

Abstract
The purpose of these studies was to identify HLA-A2+ immunogenic peptides derived from XBP1 antigens to induce a multiple myeloma (MM)-specific immune response. Six native peptides from non-spliced XBP1 antigen and three native peptides from spliced XBP1 antigen were selected and evaluated for their HLA-A2 specificity. Among them, XBP1184–192, XBP1 SP196–204 and XBP1 SP367–375 peptides showed the highest level of binding affinity, but not stability to HLA-A2 molecules. Novel heteroclitic XBP1 peptides, YISPWILAV or YLFPQLISV, demonstrated a significant improvement in HLA-A2 stability from their native XBP1184–192 or XBP1 SP367–375 peptide, respectively. Cytotoxic T lymphocytes generated by repeated stimulation of CD3+ T cells with each HLA-A2-specific heteroclitic peptide showed an increased percentage of CD8+ (cytotoxic) and CD69+/CD45RO+ (activated memory) T cells and a lower percentage of CD4+ (helper) and CD45RA+/CCR7+ (naïve) T cells, which were distinct from the control T cells. Functionally, the CTLs demonstrated MM-specific and HLA-A2-restricted proliferation, IFN-γ secretion and cytotoxic acivity in response to MM cell lines and importantly, cytotoxicty against primary MM cells. These data demonstrate the distinct immunogenic characteristics of unique heteroclitic XBP1 peptides which induce MM-specific CTLs and highlights their potential application for immunotherapy to treat the patients with MM or its pre-malignant condition.
INTRODUCTION
Multiple myeloma (MM) is a hematological malignancy characterized by the accumulation of clonogenic mature plasma cells in the bone marrow. Despite recent advances in treatment using new drugs developed in the past several years, the disease still remains incurable, thus novel therapeutic approaches are required to improve outcome.1–3 Based on the success of allogeneic transplantation as well as graft-versus-myeloma responses following donor lymphocyte infusion, other immunotherapeutic approaches are being evaluated to treat the disease. The focus has been on augmenting and directing autologous anti-MM immune responses as allogeneic immune manipulations put patients at risk of developing graft-versus-host-disease with associated significant morbidity and mortality.4,5 A number of targets have been investigated including use of patient-specific idiotype protein, MM cell lysates or MM cell-dendritic cell fusions.6–8 However, these methods require generation of a patient-specific vaccine making its general applicability more difficult. Developing a peptide-based immunotherapy against specific MM-associated antigens would offer an attractive approach for a safe and effective immunotherapy. We report here on the identification and characterization of HLA-A2+ peptides that elicit specific CTLs targeting XBP1 as the MM-specific antigen.
XBP1 is a transcription factor required for the terminal differentiation of B lymphocytes to plasma cells and is essential for immunoglobulin secretion.9, 10 This antigen is a basic leucine zipper-containing transcription factor originally identified as a protein binding to the cis-acting X box region in the promoter of human MHC class II genes.11 XBP1 mRNA is processed by IRE1, an endoplasmic reticulum (ER) transmembrane protein that contains endoribonuclease and cytoplasmic protein kinase domains in response to ER stress.12–14 The mRNA spliced by IRE1 causes a reading frame shift which is translated into a spliced form of XBP1 protein that is an active transcription factor.15 To date, XBP1 is the only transcription factor found to be essential for plasma cell differentiation. XBP1 is uniformly expressed in all MM cells and cell lines and is selectively induced by exposure to IL-6 and has been implicated in the proliferation of malignant plasma cells.16–19 Microarray analyses have identified XBP1 as a differentially expressed gene between the plasma cells and monoclonal gammopathy of undetermined significance (MGUS) and MM cells.20 Gene expression profiling studies have also confirmed the specific expression of XBP1 in MM.21 A recent study shows that a splice variant of XBP1 plays a crucial role in normal plasma cell differentiation.22 XBP1 splicing is recognized to occur in terminal B cell differentiation and correlates with plasma cell differentiation. In addition, there is evidence that spliced XBP1 supports restoration of immunoglobulin production in XBP1−/− B cells and induces IL-6 secretion in normal plasma cells development.23–25 It has also been shown that the relative mRNA expression levels of spliced XBP1 compared to XBP1 are differentially expressed in myeloma compared with normal plasma cells.20
In this study, we propose the XBP1 transcription factor as a unique therapeutic target antigen for development of a MM-specific immunotherapy. We designed and characterized two heteroclitic peptides, YISPWILAV and YLFPQLISV, with improved HLA-A2 binding and stability from their respective native peptides, XBP1184–192 (NISPWILAV) and XBP1 SP367–375 (ELFPQLISV). These heteroclitic peptides were able to induce XBP1 antigen-specific CTLs which specifically recognized, targeted, and lysed HLA-A2+ MM cell lines. These data provides evidence of the potential of HLA-A2+ XBP1 specific peptides for their clinical application as a peptide-based vaccine or in the production of XBP1-specific CTLs ex vivo for adoptive transfer therapy in patients with MM or its related pre-malignant diseases.
MATERIALS AND METHODS
Cell lines
The multiple myeloma lines, McCAR, MM1S and U266 were obtained from ATCC (Manassas, VA). The acute myeloid leukemia (AML) cell line, ML-2, was kindly provided by Dr. Y. Matsuo, Fujisaki Cell Center, Okayama, Japan. The T2 cell line, a human B and T cell hybrid expressing HLA-A2 molecules26, was provided by Dr. J. Molldrem (University of Texas M. D. Anderson Cancer Center, Houston, TX) and was used as antigen presenting cells (APC). All cell lines were cultured in RPMI-1640 medium (Gibco-Life Technologies, Rockville, MD) supplemented with 10% fetal calf serum (FCS; BioWhittaker, Walkersville, MD), 100 IU/ml penicillin and 100 µg/ml streptomycin (Gibco-Life Technologies).
Reagents
Mouse anti-human CD3, CD4, CD8, CD14, CD40, CD45RA, CD45RO, CD69, CD86, CD107a, CCR7, HLA-A2 and HLA-DR monoclonal antibodies (mAbs) conjugated with FITC, PE, PerCP or APC were purchased from Becton Dickinson/Pharmingen (San Diego, CA). Mouse anti-human CD80 or CD83 mAbs conjugated with PE were purchased from Immunotech (Hialeigha, FL). Recombinant human IL-2, IL-4, IFN-α and TNF-α were purchased from R&D Systems (Minneapolis, MN), and recombinant human GM-CSF was obtained from Immunex (Seattle, WA).
Synthetic Peptides
Six native non-spliced XBP1 peptides including XBP1117–125 (LLREKTHGL), XBP1184–192 (NISPWILAV), XBP1189–197 (ILAVLTLQI), XBP1192–200 (VLTLQIQSL), XBP1110–118 (KLLLENQLL), XBP193–101 (RMSELEQQV), three native spliced XBP1 peptides including XBP1 SP196–204 (GILDNLDPV), XBP1 SP193–201 (ILLGILDNL), XBP1 SP367–375 (ELFPQLISV), and three heteroclitic peptides including XBP1184–192 (YISPWILAV), XBP1 SP196–204 (YILDNLDPV) and XBP1 SP367–375 (YLFPQLISV) were examined as potential HLA-A2-specific peptides. Influenza virus matrix protein58–66 (GILGFVFTL) was used as a control HLA-A2-specific peptide. All peptides were synthesized by standard fmoc (9-fluorenylmethyl-oxycarbonyl) chemistry, purified to >90% using reverse-phase chromatography and validated by mass-spectrometry for molecular weight (Biosynthesis, Lewisville, TX). Lyophilized peptides were dissolved in DMSO (Sigma, St. Louis, MO), diluted in AIM-V medium (Gibco-Life Technologies) and stored at −140°C.
Peptide Binding Assay
XBP1 peptides were evaluated for HLA-A2-specific binding with the T2 cell line. In the assay, T2 cells were washed, resuspended in serum-free AIM-V medium to a final concentration of 1×106 cells/ml and transferred into wells of a 24-well tissue culture plate. The cells were pulsed with 50 µg/ml of respective XBP1 peptide or 30 µg/ml of influenza virus matrix protein58–66 (GILGFVFTL) peptide plus 3 µg/ml human β2-microglobulin (Sigma) and incubated at 37°C, 5% CO2 in humidified air. Following overnight incubation, the cells were washed, stained with mouse anti-human HLA-A2-FITC mAb for 15 minutes at 4°C, washed and analyzed using a FACSort™ flow cytometer with CellQuest™ v2.1 software (Becton Dickinson, San Jose, CA). Peptide binding to HLA-A2 was determine by the up-regulation of HLA-A2 molecules on T2 cells caused by HLA-A2 specific peptide binding and demonstrated by measuring mean fluorescence intensity (MFI) by flow cytometric analyses.
Peptide Stability Assay
XBP1 peptides were examined for HLA-A2 binding stability using the T2 cell line. T2 cells were pulsed with the respective XBP1 peptide as described above. After overnight incubation, the cells were washed to remove unbound peptide and incubated with 10 µg/ml Brefeldin A (Sigma) at 37°C and 5% CO2 for 1 hour to block cell surface expression of newly synthesized HLA-A2 molecules. Peptide/HLA-A2 binding stability was evaluated at 0, 2, 4, 6 and 18 hours post-Brefeldin A treatment. Following the incubation period, the cells were harvested, washed, stained with mouse anti-human HLA-A2-FITC mAb and analyzed by flow cytometry as described above.
Generation of Monocyte-derived Dendritic Cells
Peripheral blood mononuclear cells (PBMC) were isolated by standard density gradient centrifugation over Ficoll-PaqueTM Plus (Amersham Pharmacia Biotech AB, Uppsala Sweden) from leukopaks obtained from HLA-A2+ normal donors. Dendritic cells (DC) were generated from monocytes obtained as the adherent cell fraction. To generate DC, the monocytes were cultured for 7 days in the presence of 1,000 U/ml GM-CSF and 1,000 U/ml IL-4 in RPMI-1640 medium (Gibco-Life Technologies) supplemented with 10% FCS. Fresh media plus GM-CSF and IL-4 was added to the cultures every other day. Mature DC (mDC) were obtained by adding 1,000 U/ml IFN-α plus 10 ng/ml TNF-α along with fresh GM-CSF and IL-4 in 10% FCS-RPMI on day 7 and incubating additional three days. The mDC were harvested and used as antigen-presenting cells (APC) to generate XBP1 peptide specific-CTLs.
Isolation of CD3+ T cells
CD3+ T cells were obtained from the non-adherent cell fraction (post-monocytes adherence) using the Pan T cell isolation kit from Miltenyi Biotec (Auburn, CA). In brief, T cell enrichment was accomplished by depletion of B cells, NK cells, early erythroid cells, platelets and basophils by labeling with a cocktail of hapten-conjugated CD11b, CD16, CD19, CD36 and CD56 antibodies and MACs microbeads coupled to an anti-hapten monoclonal antibody. The effluent (negative cell fraction) was collected from the column as enriched CD3+ T cells. The purity of the enriched CD3+ T cells was examined by flow cytometry and was found to be 94±2% (mean ± standard deviation).
Induction of Peptide-specific CTLs
Peptide-specific CTLs were generated ex vivo by repeated stimulation of CD3+ T lymphocytes obtained from HLA-A2+ normal donors with XBP1 peptide-pulsed APC, either mDC or T2 cells. In brief, APC were washed and resuspended in serum-free medium AIM-V medium and pulsed overnight at 37°C and 5% CO2 in humidified air with 50 µg/ml of the appropriate XBP1 peptide. Peptide-pulsed APC were washed, counted, irradiated at 10 Gy and used to prime CD3+ T cells at a 1:20 APC/peptide (stimulator)-to-CD3+ T cell (responder) ratio in AIM-V medium supplemented with 10% human AB serum (BioWhittaker). The T cell cultures were restimulated every seven days with irradiated APC/peptide for a total of 4 cycles to generate XBP1 peptide-specific CTL (XBP1-CTL). IL-2 (50 U/ml) was added to the cultures two days after the second stimulation. Control CD3+ T cell cultures (unstimulated) were maintained in AIM-V medium supplemented with 10% human AB serum containing 50 U/ml IL-2.
Phenotypic analysis of the XBP1-CTLs or target/stimulatory cell lines
XBP1-CTLs were stained with CD4-PE or CD8-FITC mAbs for their phenotype analyses. In addition, they were stained with CD69-FITC/CD45RO-PE or CD45RA-FITC/CCR7-PE mouse mAbs. Alternatively, the multiple myeloma cell lines (McCAR, U266 and MM1S) along with an acute myeloid leukemia cell line (ML-2) were stained with HLA-A2-FITC mAb prior to use as target or stimulatory cells. CTLs or cell lines were stained with the appropriate mAbs, washed and analyzed by flow cytometry.
IFN-γ ELISA
IFN-γ secretion by XBP1-CTLs was measured using a human IFN-γ ELISA kit (BD Biosciences, San Diego, CA). For the assay, XBP1-CTLs were incubated with the MM (McCAR, MM1S) or AML (ML-2) cell lines for 24 hours at 37°C, 5% CO2 in humidified air. The supernatants were harvested and stored at −20°C until analyzed by ELISA for IFN-γ release by the XBP1-CTLs in response to the MM or AML cell lines used as stimulator cells. Briefly, purified IFN-γ as standards or supernatants were transferred into wells of a 96-well plate pre-coated with a monoclonal anti-human IFN-γ capture antibody and incubated for 2 hours at room temperature. After washing, the buffer containing detection antibody and avidin-horseradish peroxidase conjugate was added to each well and incubated for 1 hour at room temperature. The wells were washed and developed by incubation with TMB substrate solution for 30 minutes. Stop solution was added to each well and the absorbance was determined at 450 nm with a VICTOR2-1420 multilabel counter (PerkinElmer, Wellesley, MA). The amount of IFN-γ (pg/ml) present in the CTLs culture supernatant was calculated based on the standard curve
Cell proliferation by Carboxy Fluorescein Succinimidyl Ester (CFSE) tracking
XBP1-CTLs were harvested, washed twice in PBS (Gibco-Life Technologies) and resuspended at 1×106 cells/ml. CFSE (Molecular Probes, Eugene, OR), 5 mM stock solution in dimethyl sulfoxide, was added to the CTLs to give a final concentration of 5 µM and incubated for 10 minutes at 37°C, 5% CO2 protected from light. After incubation, 5 volumes of ice-cold PBS with 2% FCS (PBS-FCS) were added to the cells to quench the reaction. The cells were incubated for 5 minutes on ice, washed 3× with PBS-FCS and resuspended in fresh AIM-V media supplemented with 10% human AB serum. The CFSE labeled XBP1-CTLs (2 × 106 cells/ml) were co-incubated with McCAR, MM1S or ML-2 cells (2 × 105 cells/ml) or media alone at 37°C and 5% CO2 in humidified air. The CFSE-labeled CTLs were harvested and examined on day 4 to determine their proliferation in response to the antigen present on the tumor cell lines. CTLs that proliferate in response to antigen show a reduction in CFSE fluorescence intensity, which was measured directly by flow cytometry.
Cytotoxicity Assay
The cytotoxic activity of the XBP1-CTLs was measured using a calcein-release assay as described elsewhere.27 Briefly, 3 × 105 target cells (McCAR, U266, MM1S, ML-2 or primary CD138+ cells from HLA-A2+ MM patients) were incubated with 10 mM calcein-AM (Molecular Probes) for 30 minutes at 37°C, washed 3× and resuspended in PBS containing 5% FCS. The calcein-AM labeled target cells (5 × 103/well) were incubated with the XBP1-CTLs at various effector:target cell ratios in 96-well U-bottom microtiter plates (triplicate wells/sample). Plates were incubated for 4 hours at 37°C, 5% CO2. After incubation, the cells were pelleted by centrifugation at 1,000 rpm for 5 minutes and 100 µl of the supernatant were transferred from each well to 96-well flat-bottomed plates. The fluorescence of each supernatant was monitored at 490 nm excitation and 520 nm emission wavelength using a VICTOR2-1420 multilabel counter (Perkin-Elmer). Maximum release was obtained from detergent-released target cells and spontaneous release from target cells incubated in the absence of effector cells. The cytotoxicity of the XBP1-CTLs was calculated as follows: % Specific Lysis = [(experimental release − spontaneous release) ÷ maximum release − spontaneous release)].
CD107 assay
The CD107 assay was setup with minor modifications to evaluate the functional activity of the XBP1-CTLs.28 Primary CD138+ MM cells from bone marrow mononuclear cells were isolated from HLA-A2+ myeloma patients using RoboSep® CD138 positive immunomagnetic selection technology per manufacturer’s instructions (STEMCELL Technologies, Vancouver, BC, Canada). The XBP-1 CTLs were co-incubated with HLA-A2+/CD138+ MM cells at a ratio of 1 CTL:5 CD138+ MM cells in a 96 well round bottom plate. At the same time, mouse anti-human CD107a-FITC mAb was added to each well. After 1 hour incubation, Brefeldin A (BD) and Monensin (BD) were added to the cells and cells were incubated for an additional 4 hours. Following incubation, the cells were harvested, washed, stained with mouse anti-human CD8-PE mAb for 30 minutes at 4°C, washed and analyzed by flow cytometry. The expression of CD107a by CD8+ T cells was determined as a measure of degranulation of XBP1-CTLs in response to primary HLA-A2+/CD138+ MM cells.
Statistical Analysis
Results are presented as mean ± SE. Groups were compared using unpaired Student’s t-test. Differences were considered significant when p<0.05.
RESULTS
Non-spliced and spliced XBP1 peptides display high level of binding and stability to HLA-A2 molecules
The full length amino acid sequences from non-spliced and spliced XBP1 proteins were analyzed using the search software SYFPEITHI to predict HLA-A2-binding peptides, followed by the BIMAS and NetCTL programs to select peptides with extended half-time disassociation rates, proteasomal C terminal cleavage and TAP transport. A total of six native peptides from non-spliced XBP1 protein [XBP1117–125 (LLREKTHGL), XBP1184–192 (NISPWILAV), XBP1189–197 (ILAVLTLQI), XBP1192–200 (VLTLQIQSL), XBP1110–118 (KLLLENQLL), XBP193–101 (RMSELEQQV)] and three native peptides [XBP1 SP196–204 (GILDNLDPV), XBP1 SP193–201 (ILLGILDNL), XBP1 SP367–375 (ELFPQLISV)] from spliced XBP1 protein were selected for evaluation of HLA-A2-specific binding affinity (Table 1 and Table 2). The T2 peptide-binding assay was used to assess the HLA-A2 affinity of the XBP1 peptides.
Table 1.
Potential HLA-A2 Peptides Identified from Non-spliced XBP1 protein
| PEPTIDES | SEQUENCE | SYFPEITHI | BIMAS | NetCTL |
|---|---|---|---|---|
| Non-spliced XBP1117–125 | LLREKTHGL | 29 | 11.316 | 0.9257 |
| Non-spliced XBP1184–192 | NISPWILAV | 26 | 21.996 | 1.1996 |
| Non-spliced XBP1189–197 | ILAVLTLQI | 26 | 17.736 | 1.0263 |
| Non-spliced XBP1192–200 | VLTLQIQSL | 26 | 83.527 | 0.8323 |
| Non-spliced XBP1110–118 | KLLLENQLL | 23 | 276.643 | 1.001 |
| Non-spliced XBP193–101 | RMSELEQQV | 21 | 205 951 | 0.9445 |
| Non-spliced XBP1200–208 | LISCWAFWT | 14 | 55.567 | 0.5846 |
| Non-spliced XBP117–25 | VLLLSGQPA | 17 | 31.249 | 0.6260 |
| Non-spliced XBP1124–132 | GLVVENQEL | 23 | 21.362 | 0.7905 |
| Non-spliced XBP119–27 | LISGQPASA | 22 | 8.446 | 0.7587 |
Table 2.
Potential HLA-A2 Peptides Identified from Spliced XBP1 protein
| PEPTIDES | SEQUENCE | SYFPEITHI | BIMAS | NetCTL |
|---|---|---|---|---|
| Spliced XBP1196–204 | GILDNLDPV | 26 | 163.502 | 1.2062 |
| Spliced XBP1193–201 | ILLGILDNL | 30 | 151.434 | 1.1289 |
| Spliced XBP1367–145 | ELFPQLISV | 26 | 44.392 | 1.0892 |
| Spliced XBP1110–118 | KLLLENQLL | 23 | 276.643 | 1.001 |
| Spliced XBP193–101 | RMSELEQQV | 21 | 205.951 | 0.9445 |
| Spliced XBP1189–197 | SESDILLGI | 17 | 1.913 | 0.3759 |
| Spliced XBP1238–246 | SVGTSSAKL | 20 | 1.869 | 0.4557 |
| Spliced XBP1330–338 | CLLDAYSDC | 13 | 111.047 | 0.2422 |
| Spliced XBP1263–271 | LVLEIPSET | 16 | 16.816 | 0.5069 |
| Spliced XBP1215–223 | ASLEELPEV | 23 | 15.841 | 0.9009 |
Among the XBP1 peptides derived from non-spliced XBP1 protein, XBP1184–192 (NISPWILAV) showed the highest HLA-A2 binding (MFI = 720 ± 140) which was greater than the influenza virus matrix protein58–66 peptide (GILGFVFTL) (MFI = 604 ± 10) used as a HLA-A2-specific positive control (Figure 1a). However, initial peptide stability studies revealed that the HLA-A2 binding of the native XBP1184–192 (NISPWILAV) peptide was unstable compared to influenza virus matrix protein58–66 peptide (GILGFVFTL) (data not shown). In order to improve the stability of the native XBP1184–192 (NISPWILAV) peptide binding to HLA-A2 molecules, we designed the heteroclitic peptide (YISPWILAV) by substitution of the tyrosine (Y) for asparagine (N) in position 1 from the native peptide. The HLA-A2 binding stability of the heteroclitic XBP1184–192 peptide was significantly improved (> 6 hours) compared to the native XBP1184–192 peptide (Figure 1b). In addition, the stability of the heteroclitic XBP1184–192 peptide to HLA-A2 molecule was greater than the control influenza virus matrix protein58–66 (GILGFVFTL), demonstrating that the peptide modification improved HLA-A2 stability for the heteroclitic XBP1184–192 peptide.
Figure 1. Identification of HLA-A2-specific XBP1 peptides and improvement of MHC binding stability by peptide modification.
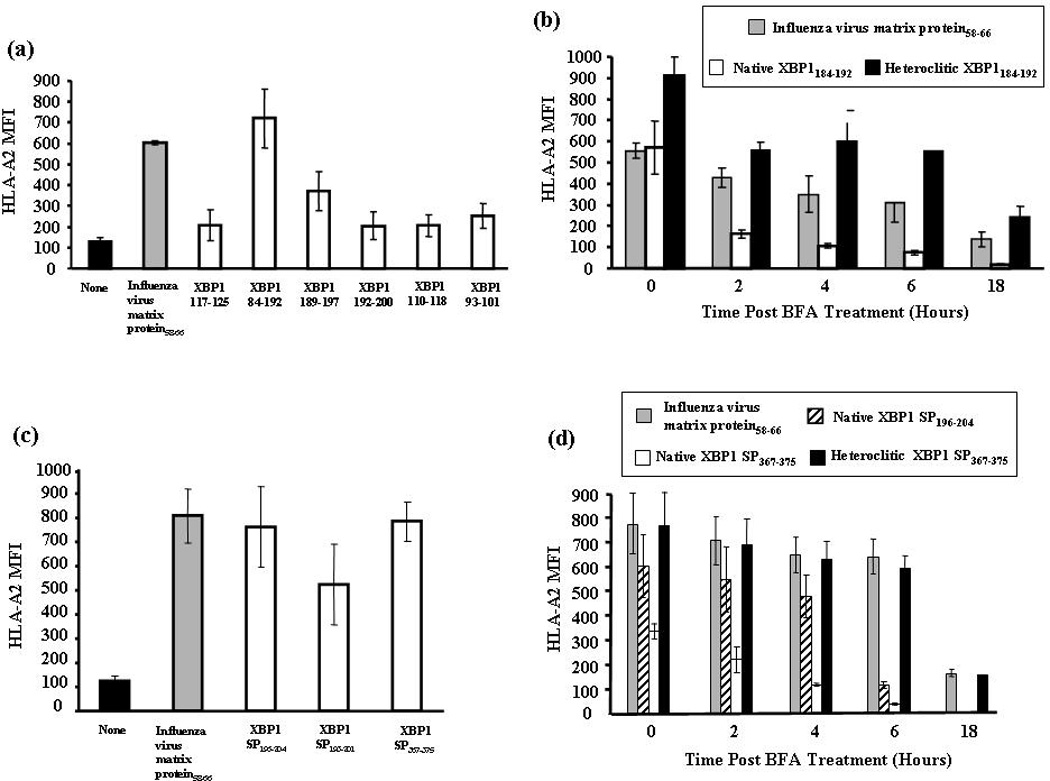
Figure 1a. HLA-A2 binding affinity of non-spliced XBP1 peptides.
T2 cells were pulsed overnight with respective XBP1 peptide (50 µg/ml) in serum-fee media. Influenza virus matrix protein58–66 was used as the positive control and T2 cell in media alone as baseline controls in these experiments. Following incubation, T2 cells were harvested, washed, and stained with HLA-A2-FITC mAb for flow cytometric analysis. HLA-A2 binding is shown as an increase in HLA-A2 mean fluorescence intensity (MFI). XBP1184–192 (NISPWILAV) showed the highest HLA-A2 binding affinity among the non-spliced XBP1 peptides. The values represent the mean ± SE of three separate experiments.
Figure 1b. HLA-A2 binding stability of non-spliced XBP1 peptides.
Native or heteroclitic XBP1 peptide (50 µg/ml)-pulsed T2 cells were washed and incubated with Brefeldin A. At 0, 2, 4, 6, and 18 hours incubation, the cells were stained with HLA-A2-FITC mAb for flow cytometric analysis. Heteroclitic XBP1184–192 (YISPWILAV) peptide showed increased HLA-A2 binding stability compared to the native XBP1184–192 (NISPWILAV) peptide. Binding stability of the heteroclitic peptide was higher than influenza virus matrix protein58–66 (GILGFVFTL), which was used as the HLA-A2-specific positive control peptide. The values represent the mean ± SE of three separate experiments.
Figure 1c. HLA-A2 binding affinity of spliced XBP1 peptides.
Spliced XBP1 peptides were evaluated for their HLA-A2 binding affinity as described in Figure 1a. XBP1 SP196–204 (GILDNLDPV) and XBP1 SP367–375 (ELFPQLISV) showed the highest HLA-A2 binding affinity among the spliced XBP1 peptides. The values represent the mean ± SE of three separate experiments.
Figure 1d. HLA-A2 binding stability of spliced XBP1 peptides.
Spliced XBP1 peptides were analyzed for their HLA-A2 binding stability as discussed in Figure 1b. Heteroclitic XBP1 SP367–375 (YLFPQLISV) peptide displayed increased HLA-A2 binding stability compared to its native XBP1 SP367–375 (ELFPQLISV) or another native XBP1 SP196–204 (GILDNLDPV) peptide. The values represent the mean ± SE of three separate experiments.
Among the peptides derived from spliced XBP1 protein, XBP1 SP196–204 (GILDNLDPV) and XBP1 SP367–375 (ELFPQLISV) showed the highest levels of HLA-A2 binding affinity (MFI = 762 ± 167, MFI = 785 ± 84, respectively) which were comparable to the control influenza virus matrix protein58–66 peptide (Figure 1c). The peptide sequences of XBP1 SP196–204 and XBP1 SP367–375 were modified to enhance their binding stability to HLA-A2 molecules. Improvement in HLA-A2 binding stability was observed with the heteroclitic peptide, YLFPQLISV, which was modified from the native XBP1 SP367–375 (ELFPQLISV) (Figure 1d). The HLA-A2 binding stability was not improved for the other heteroclitic peptide, YILDNLDPV, which was derived from the native XBP1 SP196–204 (GILDNLDPV) peptide (data not shown). Therefore, the heteroclitic XBP1184–192 (YISPWILAV) and heteroclitic XBP1 SP367–375 (YLFPQLISV) peptides were selected for further evaluation of their immunogenic potential to generate MM-specific CTLs.
The XBP1-specific CTLs display a distinct phenotype from unstimulated T cells
Antigen-specific CTLs can be phenotypically identified as activated/memory T cells from naïve T cells by their expression of distinct cell surface antigens. We analyzed the percentage of activated/memory (CD69+/CD45RO+) and naïve (CD45RA+/CCR7+) T cells along with the T cell subsets (CD4+, CD8+) in the CD3+ T cell cultures stimulated with either heteroclitic XBP1184–192 (YISPWILAV) or heteroclitic XBP1 SP367–375 (YLFPQLISV) peptide. The CD3+ T cells stimulated with respective heteroclitic XBP1 peptide induced a significantly higher percentage of CD8+ T cells (heteroclitic XBP1184–192 stimulated - 81%; heteroclitic XBP1 SP367–375 stimulated - 75%) compared to control T cells (no stimulation - 33%). We also observed a lower percentage of CD4+ T cells (heteroclitic XBP1184–192 stimulated - 14%; heteroclitic XBP1 SP367–375 stimulated - 18%) in the XBP1-CTL cultures as compared to the control CD3+ T cell cultures (no peptide stimulation = 64%) (Figure 2). The XBP1-CTLs or control T cells were further examined for naïve (CD45RA+/CCR7+) or activated memory (CD69+/CD45RO+) cell subtypes. The cell population expressing an activated memory (CD69+/CD45RO+)phenotype was significantly higher in the XBP1-CTLs (heteroclitic XBP1184–192 stimulated - 62%; heteroclitic XBP1 SP367–375 stimulated - 64%) compared to control T cells (no stimulation – 4%) obtained from the same donor (Figure 2). In addition, the control T cell cultures showed 24% naïve (CD45RA+/CCR7+) cells, whereas only 2% of the XBP1-CTLs showed this subset (data not shown). Therefore, these results demonstrate that stimulation with heteroclitic XBP1 peptides lead to an expansion of the CD8+ T cell subset with an activated/memory (CD69+/CD45RO+) phenotype characteristic of effector cells.
Figure 2. Distinct phenotype of CTLs stimulated with heteroclitic XBP1-specific peptides.
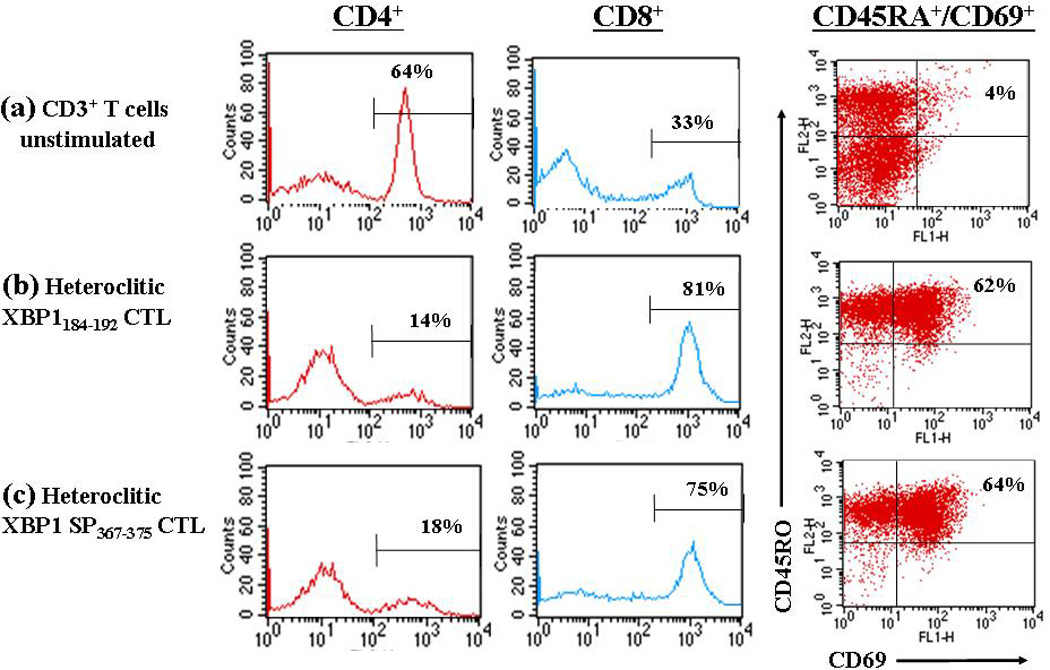
Enriched CD3+ T cells obtained from a normal HLA-A2+ donor were stimulated weekly with irradiated antigen-presenting cells pulsed with respective heteroclitic XBP1 peptide. One week after the 4th cycle of peptide stimulation, the CD3+ T cells were analyzed for effector cells by flow cytometry. The percentage of CD4+ helper T cells was decreased in the T cells stimulated with either (b) heteroclitic XBP1184–192 YISPWILAV) peptide or (c) heteroclitic XBP1 SP367–375 (YLFPQLISV) peptide, compared to (a) no peptide. In contrast, the percentage of CD8+ CTLs and CD69+/CD45RO+ (activated memory) effector cells was increased in the CD3+ T cells stimulated with respective XBP1 peptide, compared to no peptide control. The results are representative of three independent experiments.
XBP1-CTLs produce IFN-γ by stimulation with HLA-A2+ myeloma cells
To characterize the anti-tumor response, antigen-specificity and HLA-A2-restriction of the heteroclitic XBP1 peptide induced CTLs, we evaluated their ability to produce IFN-γ following stimulation with various tumor cell lines including McCAR (MM; HLA-A2+/XBP1+), MM1S (MM; HLA-A2−/XBP1+) or ML-2 (AML; HLA-A2+/XBP1−). Supernatants collected after 24 hours incubation of XBP1-CTLs with the respective tumor cell line were evaluated for IFN-γ (pg/ml) by ELISA. We observed a dramatic increase in IFN-γ secretion by both heteroclitic XBP1-CTLs stimulated McCAR (HLA-A2+/XBP1+) myeloma cells compared to the CTLs alone without stimulation (Figure 3). Stimulation with the MHC mismatched MM1S (MM; HLA-A2−/XBP1+) or the antigen mismatched ML-2 (AML; HLA-A2+/XBP1−) cell lines did not induce IFN-γ secretion by either of the heteroclitic XBP1-CTLs. These results offer evidence of an antigen-specific and HLA-A2-restricted IFN-γ secretion by the CTLs generated with heteroclitic XBP1184–192 (YISPWILAV) or heteroclitic XBP1 SP367–375 (YLFPQLISV) peptides.
Figure 3. HLA-A2 restricted and XBP1-specific IFN-γ secretion by XBP1-CTLs stimulated with heteroclitic peptides.
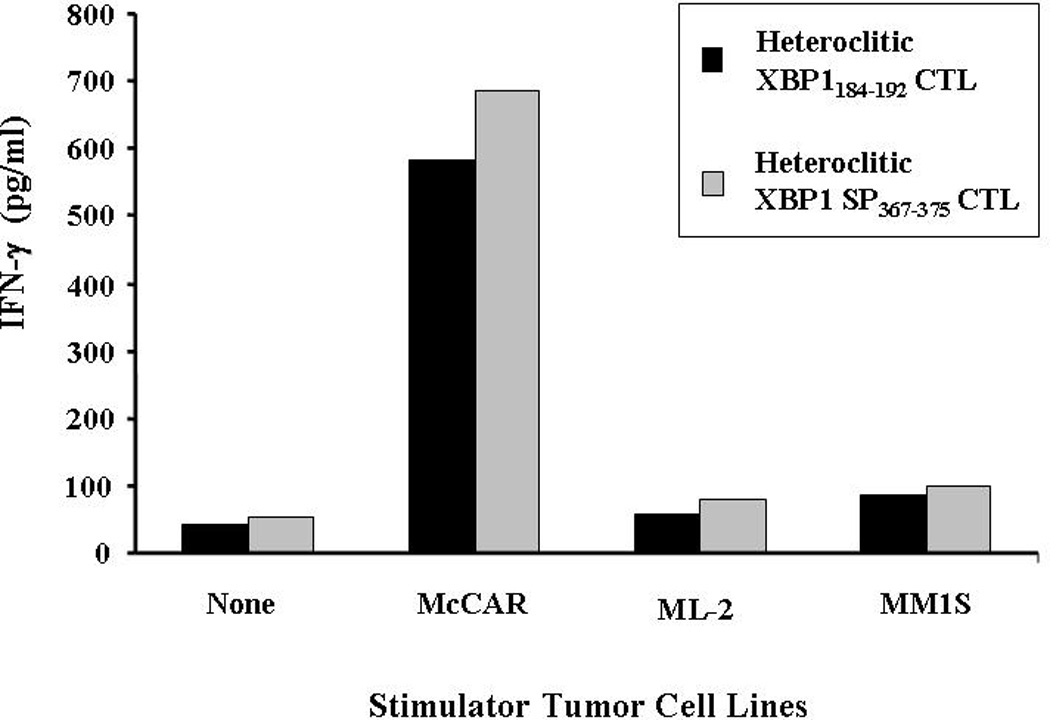
CTLs generated by repeated stimulation (4×) with heteroclitic XBP1184–192 (YISPWILAV) or heteroclitic XBP1 SP367–375 (YLFPQLISV) peptide were incubated with McCAR (MM; HLA-A2+/XBP1+), ML-2 (AML; HLA-A2+/XBP1−) or MM1S (MM; HLA-A2−/XBP1+) tumor cell lines. After 24 hours of incubation, the supernates were collected and IFN-γ secretion was measured by ELISA. An increase in IFN-γ secretion was detected in both XBP1-CTLs cultures in response to McCAR (HLA-A2+/XBP1+), but not to ML-2 (HLA-A2+/XBP1−) or MM1S (HLA-A2−/XBP1+) cells. XBP1-CTLs alone were used to examine the background IFN-γ release. The results are representative of three independent experiments.
XBP1-CTLs show specific cell proliferation in response to HLA-A2+ myeloma cells
A CFSE-based assay was used to analyze the proliferation of heteroclitic XBP1-CTLs upon stimulation with various irradiated tumor cell lines. Cell proliferation was analyzed by measuring the decrease in fluorescence of the CFSE-labeled CTLs by flow cytometry (M1-gated cells). Antigen-specific and HLA-restricted cell proliferation was observed by the XBP1-CTLs in response to HLA-A2+/XBP1+ McCAR (heteroclitic XBP1184–192–CTLs: 55%, heteroclitic XBP1 SP367–375-CTLs: 42%) compared to the control CTLs without stimulation (heteroclitic XBP1184–192–CTLs: 5%, heteroclitic XBP1 SP367–375-CTLs: 2%) (Figure 4). However, the XBP-1 CTLs did not proliferate in response to the MHC mismatched MM1S (MM; HLA-A2-/XBP1+) or the antigen mismatched ML-2 (AML; HLA-A2+/XBP1−) cell lines. Taken together, our data demonstrated the specific cell proliferation of XBP1-CTLs in response to myeloma cells in antigen-specific and HLA-A2-restricted manner.
Figure 4. Induction of HLA-A2-restricted and XBP1-specific cell proliferation of XBP1-CTLs in response to multiple myeloma cells.
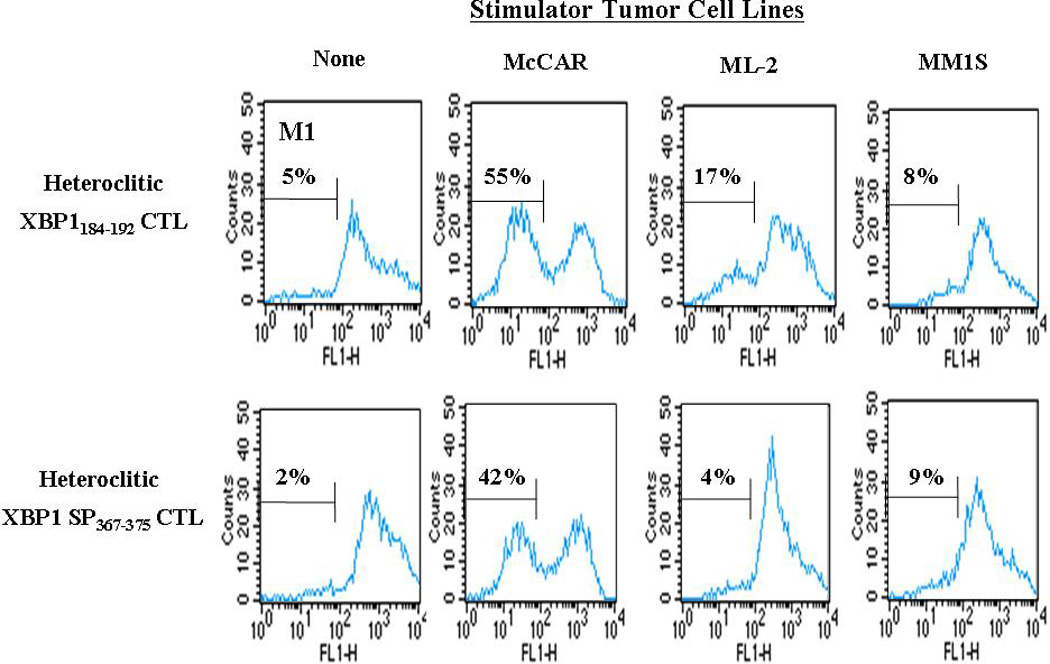
Proliferation of CFSE-labeled XBP1-CTLs was measured in response to antigen stimulation by flow cytometry on day 4 of culture. The cell proliferation of heteroclitic XBP1184–192 (YISPWILAV) and heteroclitic XBP1 SP367–375 (YLFPQLISV) CTLs were both XBP1 antigen-specific and HLA-A2-restricted in response to the McCAR (HLA-A2+/XBP1+) myeloma cell line. The CTLs did not proliferate in response to either the XBP1 antigen or MHC mismatched cell lines ML-2 (HLA-A2+/XBP1−) or MM1S (HLA-A2−/XBP1+), respectively. Background proliferation was determined using CFSE labeled heteroclitic XBP1-CTLs cultured in media alone. The results are representative of three independent experiments.
XBP1-CTLs induce specific lysis of HLA-A2+/XBP1+ myeloma cell lines
Next, we demonstrate the ability of the heteroclitic XBP1184–192 or heteroclitic XBP1 SP367–375 peptide-specific CTLs to specifically target and lyse MM cells using a calcein-release cytotoxicity assay. Heteroclitic XBP1184–192-CTLs (Figure 5a) as well as heteroclitic XBP1 SP367–375-CTLs (Figure 5b) demonstrated effective lysis of the HLA-A2+/XBP1+ McCAR cells at various effector: target cell ratios (9–50% specific lysis by heteroclitic XBP1184–192–CTLs; 1 5-69% by heteroclitic XBP1 SP367–375-CTLs). In addition, an alternative HLA-A2+/XBP1+ myeloma cell line, U266, was effectively lysed by the heteroclitic XBP1184–192–CTLs (16–74% specific lysis) and heteroclitic XBP1 SP367–375-CTLs (18–83% specific lysis). Interestingly, the cytotoxic activity against McCAR or U266 myeloma cell lines was greater for the CTLs induced with the XBP1 SP367–375 peptide derived from spliced XBP1 protein as compared to those induced with the XBP1184–192 peptide from non-spliced protein. The heteroclitic XBP1184–192-CTLs or heteroclitic XBP1 SP367–375-CTLs did not lyse the antigen mismatched ML-2 (AML; HLA-A2+/XBP1−) or MHC mismatched MM1S (MM; HLA-A2-/XBP1+) cell lines, demonstrating that the cytotoxic activity of both XBP1-CTLs is antigen-specific and HLA-restricted. The XBP1-CTLs did not lyse the NK-sensitive K562 cell line (data not shown), suggesting that the cytotoxicity of XBP1-CTLs was not due to an activated NK cell population.
Figure 5. HLA-A2-restricted and antigen-specific cytotoxicity of the XBP1-CTLs.
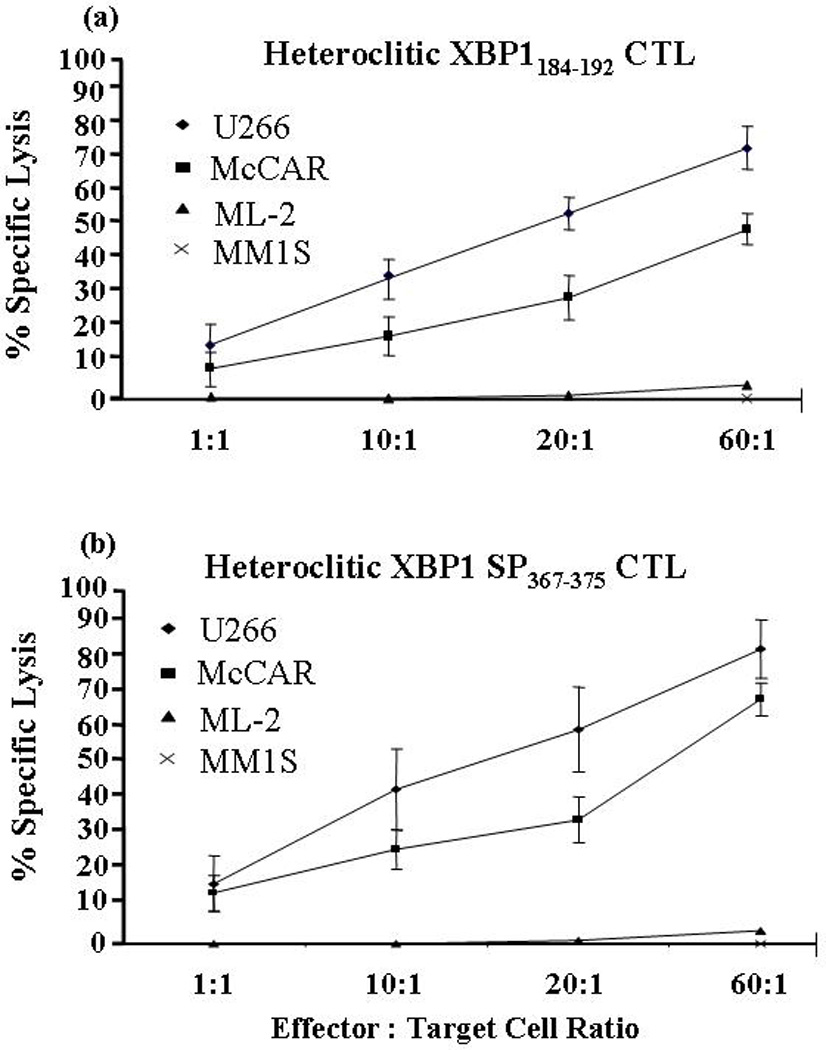
The specific cytotoxic activities of the heteroclitic XBP1-CTLs were analyzed one week after their 4th stimulation using the calcein cytotoxicity assay. A high level of cytotoxic activity against U266 (MM, HLA-A2+/XBP1+) (♦) and McCAR (MM, HLA-A2+/XBP1+) (■), but not against the ML-2 (AML, HLA-A2+/XBP1−) (▲) and MM1S (HLA-A2−/XBP1+) (X) cell lines was demonstrated by both the (a) heteroclitic XBP1184–192 CTLs and (b) heteroclitic XBP1 SP367–375 -CTLs. The values represent the mean±SE of three separate experiments.
XBP1-CTLs induce degranulation and cytotoxicity against primary HLA-A2+/CD138+ MM cells
To evaluate effectiveness of XBP1-CTLs against primary patient MM cells, we evaluated CTL degranulation in response to target cell recognition by measuring expression of CD107a, a surface antigen transiently present on the cell surface after release of cytotoxic granules. XBP1-CTLs incubated with primary HLA-A2+/CD138+ myeloma cells in the presence of CD107a-FITC mAb were stained with CD8-PE mAb and evaluated by flow cytometry. Both heteroclitic XBP1184–192–CTLs (Figure 6a) and heteroclitic XBP1 SP367–375-CTLs (Figure 6b) displayed increased CD107a expression on CD8+ T cells in response to interaction with primary CD138+ MM cells isolated from three HLA-A2+ MM patients. CD107a expression on CD8+ T cells from heteroclitic XBP1184–192 CTLs ranged between 4 – 19% while heteroclitic XBP1 SP367–375 CTLs ranged between 7 – 16%. In contrast, unstimulated T cells from the same donor showed minimal expression of CD107a on CD8+ cells in response to stimulation with the primary HLA-A2+/CD138+ cells from MM patients (0 – 1%).
Figure 6. XBP1-CTL degranulation in response to primary HLA-A2+/CD138+ patient MM cells.
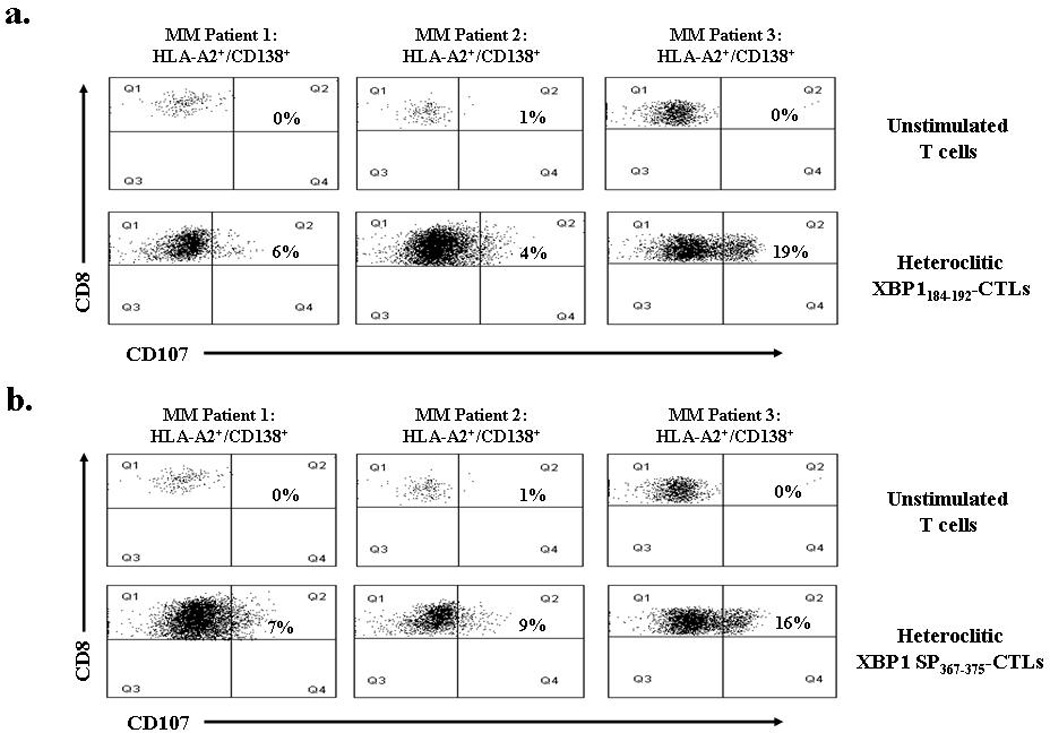
Degranulation of heteroclitic XBP1-CTLs was analyzed by flow cytometry one week after their 4th stimulation, using CD107a, a surface antigen transiently present on the cell surface after release of cytotoxic granules. CD107a expression on (a) heteroclitic XBP1184–192-CTLs and (b) heteroclitic SP367–375–CTLs following stimulation with primary CD138+ MM cells from three HLA-A2+ patients. Control unstimulated T cells did not degranulate when exposed to the primary HLA-A2+/ CD138+ MM cells. The results are expressed as the percent CD107a of CD8+ T cells.
Importantly, XBP1-CTLs demonstrated direct cytotoxicity against primary CD138+ MM cells isolated from HLA-A2+ patients using a calcein-release cytoxicity assay. We evaluated cytotoxicity of heteroclitic XBP1184–192–CTLs and heteroclitic XBP1 SP367–375-CTLs generated from two different donors against primary HLA-A2+/CD138+ MM cells from two patients (Figures 7a and 7b). XBP1-CTLs generated from both donors displayed the highest cytotoxic activity against HLA-A2+/CD138+ cells at an effector:target cell ratio of 60:1. The control unstimulated T cells did not lyse primary CD138+ MM cells from the HLA-A2+ MM patients. These data demonstrates the functional activity of the heteroclitic XBP1184–192 and XBP1 SP367–375-CTLs against the primary MM cells through degranulation and subsequent cytolytic activity.
Figure 7. Cytotoxicity of XBP1-CTLs against primary HLA-A2+/CD138+ patient MM cells.
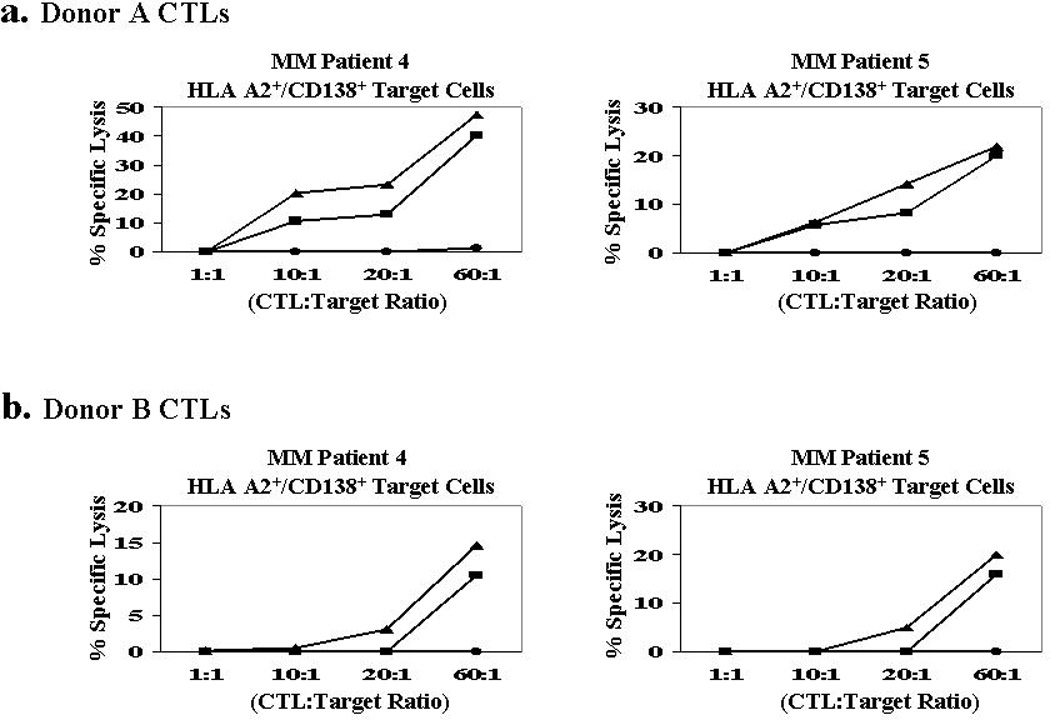
The specific cytotoxic activity of heteroclitic XBP1-CTLs were analyzed one week after their 4th stimulation using the calcein-release cytotoxicity assay. Heteroclitic XBP1184–192 CTLs (■) and heteroclitic SP367–375–CTLs (▲) generated from T cells of (a) Donor 1 and (b) Donor 2 demonstrate a high level of cytotoxic activity against primary CD138+ MM cells from two HLA-A2+ MM patients. Control unstimulated T cells (●) did not lyse primary CD138+ MM cells from the HLA-A2+ MM patients.
DISCUSSION
XBP1 is a basic leucine zipper-containing transcription factor required for the terminal differentiation of B lymphocytes to plasma cells.9, 29–30 It has been suggested that the high amount of immunoglobulin molecules produced in the plasma cells evokes ER stress, which in turn activates IRE1-mediated XBP1 expression and subsequently mRNA splicing during the plasma cell differentiation.12, 15–17 The active XBP1 intracellular protein is thus highly expressed only in the plasma cells through both transcriptional and post-transcriptional mechanisms.14, 22, 31 Additionally, XBP1 expression is higher in MM, compared to MGUS.30, 32 This unique profile of XBP1 over-expression provides an opportunity to develop an antigen-specific immunotherapy for the patients with MM or the pre-malignant diseases such as MGUS and smoldering multiple myeloma.
Here, we report on the identification and characterization of novel heteroclitic HLA-A2-specific XBP1 epitopes that induce specific anti-tumor CTLs that specifically target MM cells. The peptide identification was performed by screening the full-length sequences of non-spliced (260 amino acids) or spliced (375 amino acids) XBP1 protein to predict HLA-A2-specific peptides. We further screened the HLA-A2 anchor residues of the predicted peptides to avoid the epitopes containing amino acid which causes negative effects on HLA-A2 binding.33 Based on the examination of HLA-A2 anchor residues, optimal peptides were selected from spliced or non-spliced XBP1 protein and evaluated for their HLA-A2 affinity. Among them, non-spliced XBP1184–192 (NISPWILAV) and spliced XBP1367–375 (ELFPQLISV) peptides demonstrated a high level of binding to HLA-A2 molecules.
Previously, investigators have shown that increased peptide stability to MHC molecules enhances its immunogenicity.34–38 In these studies, we identified heteroclitic XBP1-specific peptides (YISPWILAV, YLFPQLISV) by altering the primary anchor residue of the native non-spliced XBP1184–192 (NISPWILAV) and spliced XBP1367–375 (ELFPQLISV) peptides, respectively. These heteroclitic XBP1 peptides demonstrated improved HLA-A2 binding stability on antigen-presenting cells while maintaining their recognition by T-cell receptors and ability to activate the antigen-specific T cells. In addition, the sequence of the heteroclitic analogues were confirmed not to overlap with sequences of peptides derived from other normal proteins, so that the risk of targeting the normal proteins is avoided by the heteroclitic XBP1 peptide-specific CTLs.
The challenge in targeting XBP1 antigen is to break T-cell tolerance to this plasma cell-specific antigen. Lotz et al.39 suggested that the HLA-A2-restricted XBP1-specific human T cell repertoire is affected by partial self-tolerance. We examined whether the heteroclitic XBP1 peptides are able to break T-cell tolerance to this plasma cell-specific self-antigen. Our particular additional interest was the ability of the CTLs induced by heteroclitic XBP1 peptide to recognize the native XBP1 peptide which potentially presented on MM cells. In order to address these issues, we generated CTLs specific to the respective heteroclitic XBP1 peptide and evaluated their immunogenicity against tumor cell lines. This was performed by repeated stimulation of HLA-A2+ normal CD3+ T lymphocytes with APC pulsed with either heteroclitic XBP1184–192 (YISPWILAV) or heteroclitic XBP1 SP367–375 (YLFPQLISV) peptide. The resulting cell cultures displayed an altered T cell phenotype including an increased proportion of CD8+ and activated/memory (CD69+/CD45RO+) T cells and a decreased proportion of CD4+ and naïve (CD45RA+/CCR7+) T cells (Figure 2). These observations confirm results by other investigators that ex vivo generated tumor associated antigen-specific CTLs have distinct phenotypes of effector cells.7, 40–42 In functional analysis, we demonstrated antitumor responses of the XBP1-CTLs against myeloma cells in XBP1-specific and HLA-A2 restricted manner by their IFN-γ secretion (Figure 3), cell proliferation (Figure 4) and cytotoxicity (Figure 5). Importantly, we demonstrate the functional activity of heteroclitic XBP1184–192 and XBP1 SP367–375 CTLs to both degranulate and lyse primary CD138+ cells from HLA-A2+ MM patients (Figures 6 and 7). Interestingly, the cytotoxic activity of the spliced heteroclitic SP367–375 peptide-specific CTLs was higher than the non-spliced XBP1184–192 peptide-specific CTLs against both MM cell lines (Figure 5) as well as primary MM cells (Figure 7). These results may be explained by the crucial role of spliced variant of XBP1 in plasma cell differentiation and its over-expression in malignant myeloma cells.24, 25 Taken together, the altered T cell phenotype enriched for CD8+ and activated/memory T cell subtypes support the MM-specific functional activities of the CTLs generated by the heteroclitic XBP1 peptides.
An immunotherapeutic approach using the immunogenic heteroclitic XBP1 peptides can be translated into several different clinical applications including peptide vaccinations or adoptive transfer of ex vivo generated peptide-specific CTLs.43–47 Infusion of peptide-specific CTLs in MM patients could overcome the limitation of peptide-based vaccination which requires functional immune system in the patients. In considering peptide vaccination, peptide dosage, site of injection and the selection of a suitable adjuvant are all critical factors that need to be determined in clinical trials. Alternatively, utilization of peptide-specific CTL clones could be utilized as a maintenance therapy post-autologous transplantation. A concern a self-antigen in the development of an XBP1 antigen-based immunotherapy is the potential for targeting normal plasma cells which express XBP1.30, 48 However, the higher expression of XBP1 may make MM cells more specific target to the CTLs, but this issue should be closely monitored and investigated further in clinical trials.
In summary, we report here on heteroclitic XBP1184–192 (YISPWILAV) and XBP1 SP367–375 (YLFPQLISV) peptides with immunotherapeutic potential to efficiently target and lyse MM cells. These data provides information for their clinical application as either a peptide-based vaccination or adoptive transfer of ex vivo generated XBP1-specific T cells in the patients with MM and the pre-malignant diseases.
Acknowledgments
Research Support
This work is supported by:
NIH grants PO1-155258 (NCM); P50-100707, and PO1-78378, (K.C.A. and N.C.M.), RO1-129295 and Department of Veteran’s Affairs merit review award (N.C.M.) and RO1-50947 (K.C.A.)
Footnotes
CONFLICT OF INTEREST
There are no competing financial conflicts of interest in relation to the work described.
REFERENCES
- 1.Kumar S. Multiple myeloma - current issues and controversies. Cancer Treat Rev. 2010;36(Suppl 2):S3–S11. doi: 10.1016/S0305-7372(10)70006-2. [DOI] [PubMed] [Google Scholar]
- 2.Laubach JP, Richardson PG, Anderson KC. The evolution and impact of therapy in multiple myeloma. Med Oncol. 2010;27(Suppl 1):S1–S6. doi: 10.1007/s12032-010-9442-2. [DOI] [PubMed] [Google Scholar]
- 3.Jagannath S, Kyle RA, Palumbo A, Siegel DS, Cunningham S, Berenson J. The current status and future of multiple myeloma in the clinic. Clin Lymphoma Myeloma Leuk. 2010;10:28–43. doi: 10.3816/CLML.2010.n.003. [DOI] [PubMed] [Google Scholar]
- 4.Bensinger WI. Role of autologous and allogeneic stem cell transplantation in myeloma. Leukemia. 2009;23:442–448. doi: 10.1038/leu.2008.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeiser R, Finke J. Allogeneic haematopoietic cell transplantation for multiple myeloma: reducing transplant-related mortality while harnessing the graft-versusmyeloma effect. Eur J Cancer. 2006;42:1601–1611. doi: 10.1016/j.ejca.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 6.Abdalla AO, Kokhaei P, Hansson L, Mellstedt H, Osterborg A. Idiotype vaccination in patients with myeloma reduced circulating myeloma cells (CMC) Ann Oncol. 2008;19:1172–1179. doi: 10.1093/annonc/mdn017. [DOI] [PubMed] [Google Scholar]
- 7.Lee JJ, Choi BH, Kang HK, Park MS, Park JS, Kim SK, et al. Induction of multiple myeloma-specific cytotoxic T lymphocyte stimulation by dendritic cell pulsing with purified and optimized myeloma cell lysates. Leuk Lymphoma. 2007;48:2022–2031. doi: 10.1080/10428190701583975. [DOI] [PubMed] [Google Scholar]
- 8.Vasir B, Borges V, Wu Z, Grosman D, Rosenblatt J, Irie M, et al. Fusion of dendritic cells with multiple myeloma cells results in maturation and enhanced antigen presentation. Br J Haematol. 2005;129:687–700. doi: 10.1111/j.1365-2141.2005.05507.x. [DOI] [PubMed] [Google Scholar]
- 9.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, et al. Plasma cell differentiation requires the transcription factor XBP1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 10.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/s1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 11.Liou HC, Boothby MR, Finn PW, Davidon R, Nabavi N, Zeleznik-Le NJ, et al. A new member of the leucine zipper class of proteins that binds to the HLA DR alpha promoter. Science. 1990;247:1581–1584. doi: 10.1126/science.2321018. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 13.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretary capacity by processing the XBP1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 14.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mori K. Frame switch splicing and regulated intramembrane proteolysis: key words to understand the unfolded protein response. Traffic. 2003;4:519–528. doi: 10.1034/j.1600-0854.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 16.Bagratuni T, Wu P, Gonzalez de Castro D, Davenport EL, Dickens NJ, Walker BA, et al. XBP1s levels are implicated in the biology and outcome of myeloma mediating different clinical outcomes to thalidomide-based treatments. Blood. 2010;116:250–253. doi: 10.1182/blood-2010-01-263236. [DOI] [PubMed] [Google Scholar]
- 17.Patterson J, Palombella VJ, Fritz C, Normant E. IPI-504, a novel and soluble HSP-90 inhibitor, blocks the unfolded protein response in multiple myeloma cells. Cancer Chemother Pharmacol. 2008;61:923–932. doi: 10.1007/s00280-007-0546-0. [DOI] [PubMed] [Google Scholar]
- 18.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 19.Wen XY, Stewart AK, Sooknanan RR, Henderson G, Hawley TS, Reimold AM, et al. Identification of c-myc promoter-binding protein and X-box binding protein 1 as interleukin-6 target genes in human multiple myeloma cells. Int J Oncol. 1999;15:173–178. doi: 10.3892/ijo.15.1.173. [DOI] [PubMed] [Google Scholar]
- 20.Davies FE, Dring AM, Li C, Rawstron AC, Shammas MA, O'Connor SM, et al. Insights into the multistep transformation of MGUS to myeloma using microarray expression analysis. Blood. 2003;102:4504–4511. doi: 10.1182/blood-2003-01-0016. [DOI] [PubMed] [Google Scholar]
- 21.Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99:1745–1757. doi: 10.1182/blood.v99.5.1745. [DOI] [PubMed] [Google Scholar]
- 22.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 23.Pal R, Janz M, Galson DL, Gries M, Li S, Jöhrens K, et al. C/EBPbeta regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood. 2009;114:3890–3898. doi: 10.1182/blood-2009-01-201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunsing R, Omori SA, Weber F, Bicknell A, Friend L, Rickert R, et al. B- and T-cell development both involve activity of the unfolded protein response pathway. J Biol Chem. 2008;283:17954–17961. doi: 10.1074/jbc.M801395200. [DOI] [PubMed] [Google Scholar]
- 26.Zweerink HJ, Gammon MC, Utz U, Sauma SY, Harrer T, Hawkins JC, et al. Presentation of endogenous peptides to MHC class I-restricted cytotoxic T lymphocytes in transport deletion mutant T2 cells. J Immunol. 1993;150:1763–1771. [PubMed] [Google Scholar]
- 27.Roden MM, Lee KH, Panelli MC, Marincola FM. A novel cytolysis assay using fluorescent labeling and quantitative fluorescent scanning technology. J Immunol Methods. 1999;226:29–41. doi: 10.1016/s0022-1759(99)00039-3. [DOI] [PubMed] [Google Scholar]
- 28.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 29.Shapiro-Shelef M, Calame K. Plasma cell differentiation and multiple myeloma. Curr Opin Immunol. 2004;16:226–234. doi: 10.1016/j.coi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 31.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–5947. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 32.Claudio JO, Masih-Khan E, Tang H, Gonçalves J, Voralia M, Li ZH, et al. A molecular compendium of genes expressed in multiple myeloma. Blood. 2002;100:2175–2186. doi: 10.1182/blood-2002-01-0008. [DOI] [PubMed] [Google Scholar]
- 33.Rammensee HG, Friede T, Steranovic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 34.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 35.Huang YH, Wu JC, Peng WL, Huo TI, Shih HH, Lan KH, et al. Generation of cytotoxicity against hepatitis delta virus genotypes and quasispecies by epitope modification. J Hepatol. 2009;50:779–788. doi: 10.1016/j.jhep.2008.11.028. [DOI] [PubMed] [Google Scholar]
- 36.Maeurer MJ, Necker A, Salter RD, Castelli C, Höhn H, Karbach J, et al. Improved detection of melanoma antigen-specific T cells expressing low or high levels of CD8 by HLA-A2 tetramers presenting a Melan-A/Mart-1 peptide analogue. Int J Cancer. 2002;97:64–71. doi: 10.1002/ijc.1580. [DOI] [PubMed] [Google Scholar]
- 37.Bae J, Martinson JA, Klingemann HG. Identification of novel CD33 antigen-specific peptides for the generation of cytotoxic T lymphocytes against acute myeloid leukemia. Cell Immunol. 2004;227:38–50. doi: 10.1016/j.cellimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Bae J, Martinson JA, Klingemann HG. Heteroclitic CD33 peptide with enhanced anti-acute myeloid leukemic immunogenicity. Clin Cancer Res. 2004;10:7043–7052. doi: 10.1158/1078-0432.CCR-04-0322. [DOI] [PubMed] [Google Scholar]
- 39.Lotz C, Mutallib SA, Oehlrich N, Liewer U, Ferreira EA, Moos M, et al. Targeting positive regulatory domain I-binding factor 1 and X box-binding protein 1 transcription factors by multiple myeloma-reactive CTL. J Immunol. 2005;175:1301–1309. doi: 10.4049/jimmunol.175.2.1301. [DOI] [PubMed] [Google Scholar]
- 40.Abdul-Alim CS, Li Y, Yee C. Conditional superagonist CTL ligands for the promotion of tumor-specific CTL responses. J Immunol. 2010;184:6514–6521. doi: 10.4049/jimmunol.0900448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shafer JA, Cruz CR, Leen AM, Ku S, Lu A, Rousseau A, et al. Antigen-specific cytotoxic T lymphocytes can target chemoresistant side-population tumor cells in Hodgkin lymphoma. Leuk Lymphoma. 2010;51:870–880. doi: 10.3109/10428191003713968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okada H. Brain tumor immunotherapy with type-1 polarizing strategies. Ann N Y Acad Sci. 2009;1174:18–23. doi: 10.1111/j.1749-6632.2009.04932.x. [DOI] [PubMed] [Google Scholar]
- 43.Perez SA, von Hofe E, Kallinteris NL, Gritzapis AD, Peoples GE, Papamichail M, et al. A new era in anticancer peptide vaccines. Cancer. 2010;116:2071–2080. doi: 10.1002/cncr.24988. [DOI] [PubMed] [Google Scholar]
- 44.Noguchi M, Kakuma T, Uemura H, Nasu Y, Kumon H, Hirao Y, et al. A randomized phase II trial of personalized peptide vaccine plus low dose estramustine phosphate (EMP) versus standard dose EMP in patients with castration resistant prostate cancer. Cancer Immunol Immunother. 2010;59:1001–1009. doi: 10.1007/s00262-010-0822-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patil R, Clifton GT, Holmes JP, Amin A, Carmichael MG, Gates JD, et al. Clinical and immunologic responses of HLA-A3+ breast cancer patients vaccinated with the HER2/neu-derived peptide vaccine, E75, in a phase I/II clinical trial. J Am Coll Surg. 2010;210:140–147. doi: 10.1016/j.jamcollsurg.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 46.Weber G, Karbach J, Kuçi S, Kreyenberg H, Willasch A, Koscielniak E, et al. WT1 peptide-specific T cells generated from peripheral blood of healthy donors: possible implications for adoptive immunotherapy after allogeneic stem cell transplantation. Leukemia. 2009;23:1634–1642. doi: 10.1038/leu.2009.70. [DOI] [PubMed] [Google Scholar]
- 47.Lu X, Jiang X, Liu R, Zhao H, Liang Z. Adoptive transfer of pTRP2-specific CTLs expanding by bead-based artificial antigen-presenting cells mediates anti-melanoma response. Cancer Lett. 2008;271:129–139. doi: 10.1016/j.canlet.2008.05.049. [DOI] [PubMed] [Google Scholar]
- 48.Pillai S. Birth pangs: the stressful origins of lymphocytes. J Clin Invest. 2005;115:224–227. doi: 10.1172/JCI24238. [DOI] [PMC free article] [PubMed] [Google Scholar]