

Abstract
Cancer stem cell behavior is thought to be largely determined by intrinsic properties and by regulatory signals provided by the microenvironment. Myelofibrosis (MF) is characterized by hematopoiesis occurring not only in the marrow but also in extramedullary sites such as the spleen. In order to study the effects of these different microenvironments on primitive malignant hematopoietic cells, we phenotypically and functionally characterized splenic and peripheral blood (PB) MF CD34+ cells from patients with MF. MF spleens contained greater numbers of malignant primitive HPCs than PB. Transplantation of PB MF CD34+ cells into immunodeficient (NOD/SCID/IL2Rγnull) mice resulted in a limited degree of donor cell chimerism and a differentiation program skewed toward myeloid lineages. By contrast, transplanted splenic MF CD34+ cells achieved a higher level of chimerism and generated both myeloid and lymphoid cells that contained molecular or cytogenetic abnormalities indicating their malignant nature. Only splenic MF CD34+ cells were able to sustain hematopoiesis for prolonged periods (9 months) and were able to engraft secondary recipients. These data document the existence of MF stem cells (MF-SCs) that reside in the spleens of MF patients and demonstrate that these MF-SCs retain a differentiation program identical to that of normal hematopoietic stem cells.
Introduction
The Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are thought to originate at the level of a pluripotent HSC (1–5). PMF and myelofibrosis (MF) that develop during the course of ET or PV (post-ET or PV-MF) have a similar clinical phenotype. These disorders are characterized by abnormal trafficking of HSCs and hematopoietic progenitor cells (HPCs), resulting in their constitutive mobilization and the establishment of extramedullary sites of hematopoiesis. Disease progression in MF is frequently accompanied by progressive splenomegaly due to extramedullary hematopoiesis (EMH) (6, 7).
Behavior of HSCs/HPCs is largely determined by their intrinsic properties as well as regulatory signals that are provided by the local microenvironment or niche in which they reside. The BM hematopoietic microenvironment is composed of specific matrix proteins, cytokines, as well as a variety of accessory cells including endothelial cells, osteoblasts, osteoclasts, macrophages, T cells, and stromal cells, which play a role in regulating normal hematopoiesis (8–11). Postnatally in humans the spleen is primarily a lymphoid organ that also serves as a filter for the removal of damaged or senescent circulating blood cells and contains small numbers of mature myeloid cells. Only in patients with an MPN or a variety of hemoglobinopathies, such as the thalassemia, does EMH occur in the spleen to a significant degree. For EMH to occur, a conducive microenvironment must be established that will provide the signals required in order for hematopoiesis to be sustained. Such a permissive microenvironment may be derived from endogenous splenic cells which are conditioned by hematopoietic cells that are constitutively mobilized into the PB and enter the spleen, or due to the mobilization of marrow mesenchymal cells that take residence in the spleen and create a microenvironment that provides a residence for circulating HSCs/HPCs. The possible contribution of marrow-derived mesenchymal stem cells to tumor microenvironments has been previously reported by others (12–14). Whatever its source, the cellular composition and architecture of the splenic microenvironment that supports EMH differ from those of the marrow, which has led us to hypothesize that these differences might result in the HSCs/HPCs residing within the MF spleen being functionally different from those present within the PB.
Hematopoiesis in normal individuals occurs primarily in the marrow, while in PMF and post-PV or post-ET MF patients, hematopoiesis occurs not only in the marrow but also in a variety of extramedullary sites including the spleen and liver. Since the marrow is fibrotic, in advanced forms of MF, previous studies of MF stem cells (MF-SCs) have relied on the limited numbers of PB CD34+ cells that are constitutively mobilized into the PB. The marrows of patients who require therapeutic splenectomies are frequently severely fibrotic and contain limited numbers of hematopoietic cells (15), making it impossible to compare the nature of marrow and splenic HSCs/HPCs isolated from the same individual. Patients with early phases of MF occasionally have marrows that can be aspirated, but these patients are asymptomatic and rarely require therapeutic splenectomies. These underlying features of MF have limited the accessibility to the numbers of MF CD34+ cells required to study the nature of the MF-SCs using in vivo assays. Several reports have studied mesenchymal stem cells derived from BM aspirates or bone fragments from patients with MF (16, 17), but these cell types require prolonged culture in vitro for their isolation, and they do not contribute to blood cell production. The nature of the CD34+ cells present in extramedullary sites such as spleen and liver has not been previously studied. In this report CD34+ cells isolated from the PB and surgically removed spleens provided a unique opportunity to study MF HSCs present not only in the blood but also an extramedullary site.
The cellular stage along the hematopoietic hierarchy at which PMF originates has been addressed using two approaches (15, 18, 19). Although myeloid cells have been shown to uniformly contain JAK2V617F, lymphoid cells (B, T, and natural killer cells) contain mutated JAK2 in a far smaller proportion of patients (19). These observations have led some to suggest that PMF might originate in a myeloid-lymphoid progenitor cell that generates myeloid cells but has a limited ability to generate lymphoid cells. The standard surrogate assay for human HSCs relies on the ability of a putative HSC population to establish hematopoiesis following transplantation into immunodeficient (NOD/SCID) mice (20–23). Such marrow-repopulating cells are termed SCID repopulating cells (SRCs). The SRCs assayed from the PB of PMF patients display a limited ability to generate donor-derived myeloid cells and lack the ability to generate significant numbers of B or T cells (15, 18, 19, 24). In order to test our hypothesis that the stem cells that reside within the spleens of MF patients have properties different from those that have been assayed from the PB, we have phenotypically and functionally characterized splenic and PB MF CD34+ cells from the same patients. We have shown that a greater number of true malignant HSCs capable of self-renewal and producing cells belonging to multiple hematopoietic lineages including B and T cells are present in MF spleens. These studies suggest that MF originates at the level of a cancer stem cell. These MF-SCs appear to preferentially localize to extramedullary sites such as the spleen, which likely provides a microenvironment permitting MF-SCs to retain their functional properties. Since we have not been able to study other extramedullary sites of hematopoiesis such as the liver, we cannot exclude the possibility that MF-SCs are also present in additional locations.
Results
Clinical and laboratory characterization of MF patients.
Eight patients with advanced forms of MF undergoing therapeutic splenectomy for a variety of indications, including cytopenias and painful splenomegaly, or in preparation for allogeneic stem cell transplantation were studied. The median age of these patients was 69 (range, 64–79 years); 5 subjects (63%) were male. The clinical and laboratory characteristics of these patients are outlined in Table 1. Among the 8 MF subjects, 3 patients were JAK2V617F positive, and their cells had a marker chromosomal abnormality; one patient was JAK2V617F positive but did not have a chromosomal abnormality; one patient had a marker chromosomal abnormality but was JAK2 wild type; and 3 patients were JAK2V617F negative and did not have a marker chromosomal abnormality.
Table 1.
Characteristics of MF patients

Phenotype of splenic and PB MF CD34+ cells.
We phenotypically characterized paired MF splenic and PB CD34+ cells from 7 different patients. A greater percentages of CD34+ cells was detected in the spleen than in the PB mononuclear cells (MNCs) (4.2% ± 0.9% versus 0.4% ± 0.1%; P < 0.05; Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI64397DS1). Moreover, 91.7% of splenic MF CD34+ cells and 85.3% of PB MF CD34+ cells were in the G0/G1 phase, and only 4.0% of splenic MF CD34+ cells and 5.2% of PB MF CD34+ cells were in the G2/M phase, suggesting that PB and splenic MF CD34+ cells are equally quiescent (Supplemental Figure 1B). Furthermore, a similar proportion of splenic and PB MF CD34+ cells was observed to be CD38– and CD90+ (Supplemental Table 1). In order to determine whether the expression of chemokine receptors and adhesion molecules could account for the homing and location of MF CD34+ cells to the spleen, we compared the expression of a number of such molecules (CXCR4, CD47, CD44, and CD49d) by splenic and PB MF CD34+ cells, and their expression was shown to be similar (Supplemental Table 1).
Greater numbers of malignant HPCs were assayed from splenic rather than PB MF MNCs.
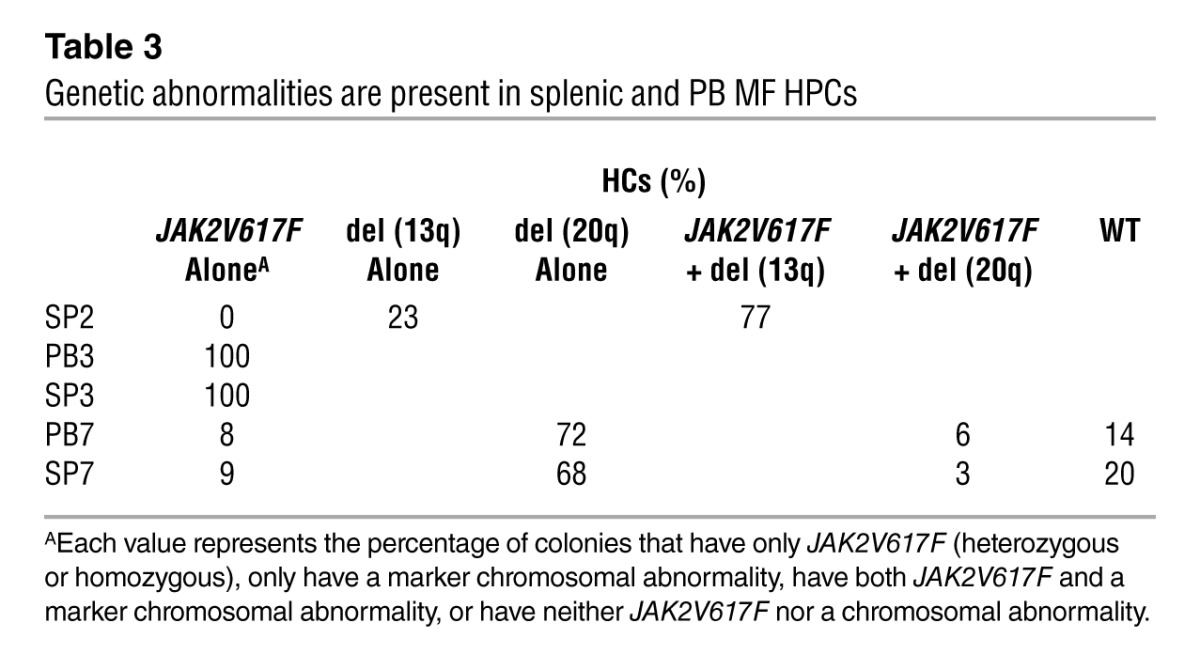
Splenic or PB MF MNCs isolated from 7 MF patients (Supplemental Table 2) were assayed for HPCs. Individual hematopoietic colonies (HCs) were generated from splenic or PB MF MNCs of 3 patients who were JAK2V617F+ and/or had a marker chromosomal abnormality. The colonies from these 3 patients were individually plucked, and half were analyzed for JAK2V617F, while the other half were analyzed for the presence of a marker chromosome. Greater numbers of HPCs were assayed from the splenic (9,839 ± 3,129/106 MNCs) compared with the PB MNCs (1,746 ± 556/106 MNCs; Table 2). However, the percentage of splenic HCs that were JAK2V617F+ or had a chromosomal abnormality was similar to that cloned from PB MNCs (Table 3). In the 3 cases of JAK2V617F+ MF studied, 12%–100% of splenic HCs were JAK2V617F+, while all the HCs from spleen (SP) 2 and 71% of HCs from SP7 harbored chromosomal abnormalities, deletion 13q [del (13q)] and deletion 20q [del (20q)], respectively (Table 3). We concluded that varying numbers of MF HPCs in individual patients were involved in the malignant process and that at least in the two cases, a small proportion of HPCs lacked the genetic abnormalities assessed. In two cases (SP2 and SP7), a proportion of HPCs were JAK2V617F negative but possessed a chromosomal abnormality, indicating that these two genetic events can occur independently.
Table 2.
Greater number of HPCs in the spleen compared with the PB of MF patients

Table 3.
Genetic abnormalities are present in splenic and PB MF HPCs
SRCs are present in the spleens of MF patients.
Currently, the standard surrogate assay for human HSCs measures the ability of a putative HSC population to establish hematopoiesis following their transplantation into immunodeficient mice. We have previously documented human marrow cell chimerism in NOD/SCID/IL2Rγnull (NSG) mice 6 months after the transplantation of PB PMF CD34+ cells, although the degree of human cell chimerism has been limited (<3% when 2 × 106 cells were transplanted) (24). The engrafted cells were mainly composed of cells belonging to multiple myeloid lineages [CD33+, glycophorin A (Gly A)+, and CD41a+] but few CD19+ and no donor-derived CD3+ cells (24). In this article, we report the behavior of splenic MF SRCs following their transplantation into NSG mice using limiting dilution analysis.
Four to 5 months after transplantation of varying numbers of MF CD34+ cells, recipient mice were evaluated for the presence of SRCs by analyzing for the presence of human cells belonging to various hematopoietic lineages. Splenic and PB grafts contained equal numbers of CD34+ cells. When 2 × 105 and 2 × 106 splenic or PB CD34+ cells were transplanted, splenic MF CD34+ cell grafts produced dramatically greater numbers of human CD45+ and CD34+ cells in the BM, spleen, thymus, and PB of recipient mice (Figure 1, A–H). Furthermore, there was a clear relationship between the numbers of splenic but not PB CD34+ cells infused and the degree of human cell chimerism achieved in the BM and spleen of these mice. These findings indicate that splenic MF CD34+ cells contain long-term marrow reconstituting cells, and that greater numbers of long-term SRCs are present in splenic than PB MF CD34+ cells.
Figure 1. Enhanced human hematopoiesis in NSG mice receiving transplants of splenic MF CD34+ cells.

(A, C, E, and G) The degree of MF hematopoietic cell engraftment is indicated by the percentage of hCD45+ cells detected within the marrow (A), spleen (C), thymus (E), and PB (G) of the NSG mice. (B, D, F, and H) Percentage of hCD34+ cells present within the hCD45+ cell population within the marrow (B), spleen (D), thymus (F), and PB (H) of the NSG mice. The x axis indicates the number of CD34+ cells (×104) injected. All data points represent the mean ± SD from 5–6 different mice, except studies in which 2 × 106 PB MF cells were infused when only 3 animals received transplants due to the limited numbers of CD34+ cells available from the PB of individual patients. P values (Student’s t test) are shown in Supplemental Table 6. Note: ≥0.3% hCD45+ cells were detected in the thymi of mice transplanted with splenic MF CD34+ cells (2 × 105 or 2 × 106) from all 6 patients, while no human cell engraftment was detected in the thymi of mice receiving PB MF CD34+ cells. (I) An increase in splenic weight was observed in mice receiving only splenic MF CD34+ cells. The y axis indicates the fold increase in spleen weight in mice receiving splenic or PB MF CD34+ cells as compared with that of mice receiving PBS alone.
The degree of engraftment achieved following the transplantation of splenic MF CD34+ cells within the spleens of the recipient mice was greater based on the degree of human CD45+ cell chimerism observed in the spleens of recipient mice (Figure 1C). The degree of human cell chimerism in the spleen was often equivalent to that observed in the marrow within the same recipient animal (Figure 1, A and C). Furthermore, the mice receiving splenic MF CD34+ cells experienced a 0.3- to 4.3-fold increase in splenic weight as compared with mice receiving equal numbers of PB MF CD34+ cells or PBS alone (Figure 1I). By contrast, there was no increase in splenic weight in animals receiving transplants of PB MF CD34+ cells as compared with those animals receiving PBS alone (Figure 1I). These findings suggest that unlike PB MF CD34+ cells, splenic MF CD34+ cells can reconstitute human hematopoiesis not only in the marrow but also in the spleen of NSG mice.
We further evaluated the engraftment of splenic and PB MF CD34+ cells in the thymus of NSG recipients. Evidence of donor-derived CD45+ and CD34+ cells was observed in the thymus of mice receiving varying numbers of splenic but not PB MF CD34+ cells (Figure 1, E and F). The human cells were composed primarily of CD3+ and a small proportion of CD19+ cells. Very few CD33+ and Gly A+ cells were observed. These findings suggest that splenic MF CD34+ cells were capable of engrafting the thymus of NSG recipients and generating human T and B cells.
Another unique characteristic of splenic MF CD34+ cell transplants was the presence of hCD45+ and hCD34+ cells in the PB of mice receiving these grafts but not in mice receiving PB MF CD34+ cells (Figure 1, G and H). The degree of PB human cell chimerism was directly related to the size of the graft from patients 3, 5, and 6 and persisted 4–9 months (Figure 1, G and H, and Supplemental Figure 2D). This constitutive mobilization of hCD34+ cells is reminiscent of the abnormal stem cell trafficking that occurs in PMF patients (25).
Differentiation program of splenic MF CD34+ cells in NSG mice.
Human marrow and splenic cells obtained from recipient mice were further analyzed for their ability to generate cells belonging to multiple hematopoietic lineages. The frequency and degree of engraftment achieved in the marrow of the mice receiving PB MF CD34+ cells were limited, and the generation of myeloid cells but few CD19+ cells and no CD3+ cells was notable (Figure 2A and ref. 24). By contrast, splenic MF CD34+ cells produced a large number of cells belonging to both myeloid and lymphoid lineages (CD33+, Gly A+, CD41a+, CD19+, and CD3+ cells) both in the marrow and spleens of recipient mice (Figure 2, A and B). These data indicate that splenic but not PB MF CD34+ cells contain true multipotent HSCs.
Figure 2. Differentiation programs of splenic and PB MF CD34+ cells.

The generation of myeloid cells (CD33+), megakaryocytes (CD41a+), B cells (CD19+), T cells (CD3+), and erythroid cells (Gly A+) in the BM (A) and spleens (B) of recipient mice receiving splenic or PB MF CD34+ cell grafts (2 × 106). Black diamonds show the percentage of human lineage cells in the BM and spleen of each individual mouse. Horizontal bars indicate the mean of percentage of human lineage cells in the BM and spleen of NSG mice. PB MF CD34+ cells were able to generate myeloid cells but few CD19+ cells and no CD3+ cells. By contrast, splenic MF CD34+ cells produced a large number of both myeloid and lymphoid cells both in the marrow and spleens of recipient mice. P < 0.01, when comparing each cell lineage generated by SP1 and PB1; P < 0.05, when comparing each cell lineage generated by SP6 and PB6.
Prolonged human cell engraftment in NSG mice receiving splenic MF CD34+ cell transplants.
The degree and site of human cell engraftment following the transplant of splenic CD34+ cells in recipient mice were evaluated over time. Robust human cell chimerism was observed in the marrow, spleens, thymi, and PB 9 months after the transplantation of exclusively splenic MF CD34+ cells (Supplemental Figure 2, A–D). Furthermore, as shown Supplemental Figure 2A, the degree of human cell chimerism in the BM of mice receiving SP1 cells was relatively diminished 9 months after transplantation (10.8%) as compared with that achieved after 5 months (42.7%). However, human cell chimerism in the spleen and thymus of these same mice was greater at 9 months than that observed after 5 months. Moreover, the degree of circulating human CD34+ cells in the PB of these mice after 9 months was significantly greater than that observed after 5 months (Supplemental Figure 2D). These findings suggest that the sites of human hematopoiesis in mice following the transplantation of splenic MF CD34+ cells shift to a pattern that more closely resembles that occurring in MF patients.
Immunohistochemical analysis of human hematopoiesis in the spleen of mice receiving transplants of splenic and PB MF CD34+ cells.
The pattern of engraftment of MF splenic and PB CD34+ cells was confirmed by immunohistochemical analyses. Rare hCD45+ cells were observed in the spleens of mice receiving PB MF CD34+ cell transplants (Figure 3B); by contrast, as shown in Figure 3, E and F and L and M, significant numbers of hCD45+ cells were detected in the spleen of mice 5–9 months after receipt of splenic MF CD34+ cells. Approximately 15% of the cells within the recipient mouse spleen (9 months) were of human origin and included both erythroid and granulocytic precursors at various stages of maturation. A large number of hCD45+ cells were observed in the white and red pulp of spleens of recipient mice 5–9 months after transplantation, and these were composed primarily of human CD5+ and CD20+ lymphocytes (Figure 3, G, H, N, and O). The red pulp of the recipient spleens contained human granulocytic and erythroid cells at various stages of maturation (Figure 3, E, I, and L).
Figure 3. Immunohistochemical analyses of spleens of NSG mice transplanted with MF CD34+ cells.

(A and B) Spleen sections 5 months after transplantation with PB1 CD34+ cells stained with H&E (A) and hCD45 (B). The image demonstrates normal splenic architecture (A); rare hCD45+ cells as indicated by arrows were detected (B). (C–I) Spleen sections 5 months after transplantation with SP1 CD34+ cells. (C) H&E staining demonstrated normal splenic architecture with a preserved white pulp and EMH in the red pulp (D). Immunohistochemical staining demonstrated the presence of human granulocytes as well as lymphocytes in the red pulp (E). The white pulp was composed primarily of hCD45+ lymphoid cells (F), which included both hCD5+ T cells (G) and hCD20+ B cells (H). Many normoblasts in the red pulp were mTER119–, indicating their human origin (I). The horizontal arrow indicates mTer119+ normoblasts, while the vertical arrow indicates mTer119– normoblast. (J–O) Spleen sections 9 months after transplantation with SP1 CD34+ cells. (J) The normal splenic architecture was preserved with a prominent white pulp. (K) H&E image demonstrated trilineage hematopoiesis in the red pulp. Immunohistochemical stains demonstrated human myeloid cells as indicated by the arrow in the red pulp (L). The white pulp was almost entirely composed of hCD45+ lymphoid cells (M), which were predominantly hCD5+ T cells (N), with fewer hCD20+ B cells (O). Original magnification, A: ×200; B, D, and I: ×400; C and F–H: ×100; J and M–O: ×40; E, K, and L: ×600. WP, white pulp; RP, red pulp; Pos, positive; Neg, negative; g, granulocytic precursors; e, erythroid precursors; m, megakaryocyte; l, lymphocyte.
Frequency of SRCs in splenic and PB MF CD34+ cells.
We next determined the frequency of SRCs in PB and splenic MF CD34+ cells. We performed a limiting dilution analysis of paired PB and splenic CD34+ cells with specimens from 5 MF patients and splenic MF CD34+ cells from an additional patient for whom corresponding PB cells were not available. The frequency of SRCs within splenic MF CD34+ cells was 70- to 205-fold greater than that calculated for PB MF CD34+ cells; the SRC frequency for PB MF CD34+ cells ranged from 1 in 6.1 × 106 CD34+ cells to 1 in 4.5 × 105 CD34+ cells (Table 4), while the SRC frequency for splenic MF CD34+ cells ranged from 1 in 8.7 × 104 CD34+ cells to 1 in 2.2 × 103 CD34+ cells (Table 4). These findings indicate that significantly greater numbers of SRCs are present in the spleens of these patients.
Table 4.
Frequency of SRCs in splenic and PB MF CD34+ cells

Splenic MF CD34+ cells contain HSCs that are capable of self-renewal in vivo.
One of the defining characteristics of HSCs is their ability to self-renew. Serial transplantation of putative stem cell populations provides the only experimental approach by which to assess the self-renewal capacity of the splenic MF CD34+ cells (26). Human cells isolated after 5 months from the BM and the spleens of NSG mice that were engrafted with PB or splenic MF CD34+ cell grafts were injected into secondary recipients, NSG mice, and the presence of donor cells in the BM and spleen was assessed by flow cytometry after 3 months. Human cell engraftment was not detected in secondary mice with cells harvested from either the BM or the spleen of mice receiving primary PB MF CD34+ cell grafts irrespective of the number of cells constituting the primary graft. However, as shown in Figure 4, both BM cells and splenic cells from recipients transplanted with as few as 2 × 104 primary splenic MF CD34+ cells were able to reconstitute donor-derived myeloid and lymphoid cells in secondary mouse recipients.
Figure 4. Secondary transplantation of splenic MF CD34+ cells.

Human cells isolated from the BM and the spleen of NSG mice 5 months after they had received PB or splenic MF CD34+ cell grafts were injected into secondary NSG mice, and the presence of donor cells in the BM and spleen was assessed by flow cytometry after 3 months. (A and B) Human cell engraftment (hCD45+) and the presence of human hCD34+ cells, myeloid cells (hCD14+), megakaryocytes (hCD41a+), B cells (hCD19+), T cells (hCD19+), and erythroid cells (hGly A+) in the BM (A) and spleen (B) of the secondary recipients. Black diamonds show the percentage of human cells in the BM and spleen of each individual secondary mouse. Numbers of splenic MF CD34+ cells injected into the primary recipients are shown in parentheses. Horizontal bars indicate the mean of percentage of human cells in the BM and spleen of secondary recipients. Both BM cells and splenic cells recovered from the primary recipients were transplanted into secondary recipients.
Splenic MF CD34+ cells are malignant.
We next determined whether the donor-derived cells generated in NSG mice were JAK2V617F+ or possessed marker chromosomal abnormalities. Four or 5 months following transplantation, human CD45+ cells and cells belonging to various hematopoietic lineages were harvested and isolated from the BM and spleens of the mice receiving splenic CD34+ cells from JAK2V617F+ MF patients (SP2 and SP3), one of which also had a marker chromosomal abnormality (SP2). As shown in Table 5, similar JAK2V617F allele burdens were observed in CD34+ cells, as well as hematopoietic cells belonging to each myeloid and lymphoid lineage analyzed from the BM and spleens of the recipient mice. The JAK2V617F allele burden in the cells belonging to the various lineages was virtually identical to that observed in the patients’ PB granulocytes. These findings indicate that malignant multipotent HSCs exist in the spleens of MF patients that are capable of generating cells belonging to both malignant myeloid and lymphoid cells.
Table 5.
The JAK2V617F allele burden of various human lineage cells within the BM and spleens of NSG mice receiving transplants of JAK2V617F+ splenic MF CD34+ cells

We further studied the malignant nature of the splenic SRCs by examining the presence of a marker chromosomal abnormality. Analysis of primary splenic cells (SP2) revealed that 89% of the splenic cells had del (13q), 3% of the cells had duplication of del (13q) and one normal chromosome 13 (as evidenced by the presence of 3 copies of the LAMP1 locus on the 13q34 chromosomal site), and 8% of the cells were normal, indicating the presence of three populations. Of the primary spleen cells studied, 7% of the CD34+ cells and 1% of the CD19+ cells had del (13q), while none of the CD3+ cells had this cytogenetic abnormality. By contrast, following transplantation of splenic CD34+ cells into NSG mice, 99%–100% of donor-derived myeloid and lymphoid cells within the BM and spleen of the recipient contained duplication of del (13q) and one normal chromosome 13 (Supplemental Figure 3 and Table 6). These data further confirm the malignant nature of the splenic SRCs capable of generating myeloid and lymphoid cells and that individual clones may predominate following their transplantation into NSG mice.
Table 6.
Chromosomal abnormalities were present in human various lineage cells within the BM and spleens of NSG mice receiving chromosomally abnormal splenic MF CD34+ cells

Gene expression profiles of splenic and PB MF CD34+ cells.
In order to understand the biological mechanisms that are responsible for the distinctive behavior of splenic and PB MF CD34+ cells following their transplantation into NSG mice, we studied their gene expression profiles. Two hundred and twenty-five genes were identified with significant differences in levels of expression between MF splenic and PB MF CD34+ cells. Among these 225 differentially expressed genes, 68 were well characterized (Supplemental Tables 3 and 4). Among the 68, 36 genes were downregulated in MF splenic CD34+ cells as compared with PB CD34+ cells. The downregulated genes included tumor suppressor genes, such as DIRAS3; genes that instruct the production of proteins that are involved in signal transduction, such as BALAP2L1, SHC4, OSMR, RGS11, and APBA2; genes that encode cytokines and cytokine receptors, such as CSF2, IL13, IL24, and IL17RB; genes that encode molecules that promote cell adhesion to laminin-332 and fibronectin, such as CLEC3A, to collagen and laminin, such as ITGA1, or cell-cell adhesion, such as NCAM1 and CDAM1; genes that encode chemokines, such as CXCL6 and CXCL13; as well as genes that encode proteases, such as TMPRSS13 and MMP20. Thirty-two genes were upregulated in MF splenic CD34+ cells as compared with PB CD34+ cells, including PTPRD, which encodes signaling molecules that regulate a variety of cellular processes including cell growth, differentiation, cell cycle, and oncogenic transformation; CLEC10A, which is involved in cell adhesion and cell-cell interaction; MAP3K9, which acts as an essential component of the MAPK signaling pathway; RGS4, which inhibits signal transduction by increasing the GTPase activity of G protein α subunits — as well as genes that encode cytokine receptors, such as IL5RA and CSF1R; chemokines, such as XCL1; and chemokine receptors, such as CCR2 and CCR9. These differentially expressed genes might account for the functional differences observed between these two sources of MF-SCs.
Discussion
In this study we capitalized on our access to both PB and splenic cells from MF patients to examine the behavior of MF-SCs. We have demonstrated that MF spleens contain greater numbers of malignant HPCs than PB. The behavior of PB and splenic MF CD34+ cells following their transplantation into NSG mice revealed functional differences between these two potential sources of MF-SCs. We and others have previously reported that PB MF CD34+ cells were able to engraft primary recipient mice, achieving a limited degree of donor chimerism (18, 19, 24, 27). The PB MF SRCs also had a restricted differentiative capacity, being able to produce myeloid cells belonging to multiple lineages but unable to generate large numbers of B cells or any T cells (18, 19, 24, 27). In addition, the human cells that engrafted the primary recipient mice were unable to repopulate secondary recipients. By contrast, the CD34+ cells isolated from the spleens of MF patients achieved a higher level of donor cell chimerism and were able to differentiate into cells belonging to both myeloid and lymphoid lineages with equal efficiency. These observations are quite distinct from those made with other leukemia stem cells, where similar in vivo functional studies have led to the generation of exclusively myeloid cells, and might represent a unique underlying feature of MF-SCs. In addition, splenic MF CD34+ cells were able to sustain hematopoiesis for prolonged periods of time, and the human cells from the primary recipients were able to engraft secondary recipients. The CD34+ cells isolated from the spleens of MF patients, therefore, possessed each of the properties that would be associated with a true MF-SC. Furthermore, the frequency of SRCs in splenic CD34+ cells was documented to be several logs higher than that observed in the PB. The availability of large numbers of splenic cells that contain greater numbers of MF-SCs should prove useful for further phenotypic characterization of MF-SCs.
The relative frequency of MF splenic and PB CD34+ cells and their phenotype were studied. Previously, it had been reported that the numbers of CD34+ cells per liter correlated with disease severity and was predictive of disease progression (25). The composition of MNCs in PB is likely different from that in spleen. Since the spleen is a solid organ, while PB is liquid, comparing the numbers of CD34+ cells per liter was not possible. With these limitations in mind, numbers of MNCs isolated from spleen or PB were used as a standard to evaluate the frequency of CD34+ cells present in these two types of tissues. Using this approach, we showed MF spleens to contain greater numbers of CD34+ cells, but splenic and PB MF CD34+ cells were equally quiescent and were characterized by a similar proportion of CD34+ that were CD38– and CD90+. Furthermore, the expression of a number of chemokine receptors and adhesion molecules (CXCR4, CD47, CD44, and CD49d) was also similar in splenic and PB MF CD34+ cells. These data indicate that the behavior of the two sources of grafts following their transplantation into immunodeficient mice cannot be attributed merely to phenotypic differences. In order to further understand the biological mechanisms that were responsible for the distinctive behavior of splenic and PB MF CD34+ cells following their transplantation into NSG mice, we studied their gene expression profiles. Sixty-eight well-characterized genes were identified with significant differences in levels of expression between MF splenic and PB MF CD34+ cells. The products of some of these differentially expressed genes might account for the functional differences observed between these two sources of MF-SCs.
With the use of molecular and cytogenetic markers (15) as well as glucose-6-phosphate dehydrogenase isoenzyme analysis and X-linked restriction fragment length polymorphisms (28), the MPNs have been shown to involve cells belonging primarily to the myeloid lineages, with occasional studies indicating involvement of B cells and more rarely T cells (18, 19). In this article, we document a potential source for the lymphoid cells reported to be involved in the malignant process in MF. When splenic but not PB MF-SCs were transplanted into the permissive environment of NSG mice, they were capable of producing similar numbers of not only malignant myeloid but also T and B cells as identified by both molecular and cytogenetic markers. These findings suggest that the splenic microenvironment is capable of preserving the pluripotentiality of MF-SCs, which is significantly restricted once they exit the spleen. These distinct differentiation programs likely occur in response to unique environmental cues present in the spleen of MF patients that prevent the silencing of MF-SC genetic programs. These MF-SCs are capable in selected patients of producing limited numbers of B and T cells belonging to the malignant clone, but in large part this potential to generate malignant T cells and B cells is silenced as hematopoietic commitment and differentiation proceeds. The potential of a stem cell from a myeloid malignancy to generate not only myeloid but also lymphoid cells has not been previously reported to such an extensive degree (29, 30).
The unique properties of MF splenic MF-SCs described here have not been extensively reported in other hematological malignancies. The distinctive characteristics of splenic and marrow HSCs have, however, recently been examined in an inducible transgenic mouse model of another MPN, chronic myeloid leukemia (CML). These studies indicated that the expansion of primitive leukemic cells occurred in the spleen and to a far lesser degree in the bone marrow and documented qualitative differences including imatinib resistance that distinguished spleen from marrow CML stem cells (31). The spleen appears to become a major site of leukemia-initiating cells in this transgenic CML model, perhaps due to either cell-autonomous factors or external influences unique to the spleen.
Although symptoms related to splenomegaly frequently dominate the clinical course of patients with MF, the role that the spleen plays in the origins of MF or disease progression has remained the subject of considerable speculation. Some investigators have demonstrated that additional cytogenetic abnormalities occur within splenic cells that are not present in cells isolated from the PB or marrow of these patients (32), indicating that disease progression might be favored by the splenic microenvironment; others have reported that splenectomy is associated with the transition to leukemia, suggesting that the spleen might somehow impair evolution to AML (33). From the studies reported, here we conclude that the spleen in MF patients influences the fate of MF-SCs and might thereby affect the natural history of MF. These findings raise the possibility that therapies capable of affecting the genetic abnormality and the function of the MF spleen microenvironment components might alter the behavior of MF-SCs. Such therapeutic alternative approaches — in which the tumor microenvironment serves as a possible target to be used in combination with agents that affect malignant cells — have been suggested by others in order to overcome tumor-protective niches provided by the tumor microenvironment (34).
The persistent high level of human malignant cell chimerism observed with the transplantation of splenic MF CD34+ cells into NSG recipients provides an opportunity to create an animal model that closely resembles the early stages of PMF (35). The creation of such a model requires the persistence of the engrafted cells for a protracted period of time so as to enable one to monitor the evolution of a chronic MPN, which is now possible due to the more prolonged lifespan of NSG mice as compared with other strains of NOD/SCID mice. The animals receiving transplants of splenic MF CD34+ cells have many features of human PMF, including progressive splenomegaly, prolonged survival, as well as multilineage hematopoietic engraftment and constitutive mobilization of cells into the PB. The animals to date have not been observed to have any ill effects that can be attributed to the persistent human malignant cell chimerism, which is consistent with early stages of the MF, during which patients are not infrequently asymptomatic. In recipient mice after 9 months following transplantation, there was no evidence of either marrow or splenic fibrosis, although there was considerable human cell chimerism. Early forms of PMF in humans can be associated with cellular atypia and characteristic genetic abnormalities without the development of fibrosis (35). The lack of marrow fibrosis observed in recipient mice can also be attributed to the need for greater periods of time for the reactive fibrosis to develop, a lack of cross-reactivity between the responsible human cytokines and the recipient marrow stromal cells, or the limited numbers of donor-derived marrow megakaryocytes being observed. MF megakaryocytes are thought to be the principal source of fibrogenic cytokines that lead to marrow fibrosis and megakaryocyte hyperplasia in MF (36–38). Splenic CD34+ cells have been shown to retain the capacity to differentiate into CD41a+ and CD61+ megakaryocytes in vitro in the presence of thrombopoietin (TPO; data not shown), indicating that the absence of megakaryocytic hyperplasia in the recipient mice is not due to an intrinsic abnormality in splenic MF-SCs but likely represents the consequence of the absence of appropriate environmental cue in this xenogeneic system.
The detection of donor-derived cells within the thymus of recipient mice was unanticipated but is consistent with the potential of MF splenic CD34+ cells to generate significant numbers of T cells. The presence of similar populations of cells in the thymus of MF patients has not to date been systematically examined due to the lack of access to thymic tissue from MF patients after necropsy. The localization of human T and B cells derived from MF splenic CD34+ cells belonging to the malignant clone to nodules within the white pulp of the recipient mice is likely the consequence of the murine splenic white pulp providing an environment that favors lymphopoiesis. The presence of cells belonging to the various myeloid lineages within the marrow and red pulp of recipient spleen more closely resembles the histopathological picture that is observed in PMF spleens.
Myeloid and lymphoid cells recovered from both the marrow and spleens of the mice receiving splenic MF CD34+ cell grafts were JAK2V617F+, suggesting that the JAK2V617F mutation initially occurs in a true HSC. We and others have previously reported the existence of additional genetic events such as marker chromosomal abnormalities that can precede the acquisition of JAK2V617F during the multistep pathogenesis of an MPN (39, 40). In this study, we further extended this observation to true HSCs; the JAK2V617F allele burden in SRCs of SP2 was less than 50%, suggesting that a population of SP2 SRCs was characterized by wild-type JAK2, while all the hematopoietic cells derived from the SRC of SP2 possessed del (13q). Furthermore, the donor-derived cells had multiple copies of del (13q), while cells with a single copy of this deletion were present in the patient’s own blood cells, providing further evidence that the spleen promotes the expansion of specific clones and thereby plays a role in disease progression. We therefore hypothesize that distinctive hematopoietic microenvironments (HMs) are present in the spleen and BM of MF patients that differentially influence MF HSC/HPC proliferation and trafficking, thereby determining disease phenotype and disease progression to a greater extent than previously appreciated.
In conclusion, our data clearly show that the stem cells that reside within the spleens of MF patients have both functional and genetic properties that differ from PB HSCs from the same patient. The splenic MF-SC might be an important source of clones leading to disease progression and leukemic transformation. The model for MF described in this report will likely prove useful in further defining the importance of the interaction between MF-SC and the microenvironment in determining the biology of MF and might be useful in identifying novel targets for therapeutic interventions. In addition, the significant degree of human chimerism achieved in NSG mice with MF splenic cells might provide a unique opportunity to further characterize the MF-SC and to evaluate the effects of potential novel therapeutic strategies on the natural history of PMF as well as the MF-SC.
Methods
Patient specimens and cell preparation
Surgically removed spleens were obtained from patients with advanced forms of MF who were undergoing therapeutic splenectomy. The disease stage for each patient was determined using the Dynamic International Prognostic Scoring System (DIPSS) (41). Single-cell preparations were prepared according to the method of Barosi and coworkers (42) from the spleens of 8 patients with PMF or PV/ET-related MF who fulfilled the WHO diagnostic criteria (ref. 43 and Table 1). The experiments performed with cells isolated from each patient are indicated in Supplemental Table 2. PB was collected from 7 of these patients at the time of splenectomy. Spleen or PB MNCs were isolated by density gradient centrifugation of splenic and PB single-cell suspensions using Ficoll-Paque (GE Healthcare Life Sciences). CD34+ cells were selected from these MNCs using a CD34+ cell selection kit (Stemcell Technologies). The purity of the CD34+ cell population was analyzed using a FACSCanto Flow Cytometer (BD). CD34+ cells with purity of ≥95% were used in all experiments.
JAK2V617F mutational analysis and cytogenetic and FISH analyses
The JAK2V617F status of the MF patients was determined by analyzing the PB granulocytes of patients utilizing a real-time allele-specific polymerase chain reaction (AS-PCR) assay previously described (24, 40). Cytogenetic and FISH analyses were performed as previously described (39, 44).
Flow cytometric analysis of splenic and PB cells
Splenic and PB MNCs were labeled with anti-human CD34 mAb conjugated to allophycocyanin (APC), and the percentage of cells expressing CD34 was determined flow cytometrically. Isolated splenic and PB CD34+ cells were labeled with anti-human CD34 mAb conjugated to APC and anti-human CD90 mAb conjugated to FITC, anti-human CD38 mAb conjugated to FITC, anti-human CXCR4 mAbs conjugated to phycoerythrin (PE), anti-human CD47 mAb conjugated to FITC, anti-human CD44 mAbs conjugated to PE, or anti-human CD49d mAbs conjugated to PE in order to determine the phenotype of PB and splenic CD34+ cells. All mAbs were purchased from BD Biosciences — Pharmingen. Each analysis was paired with a corresponding matched isotype control. Immediately prior to flow cytometric analysis, 1 μg/ml propidium iodide (PI; Sigma-Aldrich) was added in order to exclude nonviable cells. Cells were analyzed flow cytometrically, and at least 30,000 viable MNCs or 10,000 viable CD34+ cells were acquired from each sample (CellQuest software, BD).
In order to analyze the cell cycle status of splenic or PB MF CD34+ cells, these cells were fixed in chilled 70% ethanol at 4°C for 2 hours and washed with PBS. Afterward, cells were incubated with RNase A (1 mg/ml) at 37°C for 30 minutes, and then cells were incubated with PI at 4°C for an additional 30 minutes. The cell cycle status of these cells were then analyzed flow cytometrically.
HPC assays
Splenic or PB MF MNCs were assayed in semisolid medium as described previously (45). Briefly, 5 × 103 cells were plated in duplicate culture dishes containing 1 ml IMDM with 1.1% methylcellulose, 30% FBS, 5 × 10–5 mol/l 2-mercaptoethanol (Stemcell Technologies), to which SCF, TPO, IL-3, IL-6, G-CSF, each at 100 ng/ml, and 4 U/ml erythropoietin (Amgen) were added. Colonies were enumerated after 12–14 days of incubation. Individual colonies were plucked and analyzed for JA2V617F as previously described (45), and the presence of a marker chromosomal abnormality was sought using FISH as described above. The percentage of JAK2V617F-positive colonies or colonies having chromosomal abnormalities was then determined.
NSG mice marrow repopulating cell (SRC) assay
NSG mice were purchased from the Jackson Laboratory. Splenic or PB PMF CD34+ cells (2 × 103, 2 × 104, 2 × 105, and 2 × 106 CD34+ cells/mouse) were transplanted via the tail vein into 8- to 9-week-old sublethally irradiated (240 cGy) NSG mice. Four to 9 months after transplantation, mice were sacrificed, and cells were recovered from the BM, spleens, livers, thymi, and the PB of recipient mice. The presence of human CD45+, CD33+ or CD14+, Gly A+, CD41a+, CD19+, CD3+, and CD34+ cells was determined by mAb staining and flow cytometric analysis. Each analysis was paired with a corresponding matched isotype control. Cells obtained from mice not receiving human cell transplants were analyzed in a similar fashion in parallel in order to exclude the possibility of false-positive immunostaining. The antibodies utilized did not cross-react with murine cells. We considered human engraftment to have occurred if human CD45+ cells were present at ≥0.1% of the nucleated cells in the BM or spleen of a recipient mouse.
Human CD45+ cells and cells belonging to various hematopoietic lineage cells in the BM and spleen of the recipient mice were isolated by mAb staining and cell sorting using an Aria BSL2 (BD). The purity of the human cells was shown to be ≥95% when analyzed flow cytometrically. The JAK2V617F allele burden of the sorted human cells was determined by real-time AS-PCR as previously described (24, 40). Selected human cells were further analyzed for marker chromosomal abnormalities using FISH (39, 44).
For morphological and immunohistochemical evaluation, sections were obtained from formalin-fixed, decalcified, paraffin-embedded femurs and formalin-fixed, paraffin-embedded spleen tissue from mice at 5 and 9 months after transplantation. These sections were stained with H&E as well as antibodies specific for human CD34, CD45, CD20, CD5, glycophorin C, lysozyme, and CD61 or mouse CD34, TER119, B220, CD3, and lysozyme to evaluate the human origin of the cells as well as the hematopoietic cell lineages represented (46). The source of these reagents is provided in Supplemental Table 5. Following appropriate antigen retrieval, immunohistochemical stains were performed using an automated immunostainer (Bond Max). Additionally, reticulin and trichrome special stains were performed on the marrow and spleen sections to evaluate for the presence of fibrosis.
The ability of human cells in the spleen and marrows of primary recipient mice to repopulate a second immune-deficient mouse was assessed in order to further examine the relationship between the characteristics of these MF SRCs and true HSCs. Whole BM cells or splenic cells from chimeric primary mice engrafted with splenic or PB PMF CD34+ cells were harvested and injected into secondary NSG recipients. BM cells or splenic cells from a single chimeric mouse were transplanted into a single secondary mouse. Three months after transplantation, mice were sacrificed, and cells were recovered from the BM and spleens and analyzed using a panel of mAbs as described above for evidence of human cell engraftment and their ability to generate multiple hematopoietic lineages.
Microarray analysis
Total RNA extraction.
Splenic and PB CD34+ cells from patients 1, 3, and 4 were isolated by mAb staining and cell sorting using an Aria BSL2 (BD). The purity of the isolated CD34+ cells (≥95%) was determined flow cytometrically. Total RNA was isolated from the purified MF splenic and PB CD34+ cells using standard RNA extraction procedures (NucleoSpin RNA II, Macherey-Nagel).
Linear T7-based amplification of RNA.
In order to generate Cy3-labeled cRNA, 10 ng of each individual RNA sample was amplified and labeled using the Agilent Low Input Quick Amp Labeling Kit (Agilent Technologies) according to the manufacturer’s protocol. The cRNA yields and dye-incorporation rate were measured with a ND-1000 Spectrophotometer (NanoDrop Technologies).
Hybridization to Agilent Whole Genome Oligo Microarrays.
The hybridization procedure was performed using the Agilent Gene Expression Hybridization Kit (Agilent Technologies). Briefly, 600 ng of Cy3-labeled fragmented cRNA in hybridization buffer was hybridized overnight (17 hours, 65°C) to Agilent Whole Human Genome Oligo Microarrays 8 × 60K using the hybridization chamber and oven recommended by the manufacturer. The microarrays were washed initially with the Agilent Gene Expression Washing Buffer 1 for 1 minute at room temperature, followed by a second wash with preheated Agilent Gene Expression Wash Buffer 2 (37°C) for 1 minute. The third washing step was performed with acetonitrile.
Scanning results.
Fluorescence signals from the hybridized Agilent Microarrays were detected using a Microarray Scanner System (Agilent Technologies).
Image and data analysis.
Agilent Feature Extraction Software (FES) was used to read out and process the microarray image files. For determination of differential gene expression profiles, FES-derived output data files were further analyzed using the Resolver gene expression data analysis system (Rosetta Biosoftware). This software is able to compare two single intensity profiles as a ratio. Resolver Software allows the export of a gene list with all normalized sample/control log10 ratios and fold changes, sequence descriptions, P values, etc., referred to as gene ratio list (of all genes). Note that the ratios are calculated by dividing the signal intensity of splenic CD34+ cells by that of their PB counterparts; for example, a “10-fold change” in the gene ratio lists indicates 10-fold-higher gene expression in the PB CD34+ cells as compared with the splenic CD34+ cells. Well-characterized differentially expressed genes that were defined as genes with a fold change >2 and P value <0.01 between MF splenic and PB CD34+ cells are summarized in Supplemental Tables 3 and 4.
Statistics
Results are reported as the mean ± SD of data obtained from 3–7 individual experiments. Statistical significance was determined using 2 tailed Student’s t tests or paired-sample t tests. All P values were 2 sided, and P values less than 0.05 were considered significant unless otherwise specified. The limiting-dilution analyses of SRCs were performed using Poisson statistics with L-Calc software (Stemcell Technologies) as previously described (47).
Study approval
All patients provided written informed consent under a protocol approved by the Institutional Review Board of the Mount Sinai School of Medicine (MSSM) or University of Utah School of Medicine or Geisinger/Hazleton Cancer Center. All animal experiments were approved by the Animal Care Committee of the MSSM.
Supplementary Material
Acknowledgments
This study was support by grants from National Cancer Institute (1P01CA108671) to R. Hoffman. We thank Yifang Liu for performing immunohistochemical staining. Camelia Iancu-Rubin is acknowledged for critical reading of the manuscript. We also thank Joann Arandela, Michael Feldman, and Goar Mosoyan for their assistance in processing the surgically excised spleens.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):3888–3899. doi:10.1172/JCI64397.
References
- 1.Hoffman R, Ravandi F. Hematology: Basic Principles and Practice. 2004. Idiopathic myelofibrosis. In: Hoffman R, et al., eds. pp. 1255–1276. 4th ed. New York, New York, USA: Churchill Livingstone; [Google Scholar]
- 2.Kralovics R, Skoda RC. Molecular pathogenesis of Philadelphia chromosome negative myeloproliferative disorders. Blood Rev. 2005;19(1):1–13. doi: 10.1016/j.blre.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Mesa RA. Clinical and scientific advances in the Philadelphia-chromosome negative chronic myeloproliferative disorders. Int J Hematol. 2002;76(suppl 2):193–203. doi: 10.1007/BF03165117. [DOI] [PubMed] [Google Scholar]
- 4. Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002. 100 13 4272 –4290 10.1182/blood-2001-12-0349 [DOI] [PubMed] [Google Scholar]
- 5.Prchal JF, Prchal JT. Molecular basis for polycythemia. Curr Opin Hematol. 1999;6(2):100–109. doi: 10.1097/00062752-199903000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Zhang B, Lewis SM. The splenomegaly of myeloproliferative and lymphoproliferative disorders: splenic cellularity and vascularity. Eur J Haematol. 1989;43(1):63–66. doi: 10.1111/j.1600-0609.1989.tb01253.x. [DOI] [PubMed] [Google Scholar]
- 7.Barosi G, et al. Spleen neoangiogenesis in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 2004;124(5):618–625. doi: 10.1111/j.1365-2141.2004.04829.x. [DOI] [PubMed] [Google Scholar]
- 8.Raaijmakers MH, Scadden DT. Evolving concepts on the microenvironmental niche for hematopoietic stem cells. Curr Opin Hematol. 2008;15(4):301–306. doi: 10.1097/MOH.0b013e328303e14c. [DOI] [PubMed] [Google Scholar]
- 9.Frisch BJ, Porter RL, Calvi LM. Hematopoietic niche and bone meet. Curr Opin Support Palliat Care. 2008;2(3):211–217. doi: 10.1097/SPC.0b013e32830d5c12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrett RW, Emerson SG. Bone and blood vessels: the hard and the soft of hematopoietic stem cell niches. Cell Stem Cell. 2009;4(6):503–506. doi: 10.1016/j.stem.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Renström J, Kröger M, Peschel C, Oostendorp RA. How the niche regulates hematopoietic stem cells. Chem Biol Interact. 2010;184(1–2):7–15. doi: 10.1016/j.cbi.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2009;449(7162):557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 13.McGrail DJ, Ghosh D, Quach ND, Dawson MR. Differential mechanical response of mesenchymal stem cells and fibroblasts to tumor-secreted soluble factors. PLoS One. 2012;7(3):e33248. doi: 10.1371/journal.pone.0033248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klopp AH, et al. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007;67(24):11687–11695. doi: 10.1158/0008-5472.CAN-07-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James C, et al. The hematopoietic stem cell compartment of JAK2V617F-positive myeloproliferative disorders is a reflection of disease heterogeneity. Blood. 2008;112(6):2429–2438. doi: 10.1182/blood-2008-02-137877. [DOI] [PubMed] [Google Scholar]
- 16.Pieri L, Guglielmelli P, Bogani C, Bosi A, Vannucchi AM, Myeloproliferative Disorders Research Consortium (MPD-RC) Mesenchymal stem cells from JAK2 (V617F) mutant patients with primary myelofibrosis do not harbor JAK2 mutant allele. Leuk Res. 2008;32(3):516–517. doi: 10.1016/j.leukres.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 17.Bacher U, et al. Bone marrow mesenchymal stromal cells remain of recipient origin after allogeneic SCT and do not harbor the JAK2V617F mutation in patients with myelofibrosis. Clin Exp Med. 2010;10(3):205–208. doi: 10.1007/s10238-009-0058-9. [DOI] [PubMed] [Google Scholar]
- 18.Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108(9):3128–3134. doi: 10.1182/blood-2006-04-017392. [DOI] [PubMed] [Google Scholar]
- 19.Delhommeau F, et al. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. . Blood. 2007;109(1):71–77. doi: 10.1182/blood-2006-03-007146. [DOI] [PubMed] [Google Scholar]
- 20.Conneally E, Cashman J, Petzer A, Eaves C. Expansion in vitro of transplantable human cord blood stem cells demonstrated using a quantitative assay of their lympho-myeloid repopulating activity in nonobese diabeticscid/scid mice. Proc Natl Acad Sci U S A. 1997;94(18):9836–9841. doi: 10.1073/pnas.94.18.9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larochelle A, et al. Identification of primitive human hematopoietic cells capable of repopulating NOD/SCID mouse bone marrow: implications for gene therapy. Nat Med. 1996;2(12):1329–1337. doi: 10.1038/nm1296-1329. [DOI] [PubMed] [Google Scholar]
- 22.Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A. 1997;94(10):5320–5325. doi: 10.1073/pnas.94.10.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziegler BL, et al. KDR receptor: a key marker defining hematopoietic stem cells. Science. 1999;285(5433):1553–1558. doi: 10.1126/science.285.5433.1553. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, et al. Sequential treatment of CD34+ cells from patients with primary myelofibrosis with chromatin-modifying agents eliminate JAK2V617F-positive NOD/SCID marrow repopulating cells. Blood. 2010;116(26):5972–5982. doi: 10.1182/blood-2010-02-269696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barosi G, et al. Diagnostic and clinical relevance of the number of circulating CD34(+) cells in myelofibrosis with myeloid metaplasia. Blood. 2001;98(12):3249–3255. doi: 10.1182/blood.V98.12.3249. [DOI] [PubMed] [Google Scholar]
- 26.Dick JE. Human stem cell assays in immune-deficient mice. Curr Opin Hematol. 1996;3(6):405–409. doi: 10.1097/00062752-199603060-00002. [DOI] [PubMed] [Google Scholar]
- 27.Ishii T, et al. Behavior of CD34+ cells isolated from patients with polycythemia vera in NOD/SCID mice. Exp Hematol. 2007;35(11):1633–1640. doi: 10.1016/j.exphem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Tsukamoto N, et al. Clonality in chronic myeloproliferative disorders defined by X-chromosome linked probes: demonstration of heterogeneity in lineage involvement. Br J Haematol. 1994;86(2):253–258. doi: 10.1111/j.1365-2141.1994.tb04723.x. [DOI] [PubMed] [Google Scholar]
- 29.Risueño RM, et al. Identification of T-lymphocytic leukemia–initiating stem cells residing in a small subset of patients with acute myeloid leukemic disease. Blood. 2011;117(26):7112–7120. doi: 10.1182/blood-2011-01-329078. [DOI] [PubMed] [Google Scholar]
- 30.Eisterer W, et al. Different subsets of primary chronic myeloid leukemia stem cells engraft immunodeficient mice and produce a model of the human disease. Leukemia. 2005;19(3):435–441. doi: 10.1038/sj.leu.2403649. [DOI] [PubMed] [Google Scholar]
- 31.Schemionek M, et al. Leukemic spleen cells are more potent than bone marrow-derived cells in a transgenic mouse model of CML. Leukemia. 2012;26(5):1030–1037. doi: 10.1038/leu.2011.366. [DOI] [PubMed] [Google Scholar]
- 32.Mesa RA, Li CY, Schroeder G, Tefferi A. Clinical correlates of splenic histopathology and splenic karyotype in myelofibrosis with myeloid metaplasia. Blood. 2001;97(11):3665–3667. doi: 10.1182/blood.V97.11.3665. [DOI] [PubMed] [Google Scholar]
- 33.Barosi G, et al. Splenectomy and risk of blast transformation in myelofibrosis with myeloid metaplasia. Italian Cooperative Study Group on Myeloid with Myeloid Metaplasia. Blood. 1998;91(10):3630–3636. [PubMed] [Google Scholar]
- 34.Cukierman E, Bassi D. The mesenchymal tumor microenvironment: a drug resistant niche. Cell Adh Migr. 2012;6(3):285–296. doi: 10.4161/cam.20210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiele J, Kvasnicka HM. The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Curr Hematol Malig Rep. 2009;4(1):33–40. doi: 10.1007/s11899-009-0005-6. [DOI] [PubMed] [Google Scholar]
- 36.Martyré MC, et al. Transforming growth factor-beta and megakaryocytes in the pathogenesis of idiopathic myelofibrosis. Br J Haematol. 1994;88(1):9–16. doi: 10.1111/j.1365-2141.1994.tb04970.x. [DOI] [PubMed] [Google Scholar]
- 37.Martyré MC, et al. Elevated levels of basic fibroblast growth factor in megakaryocytes and platelets from patients with idiopathic myelofibrosis. Br J Haematol. 1997;97(2):441–448. doi: 10.1046/j.1365-2141.1997.292671.x. [DOI] [PubMed] [Google Scholar]
- 38.Le Bousse-Kerdilès MC, Martyré MC. Myelofibrosis: pathogenesis of myelofibrosis with myeloid metaplasia: French INSERM Research Network on Myelofibrosis with Myeloid Metaplasia. Springer Semin Immunopathol. 1999;21(4):491–508. doi: 10.1007/BF00870307. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, et al. Clonal analyses define the relationships between chromosomal abnormalities and JAK2V617F in patients with Ph-negative myeloproliferative neoplasms. Exp Hematol. 2009;37(10):1194–1200. doi: 10.1016/j.exphem.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 40.Nussenzveig RH, et al. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35(1):32–38. doi: 10.1016/j.exphem.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 41.Passamonti F, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. doi: 10.1182/blood-2009-09-245837. [DOI] [PubMed] [Google Scholar]
- 42.Barosi G, et al. Spleen neoangiogenesis in patients with myelofibrosis with myeloid metaplasia. Br J Haematol. 2004;124(5):618–625. doi: 10.1111/j.1365-2141.2004.04829.x. [DOI] [PubMed] [Google Scholar]
- 43.Thiele J, Pierre R, Imbert M, Vardiman JW, Brunning RD, Glandrin G. World Health Organization Of Tumors Of Hematopoietic And Lymphoid Tissues. 2001. Chronic idiopathic myelofibrosis. In: Jaffe ES, Harris N, Stan N, Vardiman JW, eds. pp. 35–38. Washington, DC, USA: IARC Press; [Google Scholar]
- 44.Najfeld V, Coyle T, Berk PD. Transformation of polycythemia vera to acute nonlymphocytic leukemia accompanied by t (1;3)(p36;q21) karyotype. Cancer Genet Cytogenet. 1988;33(2):193–200. doi: 10.1016/0165-4608(88)90029-5. [DOI] [PubMed] [Google Scholar]
- 45.Li Z, et al. Erlotinib effectively inhibits JAK2V617F activity and polycythemia vera cell growth. J Biol Chem. 2007;282(6):3428–3432. doi: 10.1074/jbc.C600277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prakash S, Hoffman R, Barouk S, Wang YL, Knowles DM, Orazi A. Splenic extramedullary hematopoietic proliferation in Philadelphia chromosome-negative myeloproliferative neoplasms: heterogeneous morphology and cytological composition. Mod Pathol. 2012;25(6):815–827. doi: 10.1038/modpathol.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarry JE, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121(1):384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.