

Abstract
Long-term alcohol exposure sensitizes hepatocytes to tumor necrosis factor-α (TNF) cytotoxicity. 4-Hydroxynonenal (4-HNE) is one of the most abundant and reactive lipid peroxides. Increased hepatic 4-HNE contents present in both human alcoholics and alcohol-fed animals. In the present study, we investigated the effects of intracellular 4-HNE accumulation on TNF-induced hepatotoxicity and its potential implication in the pathogenesis of alcoholic liver disease. Male C57BL/6 mice were fed an ethanol-containing or a control diet for 5 weeks. Long-term alcohol exposure increased hepatic 4-HNE and TNF levels. Cell culture studies revealed that 4-HNE, at nontoxic concentrations, sensitized hepatocytes to TNF killing, which was associated with suppressed NF-κB transactivity. Further investigation demonstrated that 4-HNE prevented TNF-induced inhibitor of κBα phosphorylation without affecting upstream IκB kinase activity. An immunoprecipitation assay revealed that increased 4-HNE content was associated with increased formation of 4-HNE–inhibitor of κBα adduction in both 4-HNE–treated hepatocytes and in the livers of alcohol-fed mice. Prevention of intracellular 4-HNE accumulation by bezafibrate, a peroxisome proliferator-activated receptor-α agonist, protected hepatocytes from TNF killing via NF-κB activation. Supplementation of N-acetylcysteine, a glutathione precursor, conferred a protective effect on alcohol-induced liver injury in mice, was associated with decreased hepatic 4-HNE formation, and improved hepatic NF-κB activity. In conclusion, increased 4-HNE accumulation represents a potent and clinically relevant sensitizer to TNF-induced hepatotoxicity. These data support the notion that removal of intracellular 4-HNE can serve as a potential therapeutic option for alcoholic liver disease.
Oxidative stress plays a central and causal role in the onset and progression of alcoholic liver disease (ALD).1,2 Long-term ethanol exposure increases production of reactive oxygen species, lowers cellular antioxidant levels, and leads to oxidative stress in the liver. Alcohol-induced liver injury is associated with enhanced lipid peroxidation, protein carbonyl formation, formation of lipid radicals, and decreased hepatic antioxidant defenses.3–5 Replacement of polyunsaturated fat (required for lipid peroxidation) with saturated fat or medium-chain triglycerides lowers or prevents lipid peroxidation and alcohol-induced liver injury.6–8 In contrast, the addition of iron, known to promote lipid peroxidation by Fenton's reaction, exacerbates liver injury.9,10 More important, supplementation with antioxidants prevents alcohol-induced liver injury.11,12
Overproduction of tumor necrosis factor-α (TNF) contributes to the pathogenesis of ALD. Patients with alcoholic hepatitis have increased systemic TNF levels, which correlate with disease severity and mortality.13–15 Moreover, a TNF promoter polymorphism has been linked with susceptibility to alcoholic hepatitis.16 Compelling data relating TNF to alcohol-induced liver injury in an experimental setting from Thurman's laboratory indicate that anti-TNF antibody prevents liver injury in alcohol-fed rats.17 Similarly, mice lacking the TNF type I receptor do not develop alcoholic liver injury.18
TNF induces both pro- and anti-apoptotic signaling. TNF activates NF-κB, inducing transcription of its target genes primarily encoding survival proteins (eg, cellular FLICE-like inhibitory protein and inhibitor of apoptosis proteins) via formation of the complex I signalosome after binding to TNF receptor-1. Depending on the cellular signaling context, complex II can be formed from complex I, which activates caspase-8 and pro-apoptotic pathways. Activation of caspases and prolonged c-Jun NH2 terminal kinase (JNK) signaling are, under normal conditions, antagonized by various NF-κB target genes. Therefore, under normal physiological conditions, hepatocytes are resistant to TNF-induced hepatotoxicity. However, several laboratories independently demonstrated that long-term alcohol exposure sensitizes hepatocytes to TNF-mediated hepatotoxicity. Subsequent investigations revealed that multiple mechanisms are implicated in the sensitizing process, including mitochondrial permeability transition pore opening,19 decreased mitochondrial glutathione (GSH) levels,20 and increased intracellular S-adenosylhomocysteine levels,21 all relevant to ALD.
Of lipid peroxides, 4-hydroxynonenal (4-HNE) is one of the most abundant and reactive aldehydic products derived from the oxidation of membrane n-6-polyunsaturated fatty acids. Although increases in both 4-HNE formation and TNF production are critically involved in the disease development of ALD, it remains unclear if 4-HNE may serve as a sensitizer to TNF-induced cell death in hepatocytes. The present studies were conducted to test this hypothesis. We report herein that exposure to 4-HNE, at nontoxic or minimally toxic concentrations, sensitizes hepatocytes to TNF-induced cell death via suppression of NF-κB activation.
Materials and Methods
Chemicals
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO), unless otherwise specified.
Hepatocytes and Culture Conditions
HepG2 and Hep3B cells, two human hepatoma cell lines, were obtained from ATCC (Manassas, VA). Primary mouse hepatocytes were obtained from Celsis In Vitro Technologies (Baltimore, MD). Hepatocytes were cultured in Dulbecco's modified Eagle's medium containing 10% (v/v) fetal bovine serum, 2 mmol/L glutamine, 5 U/mL penicillin, and 50 μg/mL streptomycin at 37°C in a humidified O2/CO2 (19:1) atmosphere.
Animal Model and Experimental Protocol
Male C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME), weighing 25 ± 0.5 g (mean ± SD) were fed ad libitum with an ethanol-containing Lieber-DeCarli diet (ethanol-derived calories were increased from 30% to 36% during the first 4 weeks, with a 2% increase each week) or an isocaloric control liquid diet (Bioserv, Frenchtown, NJ) for 5 weeks. For N-acetylcysteine (NAC) supplementation, NAC was added into the ethanol-containing diet at the dose of 0.16 mg/mL. Food intake and body weight were recorded daily and weekly, respectively. Mice were euthanized, and plasma and liver samples were harvested at the end of the experiment.
Histological Analysis and in Situ Apoptosis Detection
H&E staining of liver sections was performed as previously described.21 Apoptotic hepatocytes were detected by TUNEL assay with a TUNEL assay kit (Intergen Company, Purchase, NY), as previously described.21
Cell Death Assays
Cell death was determined by measurement of lactate dehydrogenase (LDH) release and MTT assay, as previously described.21 For Hoechst staining, 30 minutes before the end of the incubation with the indicated stimulus, Hoechst was added to each well of 24-well plates at a final concentration of 1 μmol/L. At the completion of the incubation, the cells were washed three times with ice-cold PBS, and then the fluorescence was measured by fluorescent microscope at an emission wavelength of 460 nm, using an excitation wavelength of 360 nm for the dichlorofluorescein fluorophore.
ELISA for NF-κB (p65) DNA-Binding Activity
NF-κB (p65) DNA-binding activity in the nuclear fraction of liver tissues or cultured cells was measured using enzyme-linked immunosorbent assay (ELISA) kits (Cayman Chemical, Ann Arbor, MI), in accordance with the manufacturer's instructions.
Real-Time PCR
Total RNA isolation, reverse transcription, and real-time PCR were performed as previously described.22 Briefly, total RNA, from either frozen liver tissue or cultured cells, was isolated with a phenol-chloroform extraction. For each sample, 1 μg of total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). The cDNA was amplified in MicroAmp Optical 96-well reaction plates with an SYBR Green PCR Master Mix (Applied Biosystems) on a Prism 7000 sequence detection system (Applied Biosystems). Relative gene expression was calculated after nomalization by a housekeeping gene (mouse or human 18S ribosomal RNA).
Western Blot Analysis
Western blot analysis was performed as previously described,22 and the following antibodies were used: anti-poly (ADP-ribose) polymerase (Novus Biologicals, Littleton, CO), anti-4-HNE (R&D Systems, Inc., Minneapolis, MN), anti-phospho-IκBα, anti-IκBα, anti-phosphorylated-IκB kinase (IKKβ), anti-IKKβ, anti-ubiquitin, anti-p65, anti-phosphorylated-JNK, or anti-JNK (Cell Signaling Technology, Danvers, MA).
Immunoprecipitation
Cells were lysed in an immunoprecipitation buffer (150 mmol/L NaCl, 50 mmol/L Tris-HCl, and 1% Nonidet P-40, pH 7.8) and a mammalian cell-specific protease inhibitor cocktail. Total cellular extracts (200 μg of protein) were incubated with anti-IκBα antibody (1 μg/mL) in immunoprecipitation buffer overnight at 4°C on a rocker. The antibody-protein mixture was agitated with Protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 hour at 4°C. The immunoprecipitates were washed four times with immunoprecipitation buffer. The washed immunoprecipitates were incubated in 50 μL of one times electrophoresis loading buffer and heated at 100°C for 5 minutes. The beads were spun out and the supernatant was resolved by SDS-PAGE, and the modification of IκBα by 4-HNE or ubiquitination of IκBα was analyzed by using Western blot analysis.
Statistical Analysis
All data were expressed as mean ± SD. Statistical analysis was performed using a one-way analysis of variance, and data were further analyzed by Newman-Keuls test for statistical differences. Differences between treatments were considered to be statistically significant at P < 0.05.
Results
Liver Injury Induced by Long-Term Alcohol Exposure Is Associated with Increased Hepatic 4-HNE and TNF Levels
Male C57BL/6 mice (aged 8 weeks), fed the Lieber-DeCarli alcohol-containing liquid diet for 5 weeks, were used as a model of ALD. Compared with pair-fed animals, alcohol-fed mice showed a significant increase in hepatic triglyceride contents (Figure 1A) and hepatic steatosis (Figure 1B). Alcohol exposure elevated plasma alanine aminotransferase activity compared with pair-fed animals (Figure 1C). TUNEL assays showed that alcohol feeding was associated with increased hepatocyte apoptosis (Figure 1D). Moreover, hepatic TNF levels were significantly increased after ethanol feeding (Figure 1E), whereas circulating TNF levels were not affected by long-term alcohol exposure (data not shown). To determine the effect of long-term alcohol consumption on hepatic 4-HNE contents, Western blot analysis was conducted to detect protein modification by 4-HNE using a polyclonal anti–4-HNE antibody specifically binding to 4-HNE–modified proteins. As shown in Figure 1F, 5-week alcohol feeding significantly increased hepatic 4-HNE contents when compared with pair-fed animals.
Figure 1.

Long-term alcohol exposure increases hepatic TNF and 4-HNE levels. A: Hepatic triglyceride (TG) contents. B: H&E staining. C: Plasma alanine aminotransferase (ALT) levels. D: The TUNEL assay. E: Hepatic TNF levels. F: Western blot analysis of long-term alcohol consumption on hepatic 4-HNE–protein adduct formation. Male C57BL/6 mice were pair fed liquid diets, with or without ethanol, for 5 weeks. Data are expressed as the mean ± SD (n = 5 to 8 mice per group). *P < 0.01 versus PF. AF, alcohol feeding; PF, pair feeding.
4-HNE Sensitizes Hepatocytes to TNF-Induced Cytotoxicity
The association of liver injury and elevation in hepatic 4-HNE and TNF levels in alcohol-fed mice made us determine whether 4-HNE may act as a sensitizer to TNF-induced cell death in cultured hepatocytes. We first pretreated HepG2 cells with 4-HNE (0 to 60 μmol/L) for 2 hours before TNF (0 to 40 ng/mL) stimulation. Cell death was measured 16 hours later by MTT assay. As shown in Figure 2A, 4-HNE sensitized HepG2 cells to TNF killing. The conclusion was further confirmed by using Western blot analysis for the cleaved form of poly (ADP-ribose) polymerase-1 and fluorescence microscopic examination of Hoechst 33342 staining (Figure 2, B and C). To determine its generality, cell death studies were repeated in both Hep3B cells and primary mouse hepatocytes. As shown in Figure 1D, the comparable results were observed.
Figure 2.

4-HNE sensitizes hepatocytes to TNF cytotoxicity. A: 4-HNE sensitizes HepG2 cells to TNF-induced cell death. HepG2 cells are pretreated with exogenous 4-HNE for 2 hours before TNF stimulation. Cell death is measured 16 hours later by MTT assay. Bars with different symbols (*, †, ‡, §, ¶) differ significantly from each other, all with a value of P < 0.05. B and C: 4-HNE–induced sensitization to TNF hepatotoxicity in HepG2 cells is further confirmed by an increased cleaved form of poly (ADP-ribose) polymerase (PARP)-1 and fluorescence microscopic examination of Hoechst 33342 staining. HepG2 cells are pretreated with 20 μmol/L 4-HNE for 2 hours, followed by TNF (40 ng/mL) stimulation. D: 4-HNE exposure sensitizes both Hep3B cells and primary mouse hepatocytes to TNF cytotoxicity. Both Hep3B and primary hepatocytes are pretreated with 20 μmol/L 4-HNE for 2 hours, followed by TNF (40 ng/mL) stimulation. LDH releases are measured 16 hours later. All values are denoted as the mean ± SD from three or more independent studies. *P < 0.05, **P < 0.01 versus untreated (UT).
4-HNE Suppresses TNF-Stimulated NF-κB Transactivation
Under physiological conditions, the hepatocytes were resistant to TNF-induced cell death because of NF-κB–mediated transcriptional regulation of anti-apoptotic proteins. We, therefore, investigated whether 4-HNE affected TNF-induced NF-κB transactivity. As shown in Figure 3, exposure to 4-HNE for 4 hours increased intracellular 4-HNE levels in a dose-dependent manner in both HepG2 cells and primary mouse hepatocytes (Figure 3A). Moreover, 4-HNE pretreatment dose dependently suppressed TNF-induced increases in NF-κB (p65) DNA-binding activity in HepG2 cells (Figure 3, B and C). The suppressive effect of 4-HNE on NF-κB transactivity was further corroborated by significantly decreased NF-κB–mediated gene expression after TNF stimulation (Figure 3D). To determine the in vivo relevance of 4-HNE–induced suppression of NF-κB activation in ALD, the nuclear fraction was isolated from liver tissues and the DNA-binding activity of NF-κB (p65) was measured. As shown in Figure 3E, alcohol feeding decreased hepatic nuclear p65 DNA-binding activity.
Figure 3.
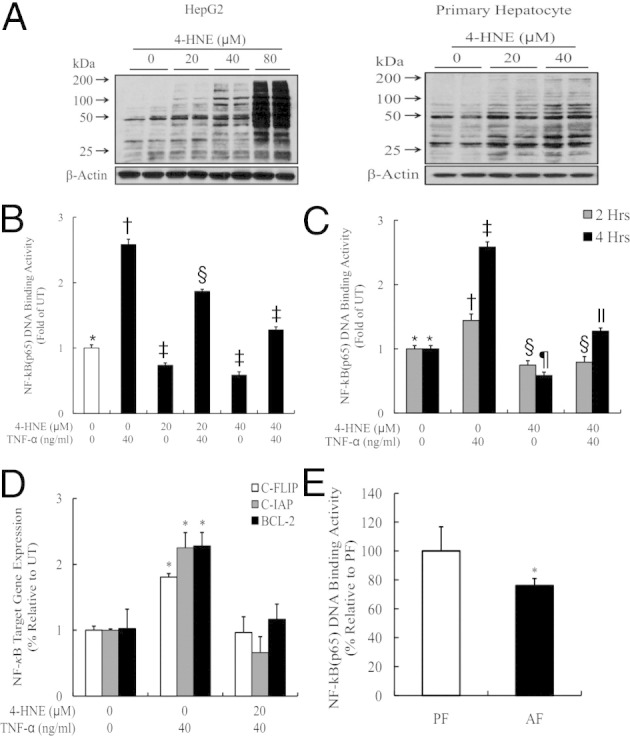
4-HNE suppresses TNF-stimulated NF-κB transactivation. A: Exogenous 4-HNE exposure increases intracellular 4-HNE levels in both HepG2 cells and primary hepatocytes. HepG2 cells are incubated with exogenous 4-HNE at the indicated doses for 8 hours. Whole cell lysates are collected and used for the detection of intracellular 4-HNE contents by using Western blot analysis. All values are expressed as the mean ± SD from three or more independent studies. *P < 0.05, **P < 0.01, and ***P < 0.001 versus untreated (UT). B and C: 4-HNE pretreatment suppresses TNF-induced increases in NF-κB (p65) DNA-binding activities in HepG2 cells. HepG2 cells are pretreated with 4-HNE for 2 hours before TNF stimulation. Four hours later, nuclear fractions are isolated and subjected to ELISA for the measurement of p65 DNA-binding activities. Bars with different symbols (*, †, ‡, §, ¶, ll) differ significantly from each other, all with a value of P < 0.05. D: 4-HNE pretreatment suppresses NF-κB–mediated gene expression after TNF stimulation. HepG2 cells are pretreated with 40 μmol/L 4-HNE for 2 hours and then incubated with 40 ng/mL TNF for 16 hours, total RNA is isolated, and NF-κB–regulated gene expression is determined by real-time RT-PCR. All values are denoted as the mean ± SD from three or more independent studies. *P < 0.05 versus UT. E: Alcohol feeding decreases hepatic nuclear p65 DNA-binding activity. Data are expressed as the mean ± SD (n = 5 to 8 mice per group). *P < 0.05 versus PF. AF, alcohol feeding; PF, pair feeding.
4-HNE Inhibits IκBα Phosphorylation
After binding to its cell membrane receptor, TNF activated NF-κB in the following order: i) IκBα phosphorylation by IKK, ii) ubiquitination and degradation of IκBα by 26S proteasome, and iii) nuclear translocation. The phosphorylation of IκBα was an obligatory step for these subsequent events. To determine the mechanisms involved in the suppressive effect of 4-HNE on NF-κB activation, we first examined the time course of 4-HNE on TNF-induced phosphorylation of IκBα. As shown in Figure 4A, TNF stimulation quickly increased IκBα phosphorylation (15 minutes after TNF exposure), and the increase was maintained during the 1-hour experimental period. Pretreatment with 4-HNE almost completely abolished TNF-induced elevation of IκBα phosphorylation in both HepG2 and primary mouse hepatocytes, which was associated with decreased nuclear p65 content (Figure 4B). Moreover, TNF stimulation induced rapid and transient IκBα degradation (15 and 30 minutes after TNF). However, at 1 hour after TNF exposure, IκBα protein levels were higher than control, suggestive of increased expression of proteins targeted by NF-κB, including IκBα protein itself. This notion was further supported by the time-matched IκBα decrease and NF-κB suppression in 4-HNE–pretreated cells (Figure 4A). These results collectively indicated that 4-HNE suppressed NF-κB transactivity via inhibition of IκBα phosphorylation. Because IKKβ was the dominant upstream kinase to phosphorylate IκBα on TNF stimulation in hepatocytes, we next determined whether decreased IκBα phosphorylation was due to altered IKKβ phosphorylation. Western blot analysis using an antibody for phosphorylated IKKα/β protein showed that 4-HNE pretreatment had no effects on IKKβ phosphorylation at 15 minutes after TNF simulation (Figure 4C). The conclusion was further confirmed by an ELISA specific for phosphorylated IKKβ (see Supplemental Figure S1 at http://ajp.amjpathol.org). To determine whether exposure to exogenous 4-HNE led to increased 4-HNE–IκBα adduct formation, immunoprecipitation analysis was conducted to evaluate the modification of IκBα by 4-HNE. As shown in Figure 4D, the 4-HNE–modified IκBα complex was significantly increased by 4-HNE in both HepG2 cells and primary mouse hepatocytes compared with untreated cells. To determine whether our observations from cell culture were relevant to ALD, we measured 4-HNE–IκBα adduct formation, nuclear NF-κB (p65) contents, and IκBα phosphorylation status using either nuclear or cytosolic proteins from pair- and alcohol-fed mice. As shown in Figure 4E, long-term alcohol exposure increased hepatic 4-HNE–IκBα adduct content, suppressed hepatic IκBα phosphorylation, and decreased hepatic nuclear p65 contents.
Figure 4.
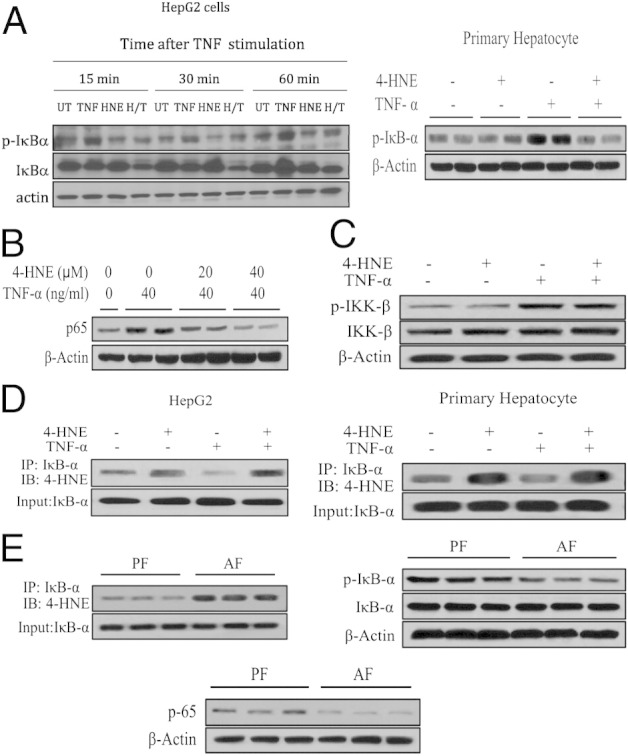
4-HNE exposure abolishes IκBα phosphorylation. A: 4-HNE pretreatment abolishes the TNF-stimulated increase in IκBα phosphorylation. Both HepG2 cells and primary mouse hepatocytes are pretreated with 40 μmol/L 4-HNE for 2 hours before TNF stimulation, and cell lysates are collected at the indicated time points for Western blot analysis of phosphorylated IκBα. B: 4-HNE pretreatment decreases nuclear p65 contents. HepG2 cells are pretreated with 4-HNE for 2 hours before TNF stimulation, and nuclear fractions are collected 4 hours later and subjected to Western blot analysis. C: 4-HNE pretreatment has no effects on the IKKβ phosphorylation. HepG2 cells are pretreated with 40 μmol/L 4-HNE for 2 hours before TNF stimulation, and cell lysates are collected at 15 minutes after TNF exposure for Western blot analysis of phosphorylated IKKβ. D: 4-HNE exposure results in increased formation of the 4-HNE–IκBα complex. HepG2 cells are pretreated with 40 μM 4-HNE for 2 hours before TNF stimulation, and cell lysates are collected 1 hour later and subjected to immunoprecipitation and Western blot analysis. E: Long-term alcohol exposure increases hepatic 4-HNE–IκBα adduct contents, decreases hepatic nuclear p65 contents, and suppresses hepatic IκBα phosphorylation. AF, alcohol feeding; H/T, 4-HNE+TNF; IB, immunoblotting; IP, immunoprecipitation; PF, pair feeding; UT, untreated.
Both Caspase Activation and Sustained JNK Activation Contribute to 4-HNE/TNF-Induced Cell Death
Both caspase activation and prolonged JNK activation contributed to TNF-induced cell death under the conditions of inhibited NF-κB activation. To determine the pathways involved in 4-HNE/TNF-induced cytotoxicity, HepG2 cells were pretreated with 10 μmol/L Z-VAD-FMK, a pan-caspase inhibitor, or SP600125, a specific JNK inhibitor, for 2 hours before exposure to 4-HNE/TNF. As shown in Figure 5, A and B, both inhibitors alleviated cell death in HepG2 cells, indicating that both caspase and JNK activation were critically involved in 4-HNE/TNF-induced hepatocyte cell death. A further examination revealed that, although TNF stimulation led to rapid and transient JNK activation, reaching peak levels at 0.5 hours and returning to basal levels at 2 hours, 4-HNE (40 μmol/L) pretreatment caused a sustained TNF-mediated JNK activation (Figure 5C).
Figure 5.
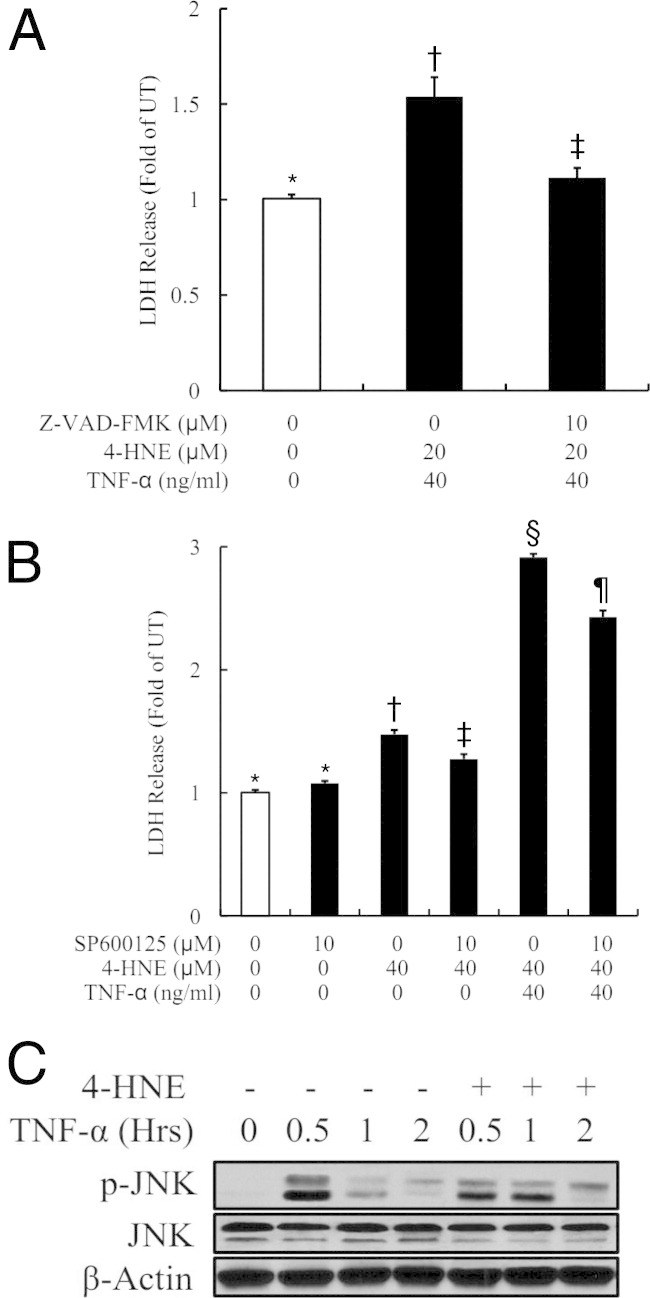
Both caspase and sustained JNK activation contribute to 4-HNE/TNF-induced cell death. A: Pan-caspase inhibitor almost completely prevents 4-HNE/TNF-induced LDH release from HepG2 cells. HepG2 cells are pretreated with 10 μmol/L Z-VAD-FMK, a pan-caspase inhibitor, for 2 hours before 4-HNE/TNF addition. LDH release is measured 16 hours later. All values are expressed as the mean ± SD from at least three experiments. Bars with different letters differ significantly (P < 0.05). B: Inhibition of JNK activation alleviates cell death in HepG2 cells treated with 4-HNE and TNF. HepG2 cells are pretreated with 10 μmol/L SP600125, a specific JNK inhibitor, for 1 hour before 4-HNE/TNF addition. LDH release is measured 16 hours later. All values are expressed as the mean ± SD from at least three experiments. Bars with different symbols (*, †, ‡, §, ¶) differ significantly from each other, all with a value of P < 0.05. C: 4-HNE pretreatment causes a sustained TNF-mediated JNK activation. HepG2 cells are pretreated with 40 μmol/L 4-HNE for 2 hours before TNF stimulation. Whole cell lysates were collected at the indicated time points and subjected to Western blot analysis for phosphorylated JNK.
Bezafibrate, a PPARα Agonist, Protects Hepatocytes against 4-HNE/TNF-Induced Cell Death
Many previous studies suggested that the deleterious effects of 4-HNE may result from induction of oxidative stress by decreased intracellular GSH levels. To determine whether 4-HNE–induced sensitization to TNF cytotoxity was mediated by inhibition of intracellular GSH, we measured GSH levels in HepG2 cells exposed to 4-HNE. As shown in Figure 6A, 4-HNE unexpectedly increased intracellular GSH levels in HepG2 cells. Furthermore, pretreatment with NAC, an antioxidant serving as a GSH precursor, did not confer protection against 4-HNE/TNF-induced cell death (Figure 6B), confirming that oxidative stress was not involved in 4-HNE–induced sensitization to TNF cytotoxicity.
Figure 6.
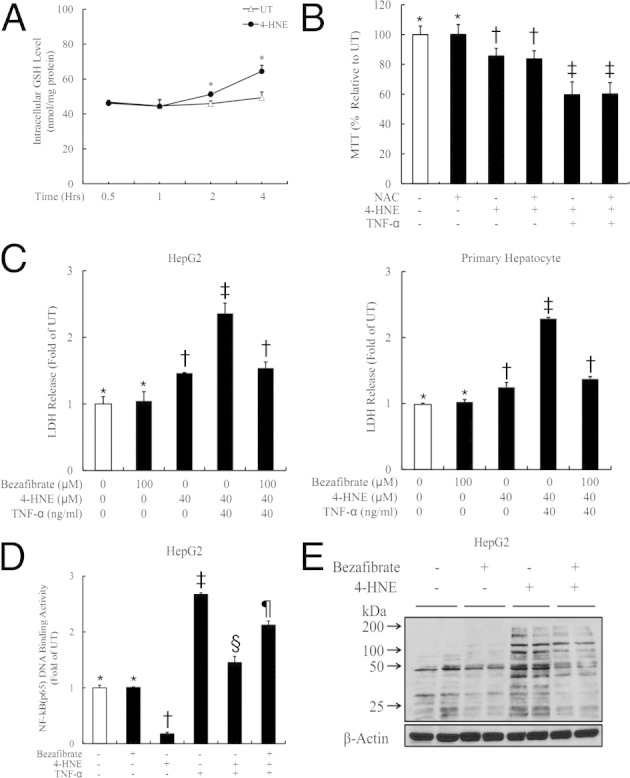
Bezafibrate protects hepatocytes against 4-HNE/TNF-induced cell death. A: Exogenous 4-HNE exposure increases intracellular GSH levels in HepG2 cells. HepG2 cells are treated with 4-HNE, and the intracellular GSH concentrations are measured at the indicated time points. All values are expressed as the mean ± SD from at least three experiments. *P < 0.05 versus untreated (UT). B: Pretreatment with NAC does not confer protection against 4-HNE/TNF-induced cell death. HepG2 cells are pretreated with 0.5 mmol/L NAC for 1 hour before 4-HNE/TNF exposure. Cell death is measured by MTT assay 16 hours later. All values are expressed as the mean ± SD from at least three experiments. Bars with different letters differ significantly (P < 0.05). C: Bezafibrate significantly suppresses LDH release induced by 4-HNE/TNF from HepG2 cells. HepG2 cells are pretreated with 100 μmol/L bezafibrate for 1 hour before 4-HNE/TNF exposure. LDH release is measured 16 hours later. All values are expressed as the mean ± SD from at least three experiments. Bars with different symbols (*, †, ‡, §, ¶) differ significantly from each other, all with a value of P < 0.05. D: Bezafibrate alleviates 4-HNE–induced suppression on TNF-mediated p65 DNA-binding activity. HepG2 cells are pretreated with 100 μmol/L bezafibrate for 1 hour before 4-HNE/TNF exposure. The nuclear fraction is isolated for 2 hours and subjected to ELISA. All values are expressed as the mean ± SD from at least three experiments. Bars with different letters differ significantly (P < 0.05). E: Bezafibrate significantly reduces the intracellular 4-HNE accumulation after exogenous 4-HNE exposure. HepG2 cells are pretreated with 100 μmol/L bezafibrate for 2 hours before 4-HNE addition. Cell lysates are collected 8 hours later for Western blot analysis. All values are expressed as the mean ± SD from at least three experiments.
Long-term alcohol exposure was associated with decreased hepatic peroxisome proliferator-activated receptor-α (PPARα) activity, and PPARα agonists conferred a beneficial effect in ALD. To determine whether the beneficial effects of PPARα agonists may result from the ability of these compounds to metabolize 4-HNE, bezafibrate was used. As shown in Figure 6C, 4-HNE/TNF-induced cell death was significantly suppressed by bezafibrate (100 μmol/L), which was associated with alleviated p65–DNA-binding suppression by 4-HNE (Figure 6D). To determine whether bezafibrate affected intracellular 4-HNE metabolism, we next measured intracellular 4-HNE levels after bezafibrate treatment. As shown in Figure 6E, bezafibrate significantly reduced the intracellular 4-HNE accumulation after 4-HNE exposure.
Preventive Effect of NAC on ALD Is Associated with Decreased Hepatic 4-HNE Formation and Improved NF-κB Activity
The NAC protected from alcohol-induced liver injury.23 To determine whether prevention of hepatic 4-HNE formation and improvement of NF-κB activation were involved in this protective process, an NAC-supplemented alcohol-containing diet was used. Five weeks later, we measured hepatic 4-HNE content, 4-HNE–IκBα adduct formation, and IκBα phosphorylation status. Our results showed that, compared with alcohol-fed mice, NAC supplementation attenuated liver injury (Figure 7A), which was associated with lowered hepatic 4-HNE content (Figure 7B), decreased 4-HNE–IκBα adduct formation (Figure 7C), and improved IκBα phosphorylation status (Figure 7D).
Figure 7.
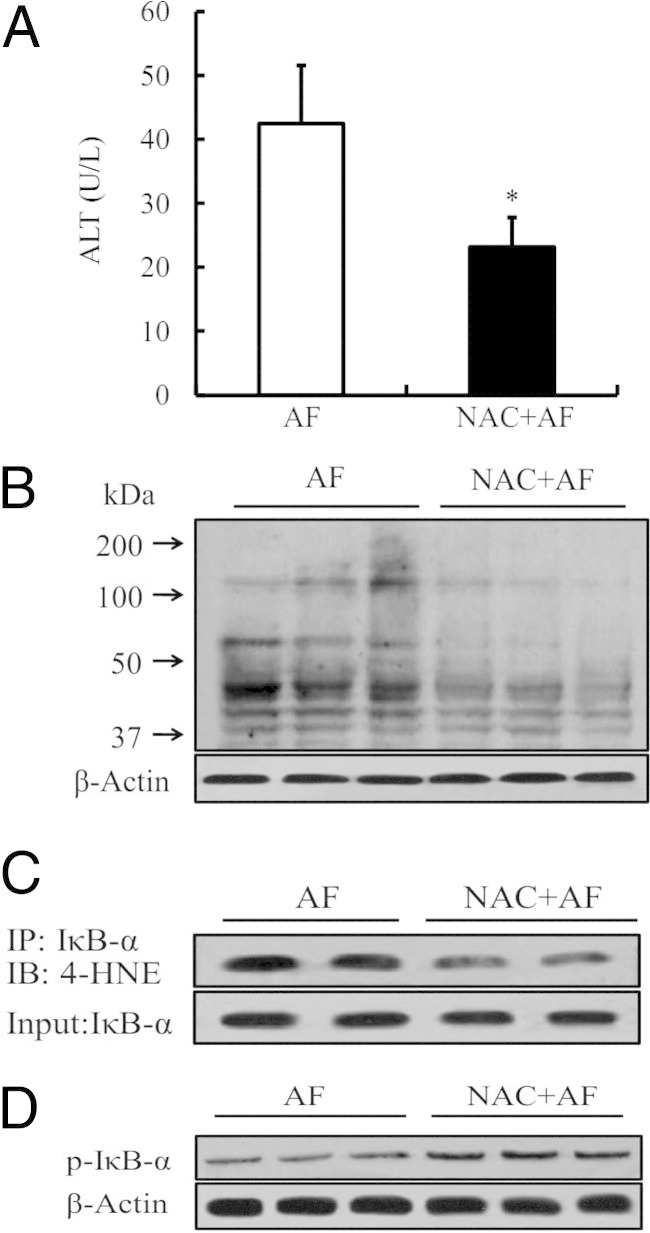
The preventive effect of NAC supplementation on ALD is associated with decreased hepatic 4-HNE formation and improved NF-κB activity. A: NAC supplementation attenuates plasma alanine aminotransferase (ALT) elevation. Data are expressed as the mean ± SD (n = 5 mice per group). *P < 0.05 versus AF. AF, alcohol feeding. B: NAC supplementation lowers hepatic 4-HNE contents. C: NAC supplementation decreases hepatic 4-HNE–IκBα adduct formation. IB, immunoblotting; IP, immunoprecipitation. D: NAC supplementation improves hepatic IκBα phosphorylation status.
Discussion
The goal of this study was to determine whether 4-HNE and TNF synergistically contribute to the pathogenesis of ALD. By using a well-established mouse model of ALD, we found that alcohol-induced liver injury was accompanied by increased hepatic 4-HNE formation and TNF production. By using cultured hepatocytes, we demonstrated that 4-HNE sensitizes hepatocytes to TNF-induced cytotoxicity. Mechanistic investigations revealed a significant increase in 4-HNE–IκBα adduct formation in both 4-HNE–treated hepatocytes and the livers from alcohol-fed mice, which were associated with suppressed IκBα phosphorylation, leading to suppressed NF-κB activation. By preventing intracellular 4-HNE accumulation, bezafibrate, a PPARα agonist, conferred a protective effect against 4-HNE/TNF-induced cytotoxicity in hepatocytes. Finally, we demonstrated that the beneficial effect of NAC supplementation on ALD was associated with decreased hepatic 4-HNE formation and improved NF-κB activation.
The liver is the major site for alcohol metabolism. Long-term alcohol exposure is associated with increased oxidative stress and subsequent lipid peroxidation in the liver. The physiological concentration of 4-HNE in plasma is reported from 0.3 to 0.7 μmol/L.24,25 A striking increase of 4-HNE levels was found in the plasma of patients with ALD, ranging from 2.8-fold in the early stage of liver insult to 8.4-fold in the cirrhotic stage.26 In the liver, local cellular concentrations may reach as high as ≥100 μmol/L in response to oxidative stress.27,28 In the present in vitro study, 20 to 60 μmol/L exogenous 4-HNE was used to mimic in vivo early-stage ALD levels. Although a direct cytotoxic effect of 4-HNE has been extensively documented in a variety of cell types, hepatocytes are relatively resistant to 4-HNE, even at pathophysiological concentrations, likely because of the relatively high capacity of the 4-HNE degradation system.29 Data from our in vitro experiments show that 4-HNE at 20 to 40 μmol/L resulted in a zero to minimal degree of cell death in both human hepatocyte cell lines (HepG2 and Hep3B) and primary mouse hepatocytes, but markedly sensitized these cells to TNF-induced killing, suggesting that 4-HNE–induced sensitization to TNF cytotoxicity represents a clinically relevant process that may account for liver injury in ALD.
Dysregulated TNF production is a pathological factor for ALD. Alcohol causes increased gut permeability, followed by increased exposure of Kupffer cells to lipopolysaccharide, leading to elevated TNF production. NF-κB represents a master switch between life and death of hepatocytes in response to TNF stimulation. Mice deficient in the p65 NF-κB subunit or essential components of the IKK complex die during midgestation as the result of massive hepatocyte apoptosis induced by TNF.30 Moreover, hepatocytes with a mutant IκBα are sensitive to TNF killing.31 Data obtained from the current study suggest that suppression of TNF-induced NF-κB activation by 4-HNE contributes to increased sensitivity of hepatocytes to TNF killing. This conclusion was supported by a series of observations, including the following: i) 4-HNE pretreatment inhibited phosphorylation of IκBα, an obligatory step for NF-κB activation; ii) 4-HNE decreased nuclear p65 content; iii) 4-HNE decreased p65 DNA-binding activity; iv) 4-HNE reduced gene expression of NF-κB–targeted anti-apoptotic molecules, including cellular FLICE-like inhibitory protein, inhibitor of apoptosis proteins-1, and manganese superoxide dismutase; and v) inhibitors for either caspases or JNK activation attenuated 4-HNE/TNF-induced cell death. Although the effects of 4-HNE on NF-κB activation have been reported, previous results were inconsistent and obviously cell type and stimulus dependent. In vascular smooth muscle cells, 4-HNE activated NF-κB,32 whereas in human acute monocytic leukemia cell line monocytes, 4-HNE suppressed NF-κB activation via inhibiting IκBα phosphorylation stimulated by lipopolysaccharide, IL-1β, and phorbol-12-myristate-13-acetate, but not by TNF.33 To our knowledge, the current study is the first to report that 4-HNE suppresses TNF-stimulated NF-κB activation in hepatocytes.
Unchanged IKK phosphorylation indicates that 4-HNE could directly act on IκBα to prevent its phosphorylation by IKK. One of the major mechanisms for 4-HNE to affect protein function is through formation of 4-HNE–protein adducts, a process called protein carbonylation. Indeed, in our study, increased 4-HNE–IκBα adduct formation was detected via immunoprecipitation. The exact mechanism underlying how 4-HNE–IκBα adduct formation prevents its phosphorylation process remains to be fully elucidated. In general, 4-HNE forms stable adducts with proteins via reacting with amino acids, such as cysteine, lysine, or histidine. Therefore, it is unlikely that 4-HNE competes with IKK, which phosphorylates serine, for its phosphorylation sites. One possibility is that 4-HNE may alter IκBα protein conformation via protein adduct formation, thereby preventing it from being accessed and phosphorylated by IKK.
Although the effects of long-term alcohol exposure on hepatic NF-κB activity have been reported, the results were inconsistent and controversial. The discrepancy could derive from differences in model selection, alcohol concentration, feeding duration, and cell types studied. In the present study, we clearly demonstrate that long-term alcohol exposure decreases hepatic NF-κB transactivity. These observations are consistent with several previous studies.34–36 It is conceivable that long-term alcohol exposure may differentially regulate NF-κB activity in hepatocytes (inhibition) and Kupffer cells (activation by lipopolysaccharide). In fact, this notion is supported by a recent study demonstrating that long-term alcohol consumption activated NF-κB in Kupffer cells, whereas NF-κB activation in hepatocytes was inhibited.37
In conclusion, our study demonstrates that long-term alcohol consumption results in a concomitant increase in hepatic 4-HNE and TNF content, and that elevation of intracellular 4-HNE sensitizes hepatocytes to TNF killing. 4-HNE–mediated suppression of NF-κB activation is mechanistically involved in 4-HNE/TNF-induced hepatocyte cell death. Our data provide important new information concerning potential interactions between lipid peroxides and TNF- induced hepatotoxicity and its critical involvement in the pathogenesis of ALD. Moreover, the study also provides indirect evidence explaining the well-established protective role of saturated fat (resistant to peroxidation) in ALD. The inability of NAC protecting against 4-HNE/TNF-induced hepatocyte cell death, along with its preventive effects in animal models of ALD, may partially explain the clinical controversies in terms of preventive versus therapeutic effects of antioxidants for ALD. Together with many other hepatoprotective effects, PPARα activation, via reducing intracellular 4-HNE accumulation, as shown in this study, deserves further investigation as a potential therapy for ALD.
Acknowledgments
We thank Drs. Alan Diamond and Giamila Fantuzzi (University of Illinois at Chicago) for their technical support and great assistance during manuscript preparation.
Footnotes
Supported by grants from the NIH National Institute on Alcohol Abuse and Alcoholism (R01 AA017442 to Z.S.; ZJNSFY12C070007 to X.D.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.08.004.
Supplementary data
4-HNE has no effect on TNF-induced IKKβ activation. HepG2 cells are pretreated with 4-HNE (20 μM) for 2 hours, followed by TNF (40 ng/mL) for 15 minutes. Whole cell lysates are collected and subjected to ELISA for p-IKKβ. Data are expressed as fold of untreated (UT) cells. Bars with different characters differ significantly (P < 0.05). H + T, 4-HNE+TNF.
References
- 1.Wu D., Cederbaum A.I. Oxidative stress and alcoholic liver disease. Semin Liver Dis. 2009;29:141–154. doi: 10.1055/s-0029-1214370. [DOI] [PubMed] [Google Scholar]
- 2.Petersen D.R. Alcohol, iron-associated oxidative stress, and cancer. Alcohol. 2005;35:243–249. doi: 10.1016/j.alcohol.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Rouach H., Fataccioli V., Gentil M., French S.W., Morimoto M., Nordmann R. Effect of chronic ethanol feeding on lipid peroxidation and protein oxidation in relation to liver pathology. Hepatology. 1997;25:351–355. doi: 10.1002/hep.510250216. [DOI] [PubMed] [Google Scholar]
- 4.French S.W., Wong K., Jui L., Albano E., Hagbjork A.L., Ingelman-Sundberg M. Effect of ethanol on cytochrome P450 2E1 (CYP2E1), lipid peroxidation, and serum protein adduct formation in relation to liver pathology pathogenesis. Exp Mol Pathol. 1993;58:61–75. doi: 10.1006/exmp.1993.1006. [DOI] [PubMed] [Google Scholar]
- 5.Polavarapu R., Spitz D.R., Sim J.E., Follansbee M.H., Oberley L.W., Rahemtulla A., Nanji A.A. Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology. 1998;27:1317–1323. doi: 10.1002/hep.510270518. [DOI] [PubMed] [Google Scholar]
- 6.You M., Cao Q., Liang X., Ajmo J.M., Ness G.C. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J Nutr. 2008;138:497–501. doi: 10.1093/jn/138.3.497. [DOI] [PubMed] [Google Scholar]
- 7.Ronis M.J., Korourian S., Zipperman M., Hakkak R., Badger T.M. Dietary saturated fat reduces alcoholic hepatotoxicity in rats by altering fatty acid metabolism and membrane composition. J Nutr. 2004;134:904–912. doi: 10.1093/jn/134.4.904. [DOI] [PubMed] [Google Scholar]
- 8.Nanji A.A., Zakim D., Rahemtulla A., Daly T., Miao L., Zhao S., Khwaja S., Tahan S.R., Dannenberg A.J. Dietary saturated fatty acids down-regulate cyclooxygenase-2 and tumor necrosis factor alfa and reverse fibrosis in alcohol-induced liver disease in the rat. Hepatology. 1997;26:1538–1545. doi: 10.1002/hep.510260622. [DOI] [PubMed] [Google Scholar]
- 9.Sadrzadeh S.M., Nanji A.A., Price P.L. The oral iron chelator, 1, 2-dimethyl-3-hydroxypyrid-4-one reduces hepatic-free iron, lipid peroxidation and fat accumulation in chronically ethanol-fed rats. J Pharmacol Exp Ther. 1994;269:632–636. [PubMed] [Google Scholar]
- 10.Tsukamoto H., Horne W., Kamimura S., Niemelä O., Parkkila S., Ylä-Herttuala S., Brittenham G.M. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith C.M., Schenker S. The role of nutritional therapy in alcoholic liver disease. Alcohol Res Health. 2006;29:296–306. [PMC free article] [PubMed] [Google Scholar]
- 12.Tome S., Lucey M.R. Current management of alcoholic liver disease. Aliment Pharmacol Ther. 2004;19:707–714. doi: 10.1111/j.1365-2036.2004.01881.x. [DOI] [PubMed] [Google Scholar]
- 13.McClain C.J., Cohen D.A. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 14.Khoruts A., Stahnke L., McClain C.J., Logan G., Allen J.I. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267–276. [PubMed] [Google Scholar]
- 15.Felver M.E., Mezey E., McGuire M., Mitchell M.C., Herlong H.F., Veech G.A., Veech R.L. Plasma tumor necrosis factor alpha predicts decreased long-term survival in severe alcoholic hepatitis. Alcohol Clin Exp Res. 1990;14:255–259. doi: 10.1111/j.1530-0277.1990.tb00482.x. [DOI] [PubMed] [Google Scholar]
- 16.Grove J., Daly A.K., Bassendine M.F., Day C.P. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic steatohepatitis. Hepatology. 1997;26:143–146. doi: 10.1002/hep.510260119. [DOI] [PubMed] [Google Scholar]
- 17.Iimuro Y., Gallucci R.M., Luster M.I., Kono H., Thurman R.G. Antibodies to tumor necrosis factor-α attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. 1997;26:1530–1537. doi: 10.1002/hep.510260621. [DOI] [PubMed] [Google Scholar]
- 18.Yin M., Wheeler M.D., Kono H., Bradford B.U., Gallucci R.M., Luser M.I., Thurman R.G. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 19.Pastorino J.G., Hock J.B. Ethanol potentiates tumor necrosis factor alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141–1152. doi: 10.1053/he.2000.7013. [DOI] [PubMed] [Google Scholar]
- 20.Colell A., Garcia-Ruiz C., Miranda M., Ardite E., Mari M., Morales A., Corrales F., Kaplowitz N., Fernández-Checa J.C. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;15:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 21.Song Z., Zhou Z., Uriarte S., Wang L., Kang Y.J., Chen T., Barve S., McClain C.J. S-adenosylhomocysteine sensitizes to TNF-α hepatotoxicity in mice and liver cells: a possible etiological factor in alcoholic liver disease. Hepatology. 2004;40:989–997. doi: 10.1002/hep.20412. [DOI] [PubMed] [Google Scholar]
- 22.Song Z., Zhou Z., Deaciuc I., Chen T., McClain C.J. Inhibition of adiponectin production by homocysteine: a potential mechanism for alcoholic liver disease. Hepatology. 2008;47:867–879. doi: 10.1002/hep.22074. [DOI] [PubMed] [Google Scholar]
- 23.Ozaras R., Tahan V., Aydin S., Uzun H., Kaya S., Senturk H. N-acetylcysteine attenuates alcohol-induced oxidative stess in rats. World J Gastroenterol. 2003;9:791–794. [PubMed] [Google Scholar]
- 24.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 25.Strohmaier H., Hinghofer-Szalkay H., Schaur R.J. Detection of 4-hydroxynonenal (HNE) as a physiological component in human plasma. J Lipid Mediat Cell Signal. 1995;11:51–61. doi: 10.1016/0929-7855(94)00027-a. [DOI] [PubMed] [Google Scholar]
- 26.Aleynik S.I., Leo M.A., Aleynik M.K., Lieber C.S. Increased circulating products of lipid peroxidation in patients with alcoholic liver disease. Alcohol Clin Exp Res. 1998;22:192–196. [PubMed] [Google Scholar]
- 27.Tsukamoto H., Horne W., Kamimura S., Niemela O., Parkkila S., Yla-Herttuala S., Brittenham G.M. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benedetti A., Comporti M., Fulceri R., Esterbauer H. Cytotoxic aldehydes originating from the peroxidation of liver microsomal lipids: identification of 4,5-dihydroxydecenal. Biochim Biophys Acta. 1984;792:172–181. doi: 10.1016/0005-2760(84)90219-4. [DOI] [PubMed] [Google Scholar]
- 29.Sakuma S., Negoro M., Kitamura T., Fujimoto Y. Xanthine oxidase-derived reactive oxygen species mediate 4-oxo-2-nonenal-induced hepatocyte cell death. Toxicol Appl Pharmacol. 2010;249:127–131. doi: 10.1016/j.taap.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka M., Fuentes M.E., Yamaguchi K., Durnin M.H., Dalrymple S.A., Hardy K.L., Goeddel D.V. Embryonic lethality, liver degeneration, and impaired NF-κB activation in IKK-β-deficient mice. Immunity. 1999;10:421–429. doi: 10.1016/s1074-7613(00)80042-4. [DOI] [PubMed] [Google Scholar]
- 31.Chaisson M.L., Brooling J.T., Ladiges W., Tsai S., Fausto N. Hepatocyte-specific inhibition of NF-κB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J Clin Invest. 2002;110:193–202. doi: 10.1172/JCI15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J.Y., Je J.H., Jung K.J., Yu B.P., Chung H.Y. Induction of endothelial iNOS by 4-hydroxyhexenal through NF-κB activation. Free Radic Biol Med. 2004;37:539–548. doi: 10.1016/j.freeradbiomed.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 33.Page S., Fischer C., Baumgartner B., Haas M., Kreusel U., Loidl G., Hayn M., Ziegler-Heitbrock H.W., Neumeier D., Brand K. 4-Hydroxynonenal prevents NF-κB activation and tumor necrosis factor expression by inhibiting IκB phosphorylation and subsequent proteolysis. J Biol Chem. 1999;274:11611–11618. doi: 10.1074/jbc.274.17.11611. [DOI] [PubMed] [Google Scholar]
- 34.Koteish A., Yang S., Lin H., Huang X., Diehl A.M. Chronic ethanol exposure potentiates lipopolysaccharide liver injury despite inhibiting Jun N-terminal kinase and caspase 3 activation. J Biol Chem. 2002;277:13037–13044. doi: 10.1074/jbc.M101632200. [DOI] [PubMed] [Google Scholar]
- 35.Diehl A.M. Effect of ethanol on tumor necrosis factor signaling during liver regeneration. Clin Biochem. 1999;32:571–578. doi: 10.1016/s0009-9120(99)00057-0. [DOI] [PubMed] [Google Scholar]
- 36.Mandrekar P., Catalano D., White B., Szabo G. Moderate alcohol intake in humans attenuates monocyte inflammatory responses: inhibition of nuclear regulatory factor kappa B and induction of interleukin 10. Alcohol Clin Exp Res. 2006;30:135–139. doi: 10.1111/j.1530-0277.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- 37.Mandrekar P., Ambade A., Lim A., Szabo G., Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. 2011;54:2185–2197. doi: 10.1002/hep.24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
4-HNE has no effect on TNF-induced IKKβ activation. HepG2 cells are pretreated with 4-HNE (20 μM) for 2 hours, followed by TNF (40 ng/mL) for 15 minutes. Whole cell lysates are collected and subjected to ELISA for p-IKKβ. Data are expressed as fold of untreated (UT) cells. Bars with different characters differ significantly (P < 0.05). H + T, 4-HNE+TNF.