

Abstract
Platforms based on thin enzyme/DNA films were used in two-tier screening of chemicals for reactive metabolites capable of producing toxicity. Microsomes were used for the first time as sources of cytochrome (cyt) P450 enzymes in these devices. Initial rapid screening involved electrochemiluminescent (ECL) arrays featuring spots containing ruthenium poly(vinylpyridine), DNA, and rat liver microsomes or bicistronically expressed human cyt P450 2E1 (h2E1). Cyt P450 enzymes were activated via the NADPH/reductase cycle. When bioactivation of substrates in the films gives reactive metabolites, they are trapped by covalent attachment to DNA bases. The rate of increase in ECL with enzyme reaction time reflects relative DNA damage rates. “Toxic hits” uncovered by the array were studied in structural detail by using enzyme/DNA films on silica nanospheres as “nanoreactors” to provide nucleobase adducts from reactive metabolites. The utility of this synergistic approach was demonstrated by estimating relative DNA damage rates of three mutagenic N-nitroso compounds and styrene. Relative enzyme turnover rates for these compounds using ECL arrays and LC-UV-MS correlated well with TD50 values for liver tumor formation in rats. Combining ECL array and nanoreactor/LC–MS technologies has the potential for rapid, high-throughput, cost-effective screening for reactive metabolites and provides chemical structure information that is complementary to conventional toxicity bioassays.
New drugs and chemicals intended for agricultural, cosmetic, or household applications must be proven safe for human exposure. In the pharmaceutical industry, over $1 billion is required at present to develop a single drug candidate, but clinical test failures approach 30% due to toxicity.1 Ultimately, the general cost of drugs is linked to how far candidates advance in the pipeline before toxicity is discovered.
In current practice, suites of biological tests are used to predict chemical and drug toxicity, but the 30% failure rate suggests the need for fresh approaches. While valuable, existing cytotoxicity bioassays2 can give false positives or negatives due to cell line limitations3 and are throughput limited.2 Animal models often fail due to fundamental interspecies physiological differences.4 Emerging methodologies are attempting to address several of these drawbacks.3,5,6 In the present paper, we demonstrate the synergism of a novel two-tier mechanism- and chemical structure-based approach to in vitro toxicity screening using novel microsome/DNA arrays and nanoreactors with the potential to ameliorate drug and chemical development and lower associated costs when implemented along with established toxicity bioassays.
The toxicity of xenobiotic chemicals is often due to the reactivity of metabolic products toward DNA, proteins, and other biomolecules.7 Human cell lines or cytochrome (cyt) P450-transformed Ames bacteria3d can be used to assay metabolism-based toxicity via cell viability, but offer limited structural details8 that are valuable for rational redesign of the target molecule.2 We recently developed metabolism-based toxicity screening approaches featuring purified cyt P450 and DNA films prepared by the versatile layer-by-layer method,6 which enables facile multi-component film fabrication of biomacromolecules.9 Electro-optical arrays utilizing these DNA/enzyme films featuring electrochemiluminescence (ECL) detection provide the potential for high-throughput reactive metabolite screening.6d, e We have also demonstrated LC–MS/MS structural and formation rate analyses,10 with the most sensitive approach employing DNA/enzyme films on nanoparticles to obtain metabolites and DNA adducts.11 Common features in these systems are that natural enzyme reactions produce reactive metabolites that are captured by a nucleophile-trapping reaction with DNA. Purified enzymes in these sensors were activated by exploiting the well-known peroxide shunt mechanism by exposure to small quantities of H2O2.7 Sensor exposure and detection takes less than a few minutes, improving upon lengthy (days) bioassays.3
Despite the demonstrated utility of ECL arrays, a significant time- and cost-related bottleneck in their fabrication is enzyme generation and purification. In order to translate these arrays into wider use, more readily available sources of enzymes are needed. One such source is tissue-specific microsomes, which are used extensively in the drug industry for conventional metabolic screening.12 Microsomes are vesicles of fragmented endoplasmic reticulum that contain lipids, collections of cyt P450 enzymes, and their natural reduction partner, cyt P450 reductase (CPR).13 The presence of the reductase allows for cyt P450 activation by protocols featuring electron donation from NADPH via the reductase, the same as in vivo.7
Information on cyt P450 isoform specificity is often necessary in order to grasp the complete metabolic landscape–i.e., to avoid drug–drug interactions from coadministered medications or to design drugs that are metabolized via multiple pathways.14 Single cyt P450 isoforms can be expressed in bacteria along with the reductase through bicistronic plasmid transformation.12 The bicistronic plasmid ensures that both a cyt P450 isoform of interest and the reductase are expressed concurrently. After bacterial lysing, the end product is a membrane material that is analogous to the mammalian microsome but contains only a single cyt P450 and reductase.
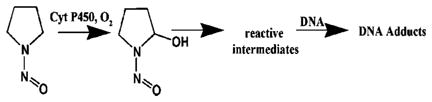
In this report, we describe the novel use of rat liver microsomes (RLM) and bicistronic human cyt P450 2E1 (h2E1) membrane films as enzyme sources in ECL arrays for toxicity screening. Combining these arrays with DNA adduct detection using RLM/DNA or h2E1/DNA nanoreactors and LC–MS (Scheme 1) identifies substrates and enzymes that make reactive intermediates and provides formation rates and structures of resulting nucleobase adducts. As model substrates, we include here N-nitroso compounds that exert in vivo carcinogenic effects after bioactivation by cyt P450-mediated α-hydroxylation. The general pathway is illustrated for N-nitrosopyrrolidine (NPYR)15 (eq 1):
Scheme 1. Microsome/DNA Films and Reactions on Nanoreactors (Left) or Array Chip (Right)a.

a A layer of cationic polymer is initially deposited (blue ribbon in circle, e.g., poly(ethyleneimine (PEI) or [Ru(bpy)2PVP10]2+ (RuPVP), where bpy = bipyridine) followed by layers of negative DNA, polycation, and microsomes (membrane is brown, CPR = red, and cyt P450 = green). NADPH generated by an enzyme reaction in solution reduces CPR, which transfers electrons to cyt P450s. O2 and Cyt P450s can combine to convert substrate to reactive metabolites that form DNA adducts in the film. ECL is detected with a CCD camera on the array upon application of +1.25 V vs Ag/AgCl and monitors DNA adduct accumulation. Hydrolysis releases labile DNA adducts from the nanoreactors for capLC–MS analysis.
 |
(1) |
Styrene is converted to styrene oxide16 by cyt P450s to form mainly guanine adducts17 and was also used as a test substrate to provide a less toxic test compound compared to the N-nitroso substrates.
EXPERIMENTAL SECTION
Reagents and Materials
Bicistronic human cytochrome P450 2E1 (h2E1) - reductase cDNA was a generous gift from Prof. T. Kamataki (Hokkaido University, Japan). XbaI/NdeI-digested human cyt P450 2E1 (h2E1) cDNA was ligated with T4 DNA ligase to the XbaI/NdeI-digested bicistronic plasmid. Proteins were expressed in transformed DH5α Escherichia coli following established protocols, and membranes were collected after French press cell lysis.18 The h2E1 membrane fractions contained 0.3 mg mL−1 cyt P450 2E1, 1.4 mg mL−1 reductase, and 13 mg mL−1 total protein.
Ruthenium metallopolymer ECL catalyst RuPVP was synthesized and characterized following established protocols.19 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) was from Toronto Research Chemicals (Ontario, Canada). N-Nitrosopiperidine (NPIP), N-nitrosopyrrolidine (NPYR), and all other chemicals were from Sigma. Rat liver microsomes purchased from BD Gentest (Woburn, MA) were accompanied with a protein analysis providing approximate enzyme activities or amounts including activity identification of different P450 families present in the microsomes. The total protein content in the RLM was 20 mg mL−l1, total P450 was measured by the method of Omura and Sato20 at 590 pmol (mg of total protein)−1; ~0.6 mg of P450 (mL RLM)−1, CPR activity (330 nmol of cyt c reduced (mg of total protein × min)−1 corresponding to 0.3 unit (mg of total protein)−1; 1 unit approximates 1 μmol of cyt c reduced/min; ~0.6 mg of CPR (mL RLM)−1) was determined from a cyt c reduction assay, and cyt b5 (760 pmol (mg of total protein)−1) was measured spectrophotometrically. The following cyt P450 families were reported to have measurable activities in the RLM (as measured using the denoted specific probe assay for each isoform in picomoles of product produced per milligram of total protein per minute (pmol (mg × min)−1): cyt P450 3A (testosterone 6α-hydroxylase, 3300), cyt P450 2C (testosterone 16α-hydroxylase, 5100), cyt P450 2E1 (p-nitrophenol hydroxylase, 1100), cyt P450 1A (7-ethoxyresorufin O-deethylase, 180), and cyt P450 4A ([14C] lauric acid-hydroxylase, 1200).
Film Fabrication
Array films containing RLM and h2E1 were formed on a pyrolytic graphite (PG) block electrode using established protocols6d from the following solutions: 1 mg mL−1 PEI (50 mM NaCl), 2 mg mL−1 calf thymus DNA (10 mM Tris pH 7.4, 50 mM NaCl), 2.5 mg mL−1 RuPVP 88 (88% H2O/12% ethanol), 2.5 mg mL−1 RuPVP 50 (50% H2O/50% ethanol), and ~15–20 mg mL−1 total protein RLM or h2E1 (in 250 mM sucrose 10 mM Tris pH 7.4). Array spots were created on the PG block serving as the array chip by exposing 1 μL of each polymer or protein adsorbate on a demarked array location for 15 min, followed by H2O rinsing and N2 drying. The first RuPVP layer was adsorbed from a solution containing higher concentrations of ethanol, RuPVP 88 (150 μL), which spreads over the entirety of the block. This step was found to be necessary to generate sufficient RuPVP coverage for eventual ECL production.6 During this application, some RuPVP adsorbs nonspecifically to the PG, but subsequent controlled-volume (i.e., localized) DNA, RuPVP, and enzyme applications ensure a localized film spot, and the analytical signal is easily distinguishable from the background. Final film formation was DNA/(RuPVP88/DNA)2(RuPVP50/DNA/RLM(or h2E1)/(RuPVP88/DNA/RLM(or h2E1))2. RLM or h2E1 as well as subsequent layers was adsorbed at 4 °C. These are denoted as RLM or h2E1 films. Similar films formed on 500-nm-diameter silica beads (Polysciences, Warrington, PA) also followed an established protocol.11 Briefly, 200 μL of silica beads was added to 800 μL of PEI for 20 min. followed by centrifugation and DI water rinsing steps.11 The beads were then resuspended in 1 mL of DNA solution, allowed to adsorb, and then removed and rinsed. Similar steps were followed for the microsome adsorption. These films had the following architecture: PEI/DNA/RLM (or h2E1).
Enzyme Reactions
Safety note: Styrene, nitrosamines, and their metabolites are suspected carcinogens. All procedures were done while wearing gloves, and under closed hoods. Substrate (1 mM) in NADPH generating system (10 mM glucose 6-phosphate, 4 units of glucose-6-phosphate dehydrogenase, 10 mM MgCl2, 0.80 mM NADP+, 50 mM MES buffer, pH 6.0) solution was exposed to the array spots (1 μL) or the silica beads (200 μL). Reaction was stopped by rinsing arrays with water or centrifuging microspheres from solution. ECL was imaged after placing the PG array in a dark gel doc housing (Syngene ChemiGeniusQ, Fredrick, MD) in an open top electrochemical cell.6d A CCD camera (QI Cam (Q-Imaging), 12-bit (0–4095 gray scales), 10 000 electron linear full-well capacity, 35% QE at 600 nm) collected signal (pixel binning high sensitivity mode) upon application of +1.25 V (CH Instruments 1232, Austin, TX) versus Ag/AgCl for 20s. Signal integration data analysis was performed using Syngene Gene Tools v3.06. Faux color was added to the b/w CCD image using Irfanview and the included Adobe8F filters followed by .tif file and additional color enhancements using Photoshop CS software. Added color is defined by user preferences and is not absolute.
LC–MS Sample Workup
Silica enzyme/DNA nanoreactors were removed from the reaction medium by centrifugation.11 Reactive metabolites were analyzed by directly injecting the supernatant medium into the capLC–MS trapping column (see below). Nucleobase adduct release was done via neutral thermal hydrolysis10 in which silica nanoreactors were resuspended in 250 μL of DI H2O and boiled for 15 min, followed by rapid cooling on ice. The nanoreactors were then centrifuged from the hydrolysate. The hydrolysate was filtered using a 3000-Da cutoff filter (Amicon, Beverly, MA) before injection.
Capillary LC–MS/MS
The capillary chromatograph (Waters capLC-XE) featured a 150-mm Atlantis dC18 column, 300-μm i.d., 5-μm particle size, and 23.5-mm Atlantis dC18 trapping column, 0.18-mm i.d., 5-μm particle size. A similar procedure as reported earlier was used.10b,11 Briefly, three 10-μL injections for each analysis were loaded into the trapping column at 4.25 μL min−1 and washed with water for 2 min at 10 μL min−1. Trapped adducts were then back-flushed onto the analytical column at 4.25 μL min−1 using the following gradient: 5 min 10% B, 10 min 10–30% B, 20 min 30% B, 5 min 30–10% B, 5 min 10% B (A, 10 mM acetate buffer pH 5.5; B, methanol). Electrospray ionization mass spectrometry (ESI-MS) employed a Micromass Quattro II (Beverly, MA) operated in positive ion mode (ESI+). The cone voltage was 15 V, collision energy 20 eV, and collision gas (Ar) pressure 5 × 10−3 mbar.
RESULTS AND DISCUSSION
Reactive Metabolite Screening with ECL Arrays
Figure 1 shows a reconstructed ECL array image of layered RLM and h2E1 film spots (with DNA and the RuPVP ECL-emitting polymer) exposed to the denoted substrates and NADPH for various times in seconds, along with negative internal controls. Comparing all responses to their respective 0-s exposure spots (first spots on the left), ECL from array spots exposed to N-nitroso substrates increased more rapidly with reaction time than those exposed to styrene. The mechanism for DNA damage detection in the ECL spots is similar to that elucidated by Thorp for electrochemical oxidation of DNA in solution by metal complexes.21 The electrode oxidizes the catalyst RuIIPVP to RuIIIPVP, which then oxidizes intact guanines on the DNA, yielding an electronically excited RuII*PVP that emits visible light (ECL).19 The key role of guanine oxidation in ECL signal development for RuIIPVP/DNA biosensors was demonstrated in previous studies with films of polynucleotides having only a single base type. Of these, only polyG reacted with RuIIIPVP,19 suggesting that only guanine reacts in the experiments with DNA. The faster the reaction of RuIIIPVP is with DNA in this electrocatalytic process (Scheme 1), the larger the ECL response. Damaged DNA gives more ECL than intact ds-DNA because, as adducts form on the bases, ds-DNA is partly disrupted and guanines are more closely approached by the redox catalyst centers, increasing the reaction rate and the associated ECL output.19 Thus, any adduct that disrupts the double helix (i.e., any nucleobase adduct positional isomer) will produce a faster RuIIIPVP-guanine redox reaction than intact ds-DNA and enhance ECL. The detection approach used in the ECL arrays has the ability to detect virtually all modes of metabolite-related DNA damage. Thus, increased ECL in the array spots after the enzyme reaction reflects DNA damage (Figure 1). In particular, a higher rate of ECL increase with enzyme reaction time indicates higher reactivity of specific metabolites with DNA.
Figure 1.

Reconstructed array data demonstrating ECL from spots of RuPVP/DNA/RLM (labeled RLM) or RuPVP/DNA/h2E1 (labeled h2E1) exposed to 1 mM concentration of denoted substrate using enzymatic NADPH regeneration for denoted time (in s). Data for respective substrates are from same array run. Control is an identical array (RLM; h2E1 was similar (not shown)) not exposed to reaction solutions. Internal controls C1 = 120-s exposure to NADPH solution only; C2 = 120-s exposure to substrate (no NADPH) only.
The controls located on the bottom of Figure 1 are spots not exposed to substrate. This control strip reveals a small amount of ECL flux across the array chip, but this is insignificant compared to that seen in the analytical spots, demonstrating that substrate exposure is necessary for increasing light emission. The negative (C1, C2) internal controls show negligible ECL increase over 2 min demonstrating the need for combined NADPH and substrate exposure to increase ECL. Taken together, these array responses suggest the production of reactive metabolites from the substrates, consistent with the activation of cyt P450 by the NADPH-dependent reductase. Results with these microsome-based arrays are comparable to those of arrays using the pure enzymes for other substrates that make reactive metabolites.10
Overall, Figure 1 demonstrates the raw ECL array data and offers merely a qualitative vantage point to gauge each substrate reaction. In order to elucidate quantitative information, the raw ECL data was integrated and the ECL increase versus reaction time charted. Figure 2 shows this percent ECL increase for each of the exposed substrates from RLM (a) and h2E1 (b) spots processed from the raw array data in Figure 1. The percent increase was measured relative to the amount of ECL produced from the 0-s spots (i.e., an internal standard) and is corrected for background ECL variation from a nonexposed control (averaged from n = 3 trials). The spots containing h2E1 generate more ECL (Figure 2b), indicative of both NNK and NPYR substrate reaction rate with cyt P450 2E1 and increased amount of protein on the array spot based on quartz crystal microbalance (QCM) measurements (Supporting Information (SI), Figure S1/Table S1).
Figure 2.

Percent ECL increase vs reaction time for (a) RLM and (b) h2E1 arrays exposed to NPYR (black circles), NNK (blue squares), NPIP (green diamonds), or styrene (red triangles). Control (open circles, dash) in (a) is the ECL increase on a RLM array not exposed to any xenobiotic solution.
The initial slopes of plots in Figure 2 are indicative of the rate of DNA damage elicited by each substrate. Initial slopes of ECL and electrochemical enzyme/DNA toxicity sensors made with pure cyt P450s were previously shown to be related to relative DNA damage rates by correlation with rates of formation of major guanine adducts measured directly by LC–MS after hydrolysis.2d,6a, d,10 These slopes measure the relative enzyme turnover rates for conversion of each compound into DNA-reactive metabolites. Of the substrates employed here, each N-nitroso compound showed similar ECL versus reaction time slopes while styrene demonstrated a much smaller ECL increase over the same time range (Figure 2). Therefore, the N-nitroso compounds show greater rates of DNA-reactive metabolite formation, while the styrene metabolite is less reactive. The array response is in accord with the expected in vivo response as N-nitroso compounds are indeed much more toxic compared to styrene, as discussed further below.
Inhibition Studies
The RLM used in the ECL array chip contains several cyt P450 isoforms each with varying substrate specificity. The use of selective inhibitors in the RLM modified ECL arrays facilitates the elucidation of the predominant microsomal enzyme(s) responsible for the metabolism of a screened compound. Figure 3a demonstrates the NPYR array response on RLM films with and without the concurrent presence of 4-methyl pyrazole (4-MP), a known selective inhibitor of cyt P450 2E1.7b Figure 3b is the corresponding % ECL response vs reaction time in the presence and absence of the inhibitor. Figure 3c–d shows the corresponding responses from h2E1 array spots, that contain only one P450 isoform, cyt P450 2E1.
Figure 3.

Inhibition by 4-MP. (a) Reconstructed data showing ECL response from RLM spots on same array exposed to 1 mM NPYR without (top) and with (bottom) 50 μM 4-MP as selective cyt P450 2E1 inhibitor for denoted times (s). (b) % ECL change for RLM spots not exposed (black squares) and exposed (blue circles) to 4-MP. (c) Similar reconstructed data as (a) using h2E1 spots. (d) % ECL change for h2E1 spots not exposed (black squares) and exposed (blue circles) to 4-MP.
Figure 3a shows that the ECL increase is effectively eliminated upon addition of the inhibitor to RLM spots. The muted ECL increase (Figure 3b) suggests that 4-MP binds in the microsomal cyt P450 2E1 active site, blocking NPYR bioactivation and further DNA adduct formation. In addition, these results demonstrate that cyt P450 2E1 is the major NPYR metabolizing enzyme in the RLM. The use of the h2E1 bicistronic material acts as a positive control for the RLM inhibitor findings as only cyt P450 2E1 is present in these films. Figure 3c, d shows a similar muting of the ECL upon the addition of 4-MP to the h2E1 spots in the presence of NPYR, confirming the role of cyt P450 2E1 in the metabolism of this compound.
LC–MS/MS Analysis of Nanoreactor Products
Substrate reactions catalyzed by “nanoreactors” consisting of microsome/DNA films on 500-nm-diameter silica beads yielded products for analysis by LC–MS. This approach provided relative rates of formation along with structures of metabolites and DNA adducts using similar films and time scales as ECL arrays. Figure 4 shows single reaction monitoring (SRM) MS–MS chromatograms of major guanine adducts on microsome/DNA nanoreactors resulting from enzymatic conversion of the N-nitroso substrates. Individual nucleobase adducts were formed and then ejected from the DNA on the nanoparticles by neutral thermal hydrolysis (NTH)10 in preparation for the analyses. Insets show an expected predominant adduct and fragmentation pattern consistent with the MS results based on previous reports,15,22 although other adducts with identical m/z are probable (SI, Scheme S2).15 Amounts of guanine adduct increased linearly with substrate exposure over several minutes, as illustrated by Figure 4d for NPYR–DNA adducts from h2E1 and RLM films. This shows that DNA adducts accumulated the reactive metabolites, in the films with time, consistent with ECL array results (Figures 1 and 2).
Figure 4.

SRM-MS chromatograms measuring total ion current for predominant guanine adduct (m/z shown in inset) produced via exposure of (a) NPYR, (b) NNK, and (c) NPIP to RLM/DNA films on silica nanospheres followed by DNA hydrolysis. Insets show examples of relevant guanine adducts and fragmentation patterns consistent with each chromatogram. (d) Integrated UV diode array measured peak area increase for NPYR–guanine adducts with reaction time from both RLM and h2E1 films.
Detailed bioactivation and DNA adduct pathways for the N-nitroso metabolites are illustrated in Scheme S2a–c, SI. Despite its equivalent ECL toxicity response, NPIP exhibits a muted MS response (Figure 4c) despite an LC-UV turnover within experimental error of the other N-nitroso compounds (Table 1). The smaller MS response can be rationalized based on the position of attachment on guanine. NPYR and NNK metabolites can form tetrahydrofuran-2-yl and 4-oxo-4-(3-pyridyl)but-1-yl (POB) adducts, respectively, primarily at N7, O6, and N2 guanine sites.15,22 N7 adducts especially cause glycosidic bond destabilization rendering these guanine bases much more prone to ejection by thermal hydrolysis. NPIP metabolites form predominantly 3,4,5,6-tetrahydro-2H-pyran-2-yl (THP)–Gua adducts at exocyclic N2 guanine sites. These are more stable linkages and thus are rendered less prone toward ejection by NTH.22 However, the adducted THP moiety itself is thermally labile and upon NTH, and the bond is hydrolyzed releasing THP-OH from the films, which can be isolated and detected. Subsequent LC–MS analysis of the neutral hydrolysate revealed the presence of THP-OH (SI, Figure S2). Taken together, these data demonstrate significant amounts of N2 adducts in the DNA films, leading to the similar sensor responses discussed above. In addition, these data add credence that the ECL sensor is indeed effective in detecting more than an isolated guanine adduct positional isomer, i.e., only N7 adducts. Rather, the elevated NPIP toxicity output shows that detection of any adducts resulting in closer approach of the oxidized Ru center to the guanine is possible.
Table 1.
Comparison of ECL Sensor and LC–MS Nanoreactor Data to in Vivo Toxicity
| substrate (film) | initial slopea | turnover rateb | LC-UV turnoverc | TD50d,e |
|---|---|---|---|---|
| NPYR (RLM) | 0.144 (±0.015) | 781(±176) | 12.9(±2.6) | 1.52 |
| NPYR (h2E1) | 0.216 (±0.021) | 779(±208) | 11.6(±2.4) | |
| NNK (RLM) | 0.116 (±0.004) | 629(±128) | 14.0(±2.8) | 1.80 |
| NNK (h2E1) | 0.230 (±0.029)) | 832(±233) | 10.9(±2.2) | |
| NPIP (RLM) | 0.110 (±0.011) | 594(±132) | 13.4(±2.7) | 1.50 |
| styrene (RLM) | 0.036 (±0.011) | 196(±126) | ndf | 23.3g |
| styrene (h2E1) | 0.051 (±0.017) | 184(±77) | nd |
In units of min−1.
Relative values from ECL arrays, in min−1 (mg of protein)−1; obtained by dividing initial slope by amount of protein based on QCM (Si, Table S1).
Relative values from LC-UV absorbance, in abs. units (normalized protein × s)−1.
TD50, chronic dose (mg/kg of body weight per day) inducing mixed liver tumors in half of test rat population at end of standard life span.23
Ad libitum in drinking water.
Not detected within 30-min reaction time.
Gavage administration.
Comparison with in Vivo Toxicity
As discussed previously, the initial slopes of Figure 2 are indicative of the turnover rate of the enzyme that results in DNA damage from a particular substrate. A more rapid ECL increase will result from a faster DNA reaction rate via increased enzyme turnover for a particular substrate. Accordingly, based on t tests, NPYR, NNK, and NPIP turnover rates from both array and LC-UV data (Table 1) do not differ significantly at the 95% confidence level. Thus, each N-nitroso compound exhibited similar initial slopes in the ECL array. The main reason for the similar turnover rates is variance stemming from variation in total protein content dictated by microsome/membrane properties–i.e., the amount of protein adsorbed in a film or nanoreactor. The sensor and MS response was slightly higher with h2E1 reflecting the larger amount of this enzyme present on the reaction surfaces. Indeed, Table 1 demonstrates similar turnover rates, which are normalized for the amount of enzyme present for both film constructions.
NNK is a prime etiolic agent in tobacco-related cancers and is more acutely toxic in vivo than NPYR and NPIP.15,22 On the other hand, each substrate acts preferentially at different tissue sites. In rats, each substrate forms liver tumors at approximately equal doses as shown by rat TD50, the chronic dose inducing mixed liver tumors in half of the test population (Table 1).23 These TD50 values are consistent with our array findings for liver cyt P450 2E1 in RLM and bicistronic membrane films. As demonstrated in Figure 3a, cyt P450 2E1 metabolizes NPYR to a great extent, and in fact, NPYR has been shown to primarily induce liver tumors.22 NNK also has been shown to be metabolized to a great extent by liver microsomes, despite its preponderance for respiratory toxicity.24 Most importantly, the array data clearly distinguished between the highly toxic N-nitrosamines and the moderately toxic styrene (Table 1), and both array and nanoreactor data turnover rates correlate well with the rat liver toxicity metric.
CONCLUSIONS
This work shows that synergistic use of ECL arrays and enzyme/DNA nanoreactors with LC–MS provides efficient screening of toxic reactive metabolites that correlates with animal liver toxicity data. The incorporation of RLM or h2E1, used here for the first time as enzyme sources in toxicity arrays, allows natural activation of cyt P450s leading to metabolism of substrates and accurate predictions of in vivo DNA-reactive metabolite formation. The use of engineered membrane fractions provided fast, inexpensive isolation of single cyt P450 with oxidoreductases in several days, circumventing a 3-week per enzyme generation and purification bottleneck in array fabrication.12
Results above demonstrate that pathways for reactive metabolite formation and subsequent DNA adduct formation can be elucidated by coupling ECL array and LC–MS results (also see SI, Scheme S1). Array studies on unknown compounds rapidly provide “hits” signaling reactive metabolite formation. DNA/microsome nanoreactors can then be used to obtain LC–MS data on resulting adduct structures and their formation rates for selected substrates. In this work, ECL arrays indicated N-nitroso compounds as major hits as expected, and LC–MS elucidated guanine adduct and reactive metabolite structures consistent with known metabolism outlined in SI, Scheme S3a–c. Extrapolating, N7 N-nitroso adducts–i.e., the NNK-derived N7-POB-Gua–can serve as in vivo exposure biomarkers.25 Their detection typically indicates the presence of highly reactive electrophilic moieties leading to toxicologically relevant N2 and O6 adducts–i.e., O6-POB–Gua. These miscoding adducts lead to G →A transitions and eventually to mutagenesis and carcinogenesis.15,22,25
In summary, ECL array–nanoreactor screening holds great promise for new drug and environmental chemical development. We envision applications involving screening of new chemicals and drugs for reactive metabolites using high-throughput ECL arrays, followed by more detailed structure and formation rate studies with the nanoreactors if warranted. Structural detail afforded by LC–MS/MS should facilitate “designing out” toxicity2a in synthetic targets while retaining the desired bioactivity.
Supplementary Material
Acknowledgments
This work was supported financially by U.S. PHS grant ES03154 from the National Institute of Environmental Health Sciences (NIEHS), NIH.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional schemes: Detailed ECL-nanoreactor proposed methodology and reaction pathways for each of the nitroso compounds used in this study. Additional data: QCM results summary and CapLC–MS data of THP-OH neutral thermal hydrolysis reaction. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kola I, Landis J. Nat Rev Drug Discovery. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kramer JA, Sagartz JE, Morris DL. Nat Rev Drug Discovery. 2007;6:636–649. doi: 10.1038/nrd2378. [DOI] [PubMed] [Google Scholar]; (b) Caldwell GW, Yan YL. Curr Opin Drug Discovery Dev. 2006;9:47–50. [PubMed] [Google Scholar]; (c) Nassar AEF, Kamel AM, Clairmont C. Drug Dev Today. 2004;9:1055–1064. doi: 10.1016/S1359-6446(04)03297-0. [DOI] [PubMed] [Google Scholar]; (d) Rusling JF, Hvastkovs EG, Schenkman JB. Curr Opin Drug Discovery Dev. 2007;10:67–73. [PubMed] [Google Scholar]
- 3.(a) Van Gompel J, Woestenborghs F, Beerens D, Mackie C, Cahill PA, Knight AW, Billinton N, Tweats DJ, Walmsley RM. Mutagenesis. 2005;20:449–454. doi: 10.1093/mutage/gei062. [DOI] [PubMed] [Google Scholar]; (b) Hastwell PW, Chai LL, Roberts KJ, Webster TW, Harvey JS, Rees RW, Walmsley RM. Mutat Res. 2006;607:160–175. doi: 10.1016/j.mrgentox.2006.04.011. [DOI] [PubMed] [Google Scholar]; (c) Kirkland D, Pfuhler S, Tweats D, Aardema M, Corvi R, Darroudi F, Elhajouji A, Glatt H, Hastwell P, Hayashi M, Kasper P, Kirchner S, Lynch A, Marzin D, Maurici D, Meuiner JR, Muller L, Nohynek G, Parry J, Parry E, Thybaud V, Tice R, van Benthem J, Vanparys P, White P. Mutat Res. 2007;628:31–55. doi: 10.1016/j.mrgentox.2006.11.008. [DOI] [PubMed] [Google Scholar]; (d) Emmert B, Bunger J, Keuch K, Mueller M, Emmert S, Hallier E, Westphal GA. Toxicology. 2006;228:66–76. doi: 10.1016/j.tox.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 4.(a) Dixit R, Boelsteri UA. Drug Discovery Today. 2007;12:336–342. doi: 10.1016/j.drudis.2007.02.018. [DOI] [PubMed] [Google Scholar]; (b) Liebler DC, Guengerich FP. Nat Rev Drug Discovery. 2005;4:410–420. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lee M, Park C, Dordick J, Clark D. Proc Natl Acad Sci U S A. 2005;102:983–987. doi: 10.1073/pnas.0406755102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kuang L, Biran I, Walt D. Anal Chem. 2004;76:2902–2909. doi: 10.1021/ac0354589. [DOI] [PubMed] [Google Scholar]; (c) Mitchell R, Gu M. Biosens Bioelectron. 2004;19:977–985. doi: 10.1016/j.bios.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 6.(a) Rusling JF, Hvastkovs EG, Hull DO, Schenkman JB. Chem Commun. 2007:141–154. doi: 10.1039/b709121b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou L, Yang J, Estavillo C, Stuart JD, Schenkman JB, Rusling JF. J Am Chem Soc. 2003;125:1431–1436. doi: 10.1021/ja0290274. [DOI] [PubMed] [Google Scholar]; (c) Wang B, Jansson I, Schenkman JB, Rusling JF. Anal Chem. 2005;77:1361–1367. doi: 10.1021/ac0485536. [DOI] [PubMed] [Google Scholar]; (d) Hvastkovs EG, So M, Krishnan S, Bajrami B, Tarun M, Jansson I, Schenkman JB, Rusling JF. Anal Chem. 2007;79:1897–1906. doi: 10.1021/ac061975q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Krishnan S, Hvastkovs EG, Bajrami B, Jansson I, Schenkman JB, Rusling JF. Chem Commun. 2007:1713–1715. doi: 10.1039/b703012f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Schenkman JB, Greim H. Cytochrome P450. Springer-Verlag; Berlin: 1993. [Google Scholar]; (b) Ortiz de Montellano PR. Cytochrome P450. 3. Kluwer/Plenum; New York: 2005. [Google Scholar]
- 8.(a) Vignati L, Turlizzi E, Monaci S, de Grossi P, Kanter P, Monshouwe M. Toxicology. 2005;216:154–167. doi: 10.1016/j.tox.2005.08.003. [DOI] [PubMed] [Google Scholar]; (b) O’Brien P, Irwin W, Diaz D, Cofield H, Krejsa C, Slaughter M, Gao B, Kaludercic N, Angeline A, Bernardi P, Brain P, Hougham C. Arch Toxicol. 2006;80:580–604. doi: 10.1007/s00204-006-0091-3. [DOI] [PubMed] [Google Scholar]
- 9.(a) Tang Z, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203–3224. [Google Scholar]; (b) Ariga K, Hill JP, Li Q. Phys Chem Chem Phys. 2007;9:2319–2340. doi: 10.1039/b700410a. [DOI] [PubMed] [Google Scholar]
- 10.(a) Tarun M, Rusling JF. Crit Rev Eukaryotic Gene Express. 2005;15:295–315. doi: 10.1615/critreveukargeneexpr.v15.i4.20. [DOI] [PubMed] [Google Scholar]; (b) Tarun M, Rusling JF. Anal Chem. 2005;77:2056–62. doi: 10.1021/ac048283r. [DOI] [PubMed] [Google Scholar]
- 11.Bajrami B, Hvastkovs EG, Jensen GC, Schenkman JB, Rusling JF. Anal Chem. 2008;80:922–932. doi: 10.1021/ac702025f. [DOI] [PubMed] [Google Scholar]
- 12.(a) Yuan R, Madani S, Wei XX, Reynolds K, Huang SM. Drug Metab Dispos. 2002;30:1311–1319. doi: 10.1124/dmd.30.12.1311. [DOI] [PubMed] [Google Scholar]; (b) Iwata H, Fujita K, Kushida H, Suzuki A, Konno Y, Nakamura K, Fujino A, Kamataki T. Biochem Pharmacol. 1998;55:1315–1325. doi: 10.1016/s0006-2952(97)00643-6. [DOI] [PubMed] [Google Scholar]
- 13.(a) Wells W. J Cell Biol. 2005;168:12–13. [Google Scholar]; (b) Birkett DJ, Mackenzie PI, Veronese ME, Miners JO. Trends Pharmacol Sci. 1993;14:292–294. doi: 10.1016/0165-6147(93)90043-j. [DOI] [PubMed] [Google Scholar]
- 14.(a) Guengerich FP. AAPS J. 2006;8:E101–E111. doi: 10.1208/aapsj080112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guengerich FP. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 15.(a) Wang M, Chen G, Sturla SJ, Shi Y, McIntee EJ, Villalta PW, Upadhyaya P, Hecht SS. Chem Res Toxicol. 2003;16:616–626. doi: 10.1021/tx034003b. [DOI] [PubMed] [Google Scholar]; (b) Hecht SS. Nat Rev Cancer. 2003;10:733–744. doi: 10.1038/nrc1190. [DOI] [PubMed] [Google Scholar]; (c) Hecht SS. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 16.McConnell EE, Swenberg JA. CRC Crit Rev Toxicol. 1994;24:S49–S55. doi: 10.3109/10408449409020141. [DOI] [PubMed] [Google Scholar]
- 17.Koskinen M, Vodicka P, Hemminki K. Chem-Biol Interact. 2000;124:13–27. doi: 10.1016/s0009-2797(99)00137-4. [DOI] [PubMed] [Google Scholar]
- 18.Gillam EMJ, Guo ZY, Guengerich FP. Arch Biochem Biophys. 1994;312:59–66. doi: 10.1006/abbi.1994.1280. [DOI] [PubMed] [Google Scholar]
- 19.Dennany L, Forster RJ, Rusling JF. J Am Chem Soc. 2003;125:5213–5218. doi: 10.1021/ja0296529. [DOI] [PubMed] [Google Scholar]
- 20.Omura T, Sato R. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 21.Thorp HH. Top Curr Chem. 2004;237:159–181. [Google Scholar]
- 22.(a) Wang M, Young-Sciame R, Chung FL, Hecht SS. Chem Res Toxicol. 1995;8:617–624. doi: 10.1021/tx00046a017. [DOI] [PubMed] [Google Scholar]; (b) Young-Sciame R, Wang M, Chung FL, Hecht SS. Chem Res Toxicol. 1995;8:607–616. doi: 10.1021/tx00046a016. [DOI] [PubMed] [Google Scholar]; (c) Wong HL, Murphy SE, Hecht SS. Chem Res Toxicol. 2003;16:1298–1305. doi: 10.1021/tx0340495. [DOI] [PubMed] [Google Scholar]
- 23.Gold LS. The Carcinogenic Potency Database. 2007 http://potency.berkeley.edu.
- 24.(a) Yamazaki H, Inui Y, Yun CH, Guengerich FP. Carcinogenesis. 1992;13:1789–1794. doi: 10.1093/carcin/13.10.1789. [DOI] [PubMed] [Google Scholar]; (b) Smith TJ, Guo Z, Gonzalez FJ, Guengerich FP. Cancer Res. 1992;52:1757–1763. [PubMed] [Google Scholar]
- 25.(a) Ziegel R, Shallop A, Jones R, Tretyakova N. Chem Res Toxicol. 2003;16:541–550. doi: 10.1021/tx025619o. [DOI] [PubMed] [Google Scholar]; (b) Rajesh M, Wang G, Jones R, Tretyakova N. Biochemistry. 2005;44:2197–2207. doi: 10.1021/bi0480032. [DOI] [PubMed] [Google Scholar]; (c) Li D. Cancer J. 2001;7:259–265. [PubMed] [Google Scholar]; (d) Saad AA, O’Conner PJ, Mostafa MH, Metwalli NE, Cooper DP, Margison GP, Povey AC. Cancer Epidemiol, Biomarkers Prev. 2006;15:740–743. doi: 10.1158/1055-9965.EPI-05-0813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.