

Abstract
Although functional asplenia from infarctions may be a major contributor to increased infectious mortality in sickle-cell disease (SCD), this relationship has not been fully defined. We used the transgenic Berkeley SCD mouse to define blood and splenic immunophenotypic differences in this model compared with C57BL/6 and hemizygous controls. In the serum of SCD mice, we found increased IgG2a and suppressed IgM, IgG2b, and IgA levels. Serum IL-6 levels in SCD mice were elevated, whereas IL-1α, CXCL10, and CCL5 levels were decreased. The blood of SCD mice had higher white blood cell counts, with an increased percentage of lymphocytes and decreases in other leukocytes. Immunophenotyping of lymphocytes revealed higher percentages of CD8+ and T-regulatory cells and lower percentages of B cells. SCD mouse spleens exhibited histological disorganization, with reduction of defined lymphoid follicles and expansion of red pulp, a greater than fourfold increase in splenic mononuclear cells, marked expansion of the nucleated red blood cell fraction, and B-cell and CD8+ T-cell lymphopenia. Within the splenic B-cell population, there was a significant decrease in B-1a B cells, with a corresponding decrease in IgA secreting plasma cells in the gut. Confocal microscopy of spleens demonstrated complete disruption of the normal lymphofollicular structure in the white pulp of SCD mice without distinct B, T, and marginal zones. Our findings suggest that altered SCD splenic morphological characteristics result in an impaired systemic immune response.
Millions worldwide live with sickle-cell disease (SCD), the most common inherited blood disorder that is caused by a single point mutation in the β-globin gene, resulting in the production of abnormal red blood cell (RBC) hemoglobin. In the deoxygenated state, hemoglobin polymerizes to form relatively stiff filaments, forcing RBCs to assume an irregular sickled shape. These sickled RBCs are thought to occlude small blood vessels, resulting in what is termed a vaso-occlusive crisis. Such episodes are recurrent, spontaneous complications of SCD in which microvascular infarction leads to extreme pain and widespread organ dysfunction. These problems often lead to several other complications of the disease, such as anemia and stroke; consequently, most patients with SCD live to between the ages of 40 and 50 years.1 Surprisingly, the most common cause of death in those with SCD is infection, not stroke or organ failure. The life expectancy of patients with SCD has increased in recent decades because of early interventions, such as universal newborn screening for the disease, the use of prophylactic penicillin, and the administration of vaccines to prevent infections.
One critical immunological organ that is affected early in individuals with SCD is the spleen. It is a complex, but highly organized, structure composed of white pulp, red pulp, and the marginal zone. The red pulp is critical for blood filtration, iron recycling from erythrophagocytosis, extramedullary hematopoiesis, and the production of antibodies from resident plasmablasts that migrate out of the splenic follicles after antigen-specific differentiation. The white pulp is organized as lymphoid sheaths with B- and T-cell compartments. Clonal expansion of activated B cells occurs in the B-cell follicles, leading to isotype switching and somatic hypermutation, whereas T cells form T-cell zones, where they interact with dendritic cells and passing B cells. The marginal zone serves as a transitory boundary between the red and white pulp and contains unique sets of macrophages and B cells that appear to be important for mounting responses against T-cell–independent antigens, such as the capsular polysaccharide of invasive bacterial species.2 Intrasplenic shunting results in reduced blood flow through the spleen in pediatric patients with SCD, rendering them functionally asplenic. Repeated infarction of the spleen throughout childhood causes severe fibrosis and calcification and eventually results in autosplenectomy and, presumably, impaired immunity. Loss of the spleen has a dramatic negative effect on natural IgM-producing B-1a B-cell numbers on splenectomy in mice3 and may also have a similar effect in homologous cells in humans.4 B-1a B cells are imperative for protection against invasive encapsulated bacteria, such as Streptococcus pneumoniae,5 and they are essential for maintaining high levels of secretory IgA in the gut.6 Much of the morbidity and mortality associated with SCD can be attributed to diseases caused by encapsulated bacteria, such as S. pneumoniae,7 making the study of the role of the SCD spleen in infectious disease pathogenesis imperative to the improvement of patient health.
SCD is a human disease that is not found in other species, making the generation of an animal model to study the disease in detail a difficult endeavor. However, several different transgenic mice have been developed, including a mouse with fully humanized hemoglobin.8 The murine globin genes have been knocked out of these mice, whereas transgenes express human hemoglobin. These mice recapitulate several aspects of disease observed in humans, including sickled erythrocytes and histological damage to multiple organ systems. These mice also demonstrate a pro-inflammatory state when administered a low-dose lipopolysaccharide challenge that produces an exaggerated inflammatory response, with elevated levels of tumor necrosis factor-α, IL-1β, and soluble vascular cell adhesion molecule-1 in the serum and bronchoalveolar lavage fluid.9 Unfortunately, there is a paucity of data about the immune system of these SCD mice at baseline, which is important because this disease in humans has many secondary, and even life-threatening, infectious complications.10
We hypothesized that altered splenic histopathological architecture from repeated vaso-occlusive infarctions leads to impaired systemic immunity. To our knowledge, detailed immunophenotyping and imaging of SCD mouse spleens have not been published. Therefore, the purpose of this study was to assess the basic immune status of the SCD transgenic mouse. In doing so, we have identified blood, gut, and splenic immunophenotypic differences that might lead to further mechanistic explanations for how the spleen is involved in increased morbidity and mortality from infection in SCD.
Materials and Methods
Mice
Three groups of female mice were used: C57BL/6, transgenic sickle-cell mice, and hemizygous sickle-cell littermates. All mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and were aged 8 to 12 weeks and weighed 15 to 25 g at sacrifice. Berkeley sickle-cell transgenic mice [Tg(Hu-miniLCRα1GγAγδβS) Hba-/- Hbb-/-] expressing human HBA and HBBS and no longer expressing mouse Hba and Hbb were used as a murine model of SCD. The stock background of this strain is a mixture of FVB/N, 129, DBA/2, black Swiss, and >50% C57BL/6 genomes. It was backcrossed to C57BL/6 one generation after importation to The Jackson Laboratory. Littermate controls of the Berkeley transgenic SCD mouse (generated on the same mixed background of strains) that express no mouse Hba, but do express one copy of mouse Hbb, human HBA, and HBBS producing a hemizygous, sickle cell trait–like genotype were used as a second control arm.
All mice were housed conventionally in plastic cages with corncob bedding. The mouse room was maintained at 22°C to 24οC with a daily light-dark cycle (light from 6 am to 6 pm). Chow and water were supplied ad libitum. The protocols for mice used were approved by the Animal Care Committee at the University of Connecticut Health Center, Farmington.
Harvesting of Tissues
On humane euthanization of mice with an i.p. ketamine-xylazine overdose, whole blood was immediately collected via cardiac puncture and divided as follows: i) into heparinized tubes for peripheral blood mononuclear cell isolation or automated complete blood cell counts and leukocyte differential counts (Charles River Laboratory, Wilmington, MA); ii) into nonheparinized tubes for serum purification; and iii) onto glass slides for peripheral blood smears. Blood in heparinized tubes that was used for peripheral blood mononuclear cell analysis was treated with Tris ammonium chloride solution (nine parts 0.83% w/v NH4Cl and one part 2.57% w/v Tris, pH 7.0) lysis at 37°C before resuspending in HBSS and before counting via hemocytometer. The blood in nonheparinized tubes was allowed to clot at room temperature. Samples were then spun at 800 × g in an Eppendorf centrifuge at room temperature. Serum was pipetted off and frozen for later use at −80°C. Peripheral blood smears were made by smearing a small aliquot of venous blood in a single layer onto a clean glass slide, allowing cells to air dry and fixing with methanol for 5 minutes before May-Grunwald staining. Spleens were harvested and placed in HBSS on ice and then mashed with a rubber tip from a 5-mL syringe through a cell strainer (Falcon 352340; BD Biosciences, Franklin Lakes, NJ) into a 50-mL tube. After rinsing with 10 mL of HBSS, mashed spleens were spun for 5 minutes at 200 × g in a Beckman TJ-6 (Beckman Coulter, Brea, CA). The supernatant was decanted, and the pellet was resuspended in TAC to lyse splenic RBCs. The cellular suspension was then put through a screen, and the tube was filled with 25 mL of HBSS and then spun for 5 minutes at 200 × g. Supernatant was then decanted. TAC lysis of splenic cells was repeated, if necessary. HBSS, 5 to 10 mL, was added, and the specimen was put through the screen again. Total nucleated cell counts from blood and spleens were obtained using a hemocytometer with nigrosin dye exclusion as a measure of viability. Aliquots containing 106 cells were resuspended in fluorescence-activated cell sorting buffer. In addition, an aliquot of this nucleated cell suspension was taken to determine cell differentials. For histological analysis, mouse spleens were harvested, fixed in 10% buffered formalin, and processed using standard techniques. For confocal microscopy, intact spleens were harvested and immersed in optimal cutting temperature solution and frozen at −80°C until they were divided into sections. For IgA ELISpot analysis, the small intestine was extracted and fat deposits and fecal matter were removed. Peyer's patches were plucked and placed in HBSS on ice and then mashed between two microscope slides to harvest cells. Peyer's patches cells were washed and counted, as previously described, and placed in RPMI 1640 medium with penicillin, streptomycin, l-glutamine, and 5% fetal calf serum (FCS).
Antibodies
The following monoclonal antibodies were used in this study for flow cytometry: CD3-Cy5.5 (145-2C11; BD Biosciences), CD4–activated protein C (APC)–Cy7 (RM4-5; BioLegend, San Diego, CA), CD8–fluorescein isothiocyanate (53-6.72; BD Biosciences), CD19-PerCP-Cy5.5 (1D3; BD Biosciences), CD19-phosphatidylethanolamine (PE) (eBio1D3; eBioscience, San Diego, CA), CD25-PE (PC61.5; eBioscience), B220-Pacific Blue (RA3-6B2; BD Biosciences), B220-APC-eFluor780 (RA3-6B2; eBioscience), Foxp3-APC (FJR-16s; eBioscience), CD21-PE (4E3; eBioscience), CD23–fluorescein isothiocyanate (B3B4; eBioscience), CD23-PE-Cy7 (B3B4; eBioscience), CD43–fluorescein isothiocyanate (S7; BD Biosciences), CD5-APC (53-7.3; eBioscience), CD11b–Alexa Fluor 700 (M1/70; BD Biosciences), and IgM-PerCP-Cy5.5 (R6-60.2; BD Biosciences). Corresponding isotype controls were also obtained and used. In addition, the following monoclonal antibodies were used for confocal microscopy: CD8-Cy5 (53-6.7; eBioscience), B220–Alexa Fluor 488 (RA3-6B2; Invitrogen, Carlsbad, CA), and MOMA-biotinylated-streptavidin 546 (MOMA1; BMA Biomedical, Augst, Switzerland).
Fluorescence-Activated Cell Sorting Analysis
Cell pellets containing lymphocytes from tissue preparations were resuspended in PBS solution (containing 0.2% bovine serum albumin and 0.1% NaN3) at a concentration of 1 × 105 to 1 × 106 white blood cells/mL, and 100 μL of the cells were diluted 1:1 with 100 μL of the appropriate monoclonal antibody and incubated at 4°C for 30 minutes. Foxp3+ T-regulatory (Treg) cells were identified by staining lymphocytes with anti-CD3, anti-CD4, and anti-CD25, followed by permeabilization using fixation/permeabilization buffer, per the manufacturer's instructions. Cells were then stained using an anti-Foxp3 antibody. After staining, cells were washed twice with PBS solution (0.2% bovine serum albumin and 0.1% NaN3), and relative fluorescence intensities were determined using an LSR-II (Becton-Dickinson, San Diego, CA) and analyzed with FlowJo flow cytometry analysis software (FlowJo, Ashland, OR). Fluorescence minus one controls were used to assist with gating.
IgA ELISpot
Anti-mouse IgA ELISpot kits were obtained from Mabtech Inc. (Cincinnati, OH) and used per the manufacturer's instructions. Briefly, ELISpot plates were wet with 70% ethanol for 2 minutes and washed three times with sterile water. Plates were then coated with 100 μL per well of 10 μg/mL anti-mouse IgA antibody and incubated overnight at room temperature. Plates were then washed three times with PBS and blocked for 30 minutes with RPMI 1640 medium and 5% FCS at room temperature. Plates were then washed with PBS, and 100,000, 20,000, 5000, and 1000 Peyer's patches cells from each mouse were plated in duplicate and incubated overnight at 37°C and 5% CO2. Plates were then washed with PBS and incubated for 2 hours with 100 μL of 1 μg/mL of anti-IgA biotin antibody with 0.5% FCS. Plates were washed again with PBS and incubated for 1 hour with 100 μL of a 1:1000 dilution of streptavidin–horseradish peroxidase with 0.5% FCS. Plates were then washed with PBS and allowed to develop for approximately 20 minutes with 100 μL of TMB substrate. The reaction was stopped with DI water, and spots were counted using a Fisher Scientific Micromaster light microscope (Hampton, NY).
Light and Confocal Microscopy
Fixed spleen tissue sections were stained with H&E and examined using light microscopy. All specimens were evaluated with a microscope-mounted Nikon Eclipse 400TM camera (Tokyo, Japan). Digital images were generated using Spot RT SliderTM Software (Spot Imaging, Sterling Heights, MI) and evaluated in Microsoft Photo EditorTM (Microsoft, Redmond, WA).
Frozen spleen sections (20 μm thick) were washed in PBS and stained overnight at 4°C with anti-CD8, anti-B220, and anti-MOMA antibodies in 2% normal goat serum and 2% FCS/PBS solution. Tissues were then fixed in 2% paraformaldehyde for 2 hours at 4°C, washed, mounted, and analyzed. Confocal images were collected using a Zeiss (Oberkochen, Germany) LSM-510-Meta confocal microscope mounted on an Axiovert 100M with automated xyz control equipped with an argon laser (emission, 458, 488, and 514 nm) and two HeNe lasers (emission wavelengths, 543 and 633 nm). Image analysis was performed using Imaris Suite (Bitplane, Inc., South Windsor, CT).
Serum Immunoglobulin and Cytokine/Chemokine Measurements
Frozen serum samples obtained from mice were thawed and used for analysis. For assessment of cytokines and chemokines, samples were processed using a Milliplex Mouse Cytokine/Chemokine 22-plex kit (catalog MPXMCYTO70KPMX22; Millipore, Billerica, MA) following the manufacturer's instructions. Assay sensitivities (minimum detectable concentrations) were as follows: granulocyte colony-stimulating factor (CSF; 0.9 pg/mL), GM-CSF (5.6 pg/mL), IL-1a (5.1 pg/mL), IL-1b (2.0 pg/mL), IL-2 (0.8 pg/mL), IL-4 (0.4 pg/mL), IL-5 (0.7 pg/mL), IL-6 (1.8 pg/mL), IL-7 (0.9 pg/mL), CXCL1 (1.4 pg/mL), IL-9 (6.0 pg/mL), IL-10 (3.3 pg/mL), IL-12 (p40) (4.9 pg/mL), IL-12 (p70) (4.1 pg/mL), IL-13 (6.3 pg/mL), IL-15 (6.5 pg/mL), IL-17 (0.5 pg/mL), CXCL10 (0.6 pg/mL), CCL2 (5.3 pg/mL), CCL3 (8.7 pg/mL), CCL5 (2.5 pg/mL), interferon-γ (0.9 pg/mL), and tumor necrosis factor-α (1.0 pg/mL). Immunoglobulin concentrations were determined using Milliplex Mouse Immunoglobulin Isotype Kit Panels (catalog MGAM-300; Millipore) following the manufacturer's instructions. Assay sensitivities (minimum detectable concentrations) were as follows: IgM (0.3 ng/mL), IgG1 (0.3 ng/mL), IgG3 (0.4 ng/mL), IgG2a (0.4 ng/mL), IgG2b (0.4 ng/mL), and IgA (0.7 ng/mL). Samples were analyzed using a Bio-Rad Bioplex-200 system (Hercules, CA), with recombinant murine cytokines as standards, and Luminex bead array software (Austin, TX) was used to analyze the data.
Statistics
Statistical comparisons between groups were made with one-way analysis of variance, and multiple pairwise comparisons were conducted using the Newman-Keuls post hoc test using GraphPad Prism version 5 (GraphPad Software, San Diego, CA). All data were expressed as mean ± SEM. Differences were considered significant if P ≤ 0.05.
Results
Peripheral Blood and Serum Analyses
An examination of the peripheral blood of C57BL/6, hemizygous, and SCD mice showed normal erythrocyte morphological characteristics in the wild-type mice. The hemizygous mice demonstrated increased target-shaped RBCs and no evidence of sickled erythrocytes. The SCD peripheral blood showed marked anisopoikilocytosis, including characteristic sickled cells. As shown in Figure 1, there was a significant increase in the concentration of white blood cells (WBCs; Figure 1A, P < 0.001) and a decrease in the percentage of eosinophils (Figure 1E, P < 0.01) and basophils (Figure 1F: P < 0.001 compared with C57BL/6 and P < 0.05 compared with hemizygous mice) in the SCD mice when compared with both hemizygous and C57BL/6 mice. Lymphocytes were increased in the SCD mice relative to the other groups. Hemizygous mice also had a higher percentage of lymphocytes when compared with C57BL/6 mice (Figure 1C, P < 0.01 for comparisons with SCD mice, and P < 0.05 for comparison between C57BL/6 and hemizygous mice). The proportion of monocytes (Figure 1B, P < 0.05) was reduced in both hemizygous and SCD mice compared with C57BL/6 mice, and there was also a significant reduction in the proportion of neutrophils when C57BL/6 and SCD mice were compared (Figure 1D, P < 0.05). No difference in platelet counts was observed, but reduced hemoglobin and hematocrit were found in the SCD mice when compared with hemizygous and C57BL/6 mice (Figure 1, H and I, P < 0.001 for hemoglobin and hematocrit), as previously described.8,11 Hemizygous mice were also anemic when compared with C57BL/6 mice, but less so than SCD mice (P < 0.01 for hemoglobin and P < 0.05 for hematocrit).
Figure 1.

SCD mice have increased peripheral blood white blood cells (WBCs) and an altered distribution of leukocytes, as well as anemic hematological parameters. A–I: Automated complete blood cell (CBC) and differential blood cell counts from C57BL/6 (n = 5), hemizygous (Hemi; n = 7), and SCD (n = 3) mice are conducted by Charles River Laboratory. Bars represent mean ± SEM levels. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
The serum immunoglobulin isotype analysis of SCD mice demonstrated suppressed IgM (135,472 ± 19,985 ng/mL) (Figure 2A, P < 0.01), IgA (6556 ± 815 ng/mL) (Figure 2B, P < 0.001), and IgG2b (165,667 ± 49,217 ng/mL) (Figure 2D, P < 0.01) levels when compared with C57BL/6 mice (IgM, 358,295 ± 60,412 ng/mL; IgA, 19,568 ± 2437 ng/mL; and IgG2b, 410,023 ± 67,005 ng/mL). However, serum IgG2a levels were significantly higher in SCD mice (482,597 ± 151,900 ng/mL) compared with C57BL/6 mice (18,034 ± 3296 ng/mL) (Figure 2C, P < 0.01). Overall, hemizygous mice demonstrated serum immunoglobulin levels (IgM, 140,386 ± 13,558 ng/mL; IgA, 8432 ± 868 ng/mL; and IgG2b, 126,693 ± 14,641 ng/mL) similar to those of SCD mice, with the exception of IgG2a (356,602 ± 81,491 ng/mL), which had a significantly lower level in hemizygous compared with SCD mice (P < 0.05), raising the possibility of an effect from mouse strain background. No significant differences in serum IgG1 and IgG3 levels were observed (data not shown).
Figure 2.
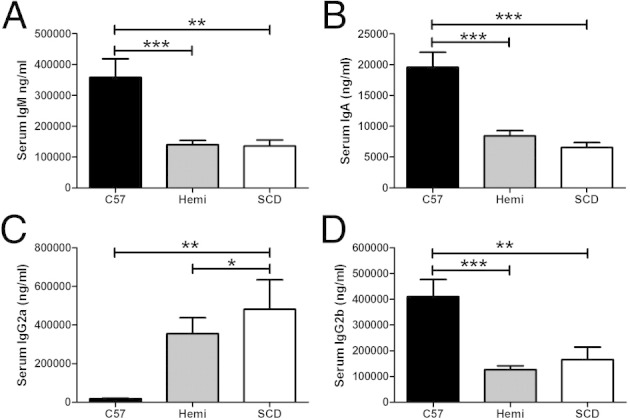
SCD and hemizygous mice have decreased serum IgG2b, IgM, and IgA levels, but increased IgG2a production. A–D: Serum immunoglobulins from C57BL/6 (n = 11), hemizygous (Hemi; n = 11), and SCD (n = 9) mice are analyzed via Luminex bead assay. Bars represent mean ± SEM levels. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
Of 17 serum cytokines that were measured, IL-1α (Figure 3A) and IL-6 (Figure 3B) were significantly different between SCD and control mice. The mean serum IL-1α levels were higher in C57BL/6 mice (894 ± 303 pg/mL) compared with SCD mice (86 ± 19 pg/mL) (P < 0.05). There was no significant difference in serum IL-1α level between hemizygous and C57BL/6 or SCD mice. The mean serum IL-6 level was significantly higher in SCD mice (15 ± 4 pg/mL) compared with both C57BL/6 mice (3 ± 0.5 pg/mL) (P < 0.01) and hemizygous controls (4 ± 0.6 pg/mL) (P = 0.001). There was no difference in serum cytokine expression of granulocyte CSF, GM-CSF, interferon-γ, IL-1b, IL-2, IL-4, IL-5, IL-7, IL-9, IL-10, IL-12, IL-13, IL-15, IL-17, and tumor necrosis factor-α (data not shown).
Figure 3.
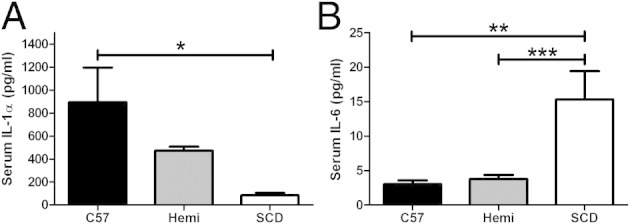
SCD mice have elevated serum IL-6 and decreased IL-1a cytokine expression. A and B: C57BL/6 (n = 11), hemizygous (Hemi; n = 11), and SCD (n = 9) mice undergo serum cytokine analysis via Luminex bead assay. Bars represent mean ± SEM levels. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
We measured five serum chemokines in SCD and control mice. Two chemokines (CXCL10 and CCL5) were significantly lower in the SCD mice (Figure 4). The mean serum CXCL10 levels (Figure 4A) were lower in SCD mice (55 ± 9 pg/mL) than in C57BL/6 mice (119 ± 15 pg/mL) (P < 0.001). There was no difference in serum CXCL10 levels between hemizygous and C57BL/6 or SCD mice. SCD mice had significantly lower serum CCL5 levels (20 ± 3 pg/mL) than C57BL/6 and hemizygous mice (36 ± 5 and 38 ± 4, respectively) (P < 0.05). There were no differences in serum chemokine levels of CXCL1, CCL2, or CCL3 (data not shown).
Figure 4.
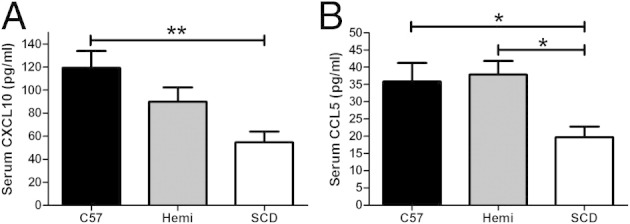
SCD mice have decreased CXCL10 and CCL5 chemokine expression. A and B: C57BL/6 (n = 11), hemizygous (Hemi; n = 11), and SCD (n = 9) mice undergo serum chemokine analysis via Luminex bead assay. Bars represent mean ± SEM levels. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. *P < 0.05, **P < 0.01.
To assess whether variations in lymphocyte subpopulations could account for the observed differences in immunoglobulin and cytokine expression patterns in SCD mice, lymphocyte flow cytometric analysis was undertaken on peripheral blood. Overall, SCD mice had a higher percentage of CD3+ (T) cells in their blood compared with both C57BL/6 and hemizygous mice (Figure 5A: 30%, 16%, and 17%, respectively; P < 0.001). More specifically, SCD mice demonstrated higher percentages of peripheral blood T cells (and a decrease in the percentage of peripheral blood B cells; Figure 5A, P < 0.001). There was no difference in blood percentage CD3+CD4+ (CD4+) cells among the three groups (P = 0.172). However, SCD mice demonstrated a significantly higher percentage of circulating CD4+CD25+FoxP3+ Treg cells than both C57BL/6 (P < 0.01) and hemizygous (P < 0.05) control mice (Figure 5C: 5%, 3%, and 3%, respectively). The percentage of CD3+CD8+ (CD8+) cells in the blood of SCD mice was higher (46%) than in the blood of hemizygous (39%; P < 0.01) and C57BL/6 (35%; P < 0.001) mice. SCD and hemizygous mice had a lower percentage of B220+CD19+ cells (B cells) than SCD mice (9%, 9%, and 27%, respectively; P < 0.001).
Figure 5.
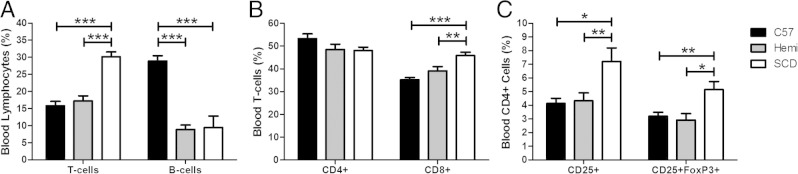
SCD mice have decreased circulating B cells and increased circulating CD8+ T cells and Tregs in peripheral blood. A–C: Lymphocyte flow cytometric analysis on peripheral blood of C57BL/6 (n = 6), hemizygous (n = 6), and SCD (n = 5) mice is performed. Single-cell suspensions are isolated and stained with anti-CD3, anti-CD4, anti-CD25, anti-FoxP3, anti-CD8, anti-CD19, and anti-B220 antibodies. T cells are CD3+ lymphocytes. B cells are B220+CD19+ lymphocytes. CD4+ T cells are CD3+CD4+ lymphocytes. CD8+ T cells are CD3+CD8+ lymphocytes. Tregs are CD4+CD25+FoxP3+ lymphocytes. Bars represent mean ± SEM percentages. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
Spleen
Gross and Histological Pathological Characteristics
On gross pathological analysis, SCD mice had greater than sixfold larger spleens (Figure 6, A and B; P < 0.001) compared with C57BL/6 and hemizygous mice. Murine C57BL/6 spleens, similar to human spleens, have highly organized areas of red and white pulp. Histologically, C57BL/6 mouse spleens had normal splenic organization (Figure 6C), with clearly identifiable regions of red and white pulp. Spleens from hemizygous mice also had normal histological characteristics (data not shown). However, SCD mouse spleens exhibited marked histological disorganization (Figure 6D). Most notable were the reduction of clearly defined splenic white pulp (including few lymphoid follicles) and expanded red pulp.
Figure 6.

The SCD mouse spleen is grossly enlarged and has distorted histological organization. SCD spleens are six times larger by wet weight than hemizygous and C57BL/6 spleens (A), which is evident on gross examination (B). Histologically, on standard H&E staining at an original magnification of ×4, SCD splenic tissue has a markedly distorted organization (D) compared with C57BL/6 mice (C), which have organized areas of white pulp and red pulp. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. ***P < 0.001.
Cellular Analysis
To elucidate the effect of SCD splenic histopathological distortion at the cellular level, we analyzed the mononuclear cell content of the SCD spleens compared with wild-type. Not surprisingly, total splenic mononuclear cells were significantly increased in SCD spleens by 4.5-fold (Figure 7A, P < 0.001), approximately the same order of magnitude of difference in weight. However, the distribution of mononuclear cells accounting for the increase in total count was dissimilar. Nucleated RBCs accounted for most of the total cellular increase in the SCD spleens (40% in SCD mouse spleens and <2% in control mouse spleens; P < 0.001) (Figure 7B). This marked increase of nucleated RBCs likely accounted for the relative lymphopenia in SCD spleens (37% in SCD spleens and approximately 90% in control mouse spleens; P < 0.001) (Figure 7B). Both T- and B-cell subsets of SCD spleens were statistically decreased compared with control groups (P < 0.001) (Figure 7C). Reductions in both T and B cells were also observed in hemizygous mice when compared with C57BL/6 mice (P < 0.01 and P < 0.001, respectively), albeit less so than was observed with the SCD mice. Although the percentage of CD4+ T cells was no different between the spleens of SCD mice and the two control groups (P = 0.3), the percentage of CD8+ T cells was statistically decreased in both SCD and hemizygous mice when compared with C57BL/6 mice (Figure 7D, P < 0.01).
Figure 7.

SCD mice have 4.5-fold more total mononuclear cells, a marked increase in nucleated RBC content, and significantly altered lymphocyte distributions. A–F: Groups of C57BL/6 (n = 5), hemizygous (Hemi; n = 6), and SCD (n = 4) mice are euthanized and their spleens are removed. Single-cell suspensions are isolated from the spleens and stained with May-Grünwald stain. Total numbers of mononuclear cells in the spleens are determined by a hemocytometer. The individual proportion of mononuclear cells (both leukocytes and nucleated RBCs) is determined by a manual differential. Lymphocytes are identified by flow cytometry. Bars represent mean ± SEM percentages. Significant differences are determined by one-way analysis of variance with multiple pairwise comparisons between groups using the Newman-Keuls post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
Most striking was the 80% reduction of splenic B-1a B cells in SCD mice compared with both control groups (Figure 7E, P < 0.01). This change was associated with a marked reduction of IgA-secreting plasma cells in the Peyer's patches of the small intestine of the SCD mice when compared with C57BL/6 mice (Figure 7F, P < 0.05).
Confocal Microscopy
To determine immune cellular localization and compartmental distribution in the splenic tissue, confocal microscopy was used. Sections of splenic tissue from C57BL/6 (data not shown), hemizygous, and SCD mice were stained to isolate the B220+ B-cell, CD8+ T-cell, and MOMA+ marginal zones (Figure 8). Normal follicular architecture of the white pulp, consisting of centralized T cells, surrounding B cells, and marginal zone macrophages, was observed in the C57BL/6 and hemizygous mice. However, complete disruption of the normal lymphofollicular structure was found in the white pulp of SCD mice, and lymphoid aggregates that could be found were sparse and much smaller than those of hemizygous or C57BL/6 mice.
Figure 8.

The cellular architecture of SCD splenic follicles is abnormal. Spleen sections are fixed and stained with B220 (Alexa Fluor 488) for B cells, CD8 (Cy 5) for CD8+ T cells, and MOMA (Alexa Fluor 546) for metallophilic macrophages. MZ, marginal zone where the MOMA-positive macrophages should be resident. B-cell follicles (B), T-cell zones (T), and MOMA-positive macrophages are not as distinct in the SCD mice compared with the hemizygous mice, as shown in B and D. Original magnification: ×10 (A and C); ×20 (B and D). The follicles are sparser in the SCD mice compared with the hemizygous controls, as seen in A and C.
Discussion
We have identified altered splenic morphological characteristics that may account for our observed aberrant serum immunoglobulin, cytokine, and chemokine levels. Taken together, these local splenic anatomical distortions may account for impaired pathogen recognition and processing, thereby leading to high rates of infectious complications in this disease. Humans living with SCD are highly susceptible to severe infections.12 Recent evidence suggests that such events may be related to dysregulated immunity associated with suppression of several immune parameters (eg, antibody responses) and increased baseline inflammation. The reasons for immune dysregulation in these patients are not clear, but the dogma has been that repeated infarction with subsequent atrophy and dysfunction of the spleen is the key mediator of this phenomenon. Indeed, patients with SCD often have splenic dysfunction beginning in the early months of life, which can contribute to impaired immunity to pathogenic insults, such as invasion by encapsulated bacteria.13 The SCD mouse spleens described herein were grossly enlarged and exhibited severely disrupted microarchitecture when compared with controls. Histological and confocal microscopy images of the spleens of SCD mice showed a dramatically attenuated white pulp in which no discernable T-cell zone could be identified. Not surprisingly then, reduced percentages of both B and T cells were observed in this tissue, as determined by flow cytometry. Such changes in lymphocyte proportions and possibly function could have a profound effect on adaptive immune responses. Fortunately, most childhood vaccinations are administered before major changes in the spleen occur, allowing for antigen-specific memory cells to be generated.14
Interestingly, natural IgM-producing B-1a B cells, which have been essential for combating some of the pathogens of high significance for SCD, do not appear to be maintained in the spleens of SCD mice. Given that these cells are innate-like, vaccination at an early age may not help abrogate the loss of this important cell type. Furthermore, splenic B-1a B cells appear to have a direct link to natural IgA-secreting plasma cells in the gut, which are imperative for protection against many pathogens of the gastrointestinal tract. Indeed, individuals with SCD appear to be more prone to invasive infection with Salmonella species,15 opening the possibility that the increased susceptibility to infection may, in fact, be associated with changes in the spleen and the subsequent loss of B-1a and IgA-secreting plasma B cells.
In addition to the disruption in lymphocyte populations in the white pulp of the spleen, the marginal zone was not distinct in the SCD mice by confocal microscopy. Proper function of these cells in the marginal zone is imperative for defense against encapsulated bacteria.16,17 The lack of an organized splenic marginal zone around lymphoid follicles may explain why patients with SCD are highly susceptible to such infections. Not only will such alterations in these cells likely affect antigen presentation and immunity associated with the integrity of the marginal zone, but loss of macrophages from this region can also negatively affect the removal of atypical erythrocytes from the circulation,18 possibly resulting in enlargement or sequestration of the spleen. Taken together, the aberrant splenic structure observed in SCD mice appears to closely mimic changes found in human patients and likely contributes to immune dysregulation observed in this disease.
In addition to localized immune dysregulation in the SCD spleen, patients also display aberrant immune phenotypes in peripheral blood,19,20 and this trend held true for the SCD mice used in this study. Immunophenotyping of lymphocytes in the SCD mice indicated that the percentage of B cells was drastically reduced, with a concomitant increase in T cells (driven by a predominant increase in T-regulatory and CD8+ cells). This shift is in contrast to several reports of lymphocyte immunophenotypic profiling in patients with SCD, showing an increase in the number of B cells with a reduction in T cells.21,22 The increase in B-cell number in patients with SCD does not result in a corresponding increase in antibodies; in fact, these patients exhibit reduced antibody and lymphoproliferative responses to stimuli. There was a significant reduction in serum IgM, IgA, and IgG2b antibodies in SCD mice, which may be explained by the observed reduction in circulating B cells and the disorganization of the splenic white pulp. The white pulp is critical for clonal expansion, isotype switching, and somatic hypermutation of activated B cells. However, serum IgG2a levels were much higher in SCD mice, which is in stark contrast to the other antibodies. The reasons for this increase and the mechanism by which it happens are not evident from this work; therefore, it is imperative to elucidate the role IgG2a in SCD to ensure that these antibodies are not pathological. Regardless of the differences in lymphocyte profiles between patients with SCD and SCD mice, both exhibit striking changes that likely affect adaptive immunity, which could have profound negative implications for vaccination in this disease.
Lymphocyte function is heavily dependent on the cytokine milieu present in the circulation and tissues, and slight changes can have profound effects on their phenotype and proliferation. Measurements of serum cytokines demonstrated reduced concentrations of CXCL10 and IL-1α in SCD mice when compared with C57BL/6 mice. Several functions can be ascribed to these cytokines, but both can have an effect on the function of T cells, indicating that SCD mice may have altered responses to presented antigens.23 Perhaps more important, SCD mice had a reduced level of CCL5 when compared with both hemizygous and C57BL/6 mice. This chemokine is important for the chemotaxis of several leukocytes, including T cells. CCL5 also appears to regulate CD8+ antiviral responses,24 indicating that these mice may have a deficiency in cell-mediated immunity. Whether there are reductions in these three cytokines in patients with SCD has not been determined, but if such changes are conserved in the human model, then this may explain why patients with SCD are particularly prone to severe viral infections, such as parvovirus B19 and pandemic H1N1 influenza A.25,26 In addition to the reduced CXCL10, IL-1α, and CCL5 concentrations in SCD mice, these animals also have increased levels of IL-6 when compared with hemizygous littermates and controls. This cytokine can be pro- or anti-inflammatory, depending on several factors, and has been found at increased levels in splenectomized patients with thalassemia.27 Our results are in stark contrast to those reported by Holtzclaw et al,9 which indicated that these mice do not have an elevated IL-6 level at baseline measurements. The differences in our results may be the result of the different methods used to measure IL-6 (they used enzyme-linked immunosorbent assay, whereas we used Luminex). Furthermore, there is debate in the literature about the levels of serum IL-6 in patients with SCD, because some groups have observed no difference when compared with controls,19,28 whereas others have determined that there is an increase in patients with SCD.29-31 These discrepancies may be attributed to the different ages of the patients tested (and the subsequent different stages of splenic atrophy), but our results are in line with the group that observed increased serum IL-6 concentrations in SCD. Taken together, SCD mice appear to have a cytokine skew that may contribute to immune dysregulation and may help explain the weak immunity observed clinically in patients with SCD.
Herein, we have reported several immune parameters that are aberrant in SCD mice (and, in some cases, in their hemizygous littermates). Some of these changes have been observed in their human counterparts, some are different, and others have not been studied in humans at all. Even when differences between the SCD mice and humans were noted, the overall functional phenotype appears to be conserved (ie, reduced antibody responses in both, even though B-cell numbers tend to increase in patients with SCD but decrease in SCD mice). Altogether, these findings support the contention that SCD is a state of altered (and perhaps impaired) immunity, which is exemplified by the finding of a reduced percentage of splenic B-1a B cells and gut IgA-secreting B cells in SCD mice. Furthermore, SCD mice also appear to live with heightened inflammation, as was demonstrated by the high levels of serum IL-6. The finding that hemizygous mice also have altered immune responses at baseline is intriguing and requires further investigation.
Acknowledgment
We thank Justin Hummel for technical assistance with mouse handling.
Footnotes
Supported by the Connecticut Institute for Clinical and Translational Science Clinical and Translational Scholars K12 Award, Lea's Foundation for Leukemia Research, Inc., and a grant from the National Institute of Allergy and Infectious Diseases (RO1 AI04357-11).
References
- 1.Platt O.S., Brambilla D.J., Rosse W.F., Milner P.F., Castro O., Steinberg M.H., Klug P.P. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 2.Harms G., Hardonk M.J., Timens W. In vitro complement-dependent binding and in vivo kinetics of pneumococcal polysaccharide TI-2 antigens in the rat spleen marginal zone and follicle. Infect Immun. 1996;64:4220–4225. doi: 10.1128/iai.64.10.4220-4225.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wardemann H., Boehm T., Dear N., Carsetti R. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J Exp Med. 2002;195:771–780. doi: 10.1084/jem.20011140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cameron P.U., Jones P., Gorniak M., Dunster K., Paul E., Lewin S., Woolley I., Spelman D. Splenectomy associated changes in IgM memory B cells in an adult spleen registry cohort. PLoS One. 2011;6:e23164. doi: 10.1371/journal.pone.0023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haas K.M., Poe J.C., Steeber D.A., Tedder T.F. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity. 2005;23:7–18. doi: 10.1016/j.immuni.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Rosado M.M., Aranburu A., Capolunghi F., Giorda E., Cascioli S., Cenci F., Petrini S., Miller E., Leanderson T., Bottazzo G.F., Natali P.G., Carsetti R. From the fetal liver to spleen and gut: the highway to natural antibody. Mucosal Immunol. 2009;2:351–361. doi: 10.1038/mi.2009.15. [DOI] [PubMed] [Google Scholar]
- 7.Battersby A.J., Knox-Macaulay H.H., Carrol E.D. Susceptibility to invasive bacterial infections in children with sickle cell disease. Pediatr Blood Cancer. 2010;55:401–406. doi: 10.1002/pbc.22461. [DOI] [PubMed] [Google Scholar]
- 8.Paszty C., Brion C.M., Manci E., Witkowska H.E., Stevens M.E., Mohandas N., Rubin E.M. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 9.Holtzclaw J., Jack D., Aguayo S., Eckman J., Roman J., Hsu L. Enhanced pulmonary and systemic response to endotoxin in transgenic sickle cell mice. Am J Respir Crit Care Med. 2004;169:687–695. doi: 10.1164/rccm.200302-224OC. [DOI] [PubMed] [Google Scholar]
- 10.Paul R.N., Castro O.L., Aggarwal A., Oneal P.A. Acute chest syndrome: sickle cell disease. Eur J Haematol. 2011;87:191–207. doi: 10.1111/j.1600-0609.2011.01647.x. [DOI] [PubMed] [Google Scholar]
- 11.Campbell A.D., Cui S., Shi L., Urbonya R., Mathias A., Bradley K., Bonsu K.O., Douglas R.R., Halford B., Schmidt L., Harro D., Giacherio D., Tanimoto K., Tanabe O., Engel J.D. Forced TR2/TR4 expression in sickle cell disease mice confers enhanced fetal hemoglobin synthesis and alleviated disease phenotypes. Proc Natl Acad Sci U S A. 2011;108:18808–18813. doi: 10.1073/pnas.1104964108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Booth C., Inusa B., Obaro S.K. Infection in sickle cell disease: a review. Int J Infect Dis. 2010;14:e2–e12. doi: 10.1016/j.ijid.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 13.Khatib R., Rabah R., Sarnaik S.A. The spleen in the sickling disorders: an update. Pediatr Radiol. 2009;39:17–22. doi: 10.1007/s00247-008-1049-9. [DOI] [PubMed] [Google Scholar]
- 14.Overturf G.D. Pneumococcal vaccination of children. Semin Pediatr Infect Dis. 2002;13:155–164. doi: 10.1053/spid.2002.125858. [DOI] [PubMed] [Google Scholar]
- 15.Burnett M.W., Bass J.W., Cook B.A. Etiology of osteomyelitis complicating sickle cell disease. Pediatrics. 1998;101:296–297. doi: 10.1542/peds.101.2.296. [DOI] [PubMed] [Google Scholar]
- 16.Kang Y.S., Kim J.Y., Bruening S.A., Pack M., Charalambous A., Pritsker A., Moran T.M., Loeffler J.M., Steinman R.M., Park C.G. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc Natl Acad Sci U S A. 2004;101:215–220. doi: 10.1073/pnas.0307124101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppel E.A., Litjens M., van den Berg V.C., van Kooyk Y., Geijtenbeek T.B. Interaction of SIGNR1 expressed by marginal zone macrophages with marginal zone B cells is essential to early IgM responses against Streptococcus pneumoniae. Mol Immunol. 2008;45:2881–2887. doi: 10.1016/j.molimm.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 18.Mebius R.E., Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 19.Rautonen N., Martin N.L., Rautonen J., Rooks Y., Mentzer W.C., Wara D.W. Low number of antibody producing cells in patients with sickle cell anemia. Immunol Lett. 1992;34:207–211. doi: 10.1016/0165-2478(92)90215-a. [DOI] [PubMed] [Google Scholar]
- 20.Venkataraman M., Westerman M.P. B-cell changes occur in patients with sickle cell anemia. Am J Clin Pathol. 1985;84:153–158. doi: 10.1093/ajcp/84.2.153. [DOI] [PubMed] [Google Scholar]
- 21.Adedeji M.O. Lymphocyte subpopulations in homozygous sickle cell anaemia. Acta Haematol. 1985;74:10–13. doi: 10.1159/000206155. [DOI] [PubMed] [Google Scholar]
- 22.Kaaba S.A., al-Harbi S.A. Reduced levels of CD2+ cells and T-cell subsets in patients with sickle cell anaemia. Immunol Lett. 1993;37:77–81. doi: 10.1016/0165-2478(93)90135-o. [DOI] [PubMed] [Google Scholar]
- 23.Taub D.D., Lloyd A.R., Conlon K., Wang J.M., Ortaldo J.R., Harada A., Matsushima K., Kelvin D.J., Oppenheim J.J. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crawford A., Angelosanto J.M., Nadwodny K.L., Blackburn S.D., Wherry E.J. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011;7:e1002098. doi: 10.1371/journal.ppat.1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrison C., Maurtua-Neumann P., Myint M.T., Drury S.S., Bégué R.E. Pandemic (H1N1) 2009 outbreak at camp for children with hematologic and oncologic conditions. Emerg Infect Dis. 2011;17:87–89. doi: 10.3201/eid1701.091499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs J.E., Quirolo K., Vichinsky E. Novel influenza A (H1N1) viral infection in pediatric patients with sickle-cell disease. Pediatr Blood Cancer. 2011;56:95–98. doi: 10.1002/pbc.22656. [DOI] [PubMed] [Google Scholar]
- 27.Chuncharunee S., Archararit N., Hathirat P., Udomsubpayakul U., Atichartakarn V. Levels of serum interleukin-6 and tumor necrosis factor in postsplenectomized thalassemic patients. J Med Assoc Thai. 1997;80(Suppl 1):S86–S91. [PubMed] [Google Scholar]
- 28.Graido-Gonzalez E., Doherty J.C., Bergreen E.W., Organ G., Telfer M., McMillen M.A. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1998;92:2551–2555. [PubMed] [Google Scholar]
- 29.Pathare A., Al Kindi S., Alnaqdy A.A., Daar S., Knox-Macaulay H., Dennison D. Cytokine profile of sickle cell disease in Oman. Am J Hematol. 2004;77:323–328. doi: 10.1002/ajh.20196. [DOI] [PubMed] [Google Scholar]
- 30.Taylor S.C., Shacks S.J., Mitchell R.A., Banks A. Serum interleukin-6 levels in the steady state of sickle cell disease. J Interferon Cytokine Res. 1995;15:1061–1064. doi: 10.1089/jir.1995.15.1061. [DOI] [PubMed] [Google Scholar]
- 31.Taylor S.C., Shacks S.J., Qu Z., Wiley P. Type 2 cytokine serum levels in healthy sickle cell disease patients. J Natl Med Assoc. 1997;89:753–757. [PMC free article] [PubMed] [Google Scholar]