem.scand.42b-0175. [DOI] [PubMed] [Google Scholar]; b Watson BD, Haynes DH. Chem Biol Interact. 1982;41(3):313–325. doi: 10.1016/0009-2797(82)90108-9. [DOI] [PubMed] [Google Scholar]
- 16.Sianidis G, Karamanou S, Vrontou E, Boulias K, Repanas K, Kyrpides N, Politou AS, Economou A. EMBO J. 2001;20(5):961–970. doi: 10.1093/emboj/20.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz N, Falcone B, Kahne D, Silhavy TJ. Cell. 2005;121(2):307–317. doi: 10.1016/j.cell.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 18.a Kim YS, Park SJ, Lee EJ, Cerbo RM, Lee SM, Ryu CH, Kim GS, Kim JO, Ha YL. J Food Sci. 2008;73(7):C540–C545. doi: 10.1111/j.1750-3841.2008.00879.x. [DOI] [PubMed] [Google Scholar]; b Banks JG, Board RG, Carter J, Dodge AD. J Appl Bacteriol. 1985;58(4):391–400. doi: 10.1111/j.1365-2672.1985.tb01478.x. [DOI] [PubMed] [Google Scholar]; c Rasooly A, Weisz A. Antimicrob Agents Chemother. 2002;46(11):3650–3653. doi: 10.1128/AAC.46.11.3650-3653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Waite JG, Yousef AE. Adv Appl Microbiol. 2009;69:79–98. doi: 10.1016/S0065-2164(09)69003-1. [DOI] [PubMed] [Google Scholar]
- 19.a Jacobsberg LB, Kantrowitz ER, Lipscomb WN. J Biol Chem. 1975;250(24):9238–9249. [PubMed] [Google Scholar]; b Yip BP, Rudolph FB. J Biol Chem. 1976;251(22):7157–7161. [PubMed] [Google Scholar]
- 20.Zheng S, Kaur G, Wang H, Li M, Macnaughtan M, Yang X, Reid S, Prestegard J, Wang B, Ke H. J Med Chem. 2008;51(24):7673–7688. doi: 10.1021/jm701635j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephens C, Shapiro L. Chem Biol. 1997;4(9):637–641. doi: 10.1016/s1074-5521(97)90217-9. [DOI] [PubMed] [Google Scholar]
- 22.Economou A. Expert Opin Ther Targets. 2001;5(2):141–153. doi: 10.1517/14728222.5.2.141. [DOI] [PubMed] [Google Scholar]
- 23.Akula N, Zheng H, Han FQ, Wang N. Bioorg Med Chem Lett. 21(14):4183–4188. doi: 10.1016/j.bmcl.2011.05.086. [DOI] [PubMed] [Google Scholar]
- 24.a Thompson JF, Hersey P, Wachter E. Melanoma Res. 2008;18(6):405–411. doi: 10.1097/CMR.0b013e32831328c7. [DOI] [PubMed] [Google Scholar]; b Foote MC, Burmeister BH, Thomas J, Mark Smithers B. Melanoma Res. 2010;20(1):48–51. doi: 10.1097/CMR.0b013e328331caa2. [DOI] [PubMed] [Google Scholar]
- 25.Anonymous. Vol. 1. U.s Government Printing Office; Washington, D.C.: 2011. [Google Scholar]
- 26.Casadaban MJ. J Mol Biol. 1976;104(3):541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 27.Cabelli RJ, Chen LL, Tai PC, Oliver DB. Cell. 1988;55(4):683–692. doi: 10.1016/0092-8674(88)90227-9. [DOI] [PubMed] [Google Scholar]
- 28.McNicholas P, Rajapandi T, Oliver D. J Bacteriol. 1995;177(24):7231–7237. doi: 10.1128/jb.177.24.7231-7237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.a Chen X, Brown T, Tai PC. J. Bacteriol. 1998;180(3):527–537. doi: 10.1128/jb.180.3.527-537.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen X, Xu H, Tai PC. J Biol Chem. 1996;271(47):29698–29706. doi: 10.1074/jbc.271.47.29698. [DOI] [PubMed] [Google Scholar]
- 30.Kanazawa H, Miki T, Tamura F, Yura T, Futai M. Proc Natl Acad Sci U S A. 1979;76(3):1126–1130. doi: 10.1073/pnas.76.3.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster DL, Fillingame RH. J Biol Chem. 1979;254(17):8230–8236. [PubMed] [Google Scholar]
- 32.Tai PC, Tian G, Xu H, Lian JP, Yu JN. Methods Cell Biol. 1991;34:167–187. [PubMed] [Google Scholar]
- 33.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. Anal Biochem. 1979;100(1):95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Na B, Yang H, Tai PC. J Bacteriol. 2008;190(4):1413–1418. doi: 10.1128/JB.01633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a Ewing TJ, Makino S, Skillman AG, Kuntz ID. Journal of computer-aided molecular design. 2001;15(5):411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]; b Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. J. Comput. Aided Mol. Des. 2006;20(10-11):601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 36.a Li M, Wang B. Biochem. Biophys. Res. Commun. 2006;347(3):662–668. doi: 10.1016/j.bbrc.2006.06.179. [DOI] [PubMed] [Google Scholar]; b Li M, Wang B. J. Mol. Model. 2007;13(12):1237–1244. doi: 10.1007/s00894-007-0245-0. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 38.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. Journal of computational chemistry. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]