

Abstract
Hypertension occurs in approximately 30% of individuals in Western populations and is known to be a major cause of stroke, heart failure, and myocardial infarction. Despite this, the molecular etiology of hypertension remains poorly understood. In this issue of the JCI, Young et al. show that endoplasmic reticulum (ER) stress is an essential signaling event for angiotensin II–induced hypertension in cells of the central nervous system. This provides new insight into the molecular mechanisms that drive hypertension and suggests a potential target for future therapy.
In 1940, Irvine Page described a crystalline substance purified from the reaction of renin and renin activator, which he named angiotonin (1). Simultaneously, Braun-Menendez and coworkers identified a similar substance, which they called hypertensin (2). It is remarkable that three-quarters of a century later, we are still learning the actions of this molecule that ultimately came to be known as angiotensin II. It is now understood that this octapeptide has pleiotropic actions, including promotion of renal tubular sodium reuptake, aldosterone release, vasoconstriction, vascular remodeling, cardiac hypertrophy, cellular oxidative stress, and inflammation. Ongoing research is constantly refining and expanding this list. These actions allow survival during stresses such as dehydration or hemorrhage and have pathological roles in numerous diseases including hypertension and heart failure.
In addition to its peripheral effects, angiotensin II and its related peptides have potent actions on the central nervous system (3). Certain brain nuclei, predominantly in the hypothalamus and the brain stem, possess all of the components of the renin-angiotensin system (RAS) and produce angiotensin peptides locally (4). These nuclei are physically separated from the peripheral RAS by the blood-brain barrier. In addition, circulating angiotensin II can activate specialized regions of the brain that are adjacent to the cerebral ventricles and lack a well-formed blood-brain barrier. These “circumventricular organs” include the subfornical organ (SFO), the median eminence, the organum vasculosum of the lamina terminalis, and the area postrema (Figure 1). The predominant CNS effects of angiotensin II are to increase sympathetic outflow, promote vasopressin release, and stimulate thirst.
Figure 1. Role of central nervous system ER stress in hypertension.
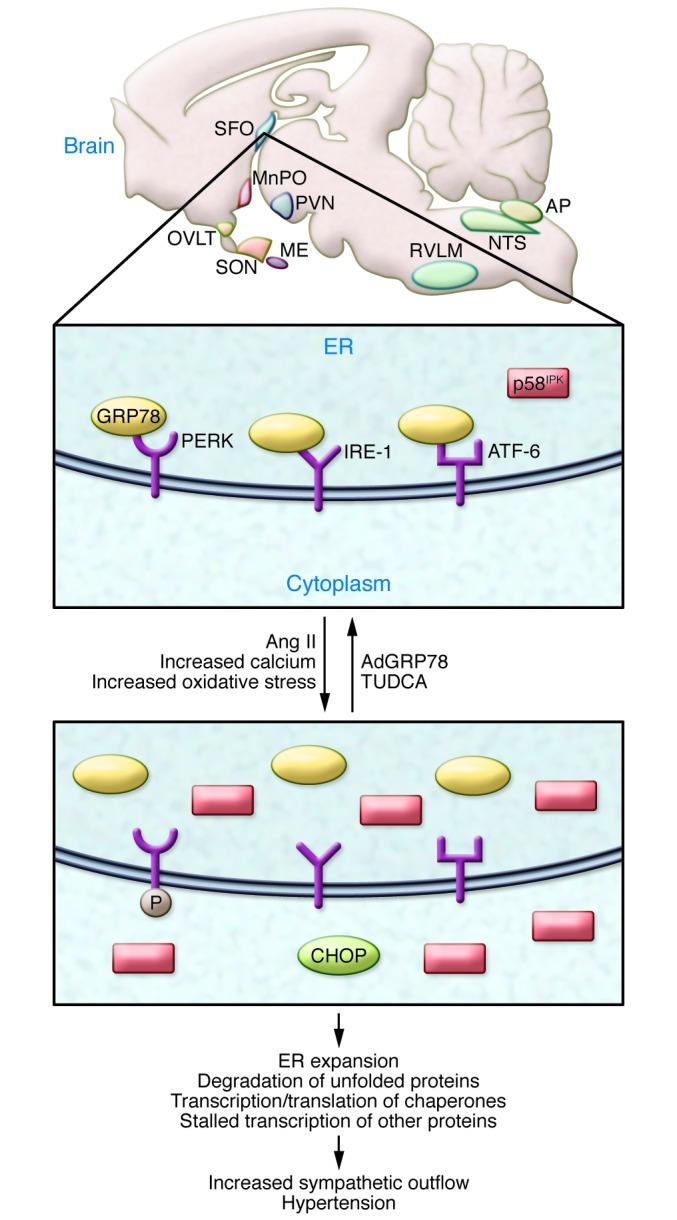
Angiotensin II acts on the circumventricular organs, including the subfornical organ (SFO), the organum vasculosum of the lamina terminalis (OVLT), the median eminence (ME), and the area postrema (AP). Also shown for orientation are the median preoptic nucleus (MnPO), the rostral ventral lateral medulla (RVLM), and the nucleus tractus solitarius (NTS). SON, supraoptic nucleus. In the SFO, angiotensin II promotes ER stress, documented by distension and disorganization of ER cisternae, increased inhibitor of interferon-induced and double-stranded RNA-activated protein kinase (p58IPK), increased C/EBP homologous protein (CHOP), and phosphorylation of PKR-like endoplasmic reticulum kinase (PERK). ER stress causes dissociation of 78 kDa glucose-regulated protein (GRP78) from PERK, inositol requiring protein (IRE-1), and activating reticulum factor-6 (ATF-6). Ultimately, ER stress in the SFO leads to increased sympathetic outflow and hypertension. Local administration of thapsigargin (TG), which also promotes ER stress, mimics these effects. Treatment with the chemical chaperone tauroursodeoxycholic acid (TUDCA) or overexpression of GRP78 prevents ER stress in the SFO and abrogates angiotensin II–induced hypertension.
A central role for angiotensin II
Angiotensin II promotes renal sodium reabsorption, adrenal aldosterone production, and vasoconstriction, all of which promote blood pressure elevation. In addition to these peripheral actions, substantial data indicate that the central effects of angiotensin II are also essential for the development of hypertension. Lesions in the anteroventral third ventricle (AV3V) region, which includes the inferior aspects of the lamina terminalis, prevent most forms of experimental hypertension (5). Intracerebroventricular injections of angiotensin II cause neuronal activation in the circumventricular organs, and these effects are blocked by angiotensin II receptor type 1 (AT1) antagonists (6). The SFO directs many of these central actions of angiotensin II via projections to the paraventricular nucleus of the hypothalamus (7), which in turn relays signals to the brain stem, increasing sympathetic outflow.
Despite the wealth of information regarding the central actions of angiotensin II, the cellular signaling events that that mediate its effects remain unclear. In this issue of the JCI, Young et al. (8) provide evidence for a unique mechanism of action of this octapeptide. They show that ER stress occurs in the SFO during angiotensin II–induced hypertension and, importantly, that blocking this prevents hypertension (Figure 1). Interestingly, the authors demonstrate that local administration of thapsigargin to pharmacologically induce ER stress increases blood pressure and renal sympathetic nerve activity (8). The idea that ER stress has such a signaling role in hypertension is extremely novel and provides much room for speculation, new experimentation, and possibly new avenues for therapy.
ER stress and hypertension
The ER is responsible for folding proteins in preparation for their transport through the Golgi and, ultimately, cellular secretion. When unfolded proteins accumulate in excess of the ER’s capacity, an unfolded protein response (UPR) is activated to restore homeostasis. The UPR involves expansion of ER membranes, accelerated degradation of unfolded proteins, increased translation of folding chaperones, and decreased transcription and translation of most other proteins. The UPR is initiated by activation of three ER transmembrane proteins: inositol-requiring enzyme 1 (IRE-1), protein kinase R–like ER kinase (PERK), and activating transcription factor–6 (ATF-6) (9). In unstressed cells, IRE-1, PERK, and ATF-6 bind to the chaperone GPR78, also known as binding immunoglobulin protein, or BiP. Upon UPR activation, GPR78 dissociates from these ER transmembrane proteins, initiating the UPR. Extended activation of the UPR is termed “ER stress” and can result in promotion of proapoptotic pathways mediated by proteins such as caspases, C/EBP homologous protein (CHOP), and JNK. Thus, the UPR and ER stress pathways exquisitely control cell fate decisions. Overactivation of ER stress contributes to diseases such as diabetes, viral infections, neurodegeneration, and cancer (reviewed in ref. 10).
Young et al. demonstrate upregulation of many ER stress pathway members in the SFO during development of hypertension, including GRP78, PERK, CHOP, and p58IPK (8). The last of these is a noncanonical ER stress protein that appears to act as a co-chaperone with GRP78 to promote degradation of translocationally stalled polypeptides, preventing their ER accumulation (11, 12). Using electron microscopy, the authors demonstrated that angiotensin II infusion causes ER membrane distension. Most importantly, Young et al. showed that inhibition of ER stress either by the chemical chaperone TUDCA or by local injection of an adenovirus expressing GRP78 (AdGRP78) prevented angiotensin II–induced hypertension. Thus, hypertension can be added to the list of diseases to which ER stress is a known contributor.
Stressing the details
One conundrum in studying the UPR is that both pro-survival and pro-death signals can be indiscriminately activated, and the fine-tuned mechanisms by which cell fate is determined are not well understood. This is relevant to the work by Young et al., because other biological stressors might also activate ER stress pathways to generate neuronal signals. It is possible that the timing of events for ER stress pathway regulation is important for the balance of life and death such that pathways activated early in the UPR promote cell survival, while those activated later lead to cell death. This temporal regulation has potential implications for the studies by Young et al. Their data show that p58IPK and p-PERK are upregulated at 7 days after angiotensin II infusion, while GRP78 and CHOP are upregulated at 14 days after infusion (8). A thorough analysis of the temporal regulation of all components of the UPR could provide insight into the homeostatic role of ER stress in the SFO response to angiotensin II.
While Young et al. present clear evidence for a role of SFO-localized ER stress in hypertension, several intriguing questions persist. Importantly, it is not clear how or why angiotensin II would initiate such a response and whether ER stress is actually mediated by the UPR in this setting. Several years ago, this same group made the important discovery that the superoxide radical mediates the effect of angiotensin II in the circumventricular organs (13). Their subsequent work showed that superoxide promotes neuronal calcium influx in response to angiotensin II (14). Indeed, the authors found that angiotensin II–induced superoxide production was blocked by TUDCA in freshly dispersed cells of the SFO and by AdGRP78 in the intact SFO. These findings suggest that ER stress might be upstream of oxidant stress in hypertension. In addition, ER stress is often associated with autophagy (15), and the relevance of this for cells in the SFO is unknown. Finally, ER stress in pathological settings is usually associated with apoptotic cell death. While Young et al. demonstrate upregulation of CHOP in the SFO of hypertensive mice, it is not clear which cells in the SFO are undergoing ER stress, or whether specific cell populations are dying.
Conclusions
In summary, the study by Young et al. provides us with a new paradigm, indicating that ER stress contributes to neuronal activation, the effects of angiotensin II, and the pathogenesis of hypertension. An obvious implication is that despite extensive study, we still do not understand the etiology of so-called essential hypertension, which affects 30% of the Western population and is a major cause of stroke, myocardial infarction, and heart failure. The idea that ER stress might be involved provides new avenues for investigation and may lead to new therapeutic approaches for this disease.
Acknowledgments
This work is supported by NIH R01HL039006, P01HL058000, P01HL095070, P01GM015431, R01HL105294-02, R01HL089466, R21DK95456, American Diabetes Association 7-11-AEC-13, and American Heart Association 12EIA827.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):3859–3861. doi:10.1172/JCI65173.
See the related article beginning on page 3960.
References
- 1.Page IH, Helmer OM. A crystalline pressor substance (angiotonin) resulting from the reaction between renin and renin-activator. J Exp Med. 1940;71(1):29–42. doi: 10.1084/jem.71.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braun-Menendez E, Fasciolo JC, Leloir LF, Munoz JM. The substance causing renal hypertension. J Physiol. 1940;98(3):283–298. doi: 10.1113/jphysiol.1940.sp003850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips MI, Sumners C. Angiotensin II in central nervous system physiology. Regul Pept. 1998;78(1–3):1–11. doi: 10.1016/S0167-0115(98)00122-0. [DOI] [PubMed] [Google Scholar]
- 4.Phillips MI, de Oliveira EM. Brain renin angiotensin in disease. J Mol Med (Berl). 2008;86(6):715–722. doi: 10.1007/s00109-008-0331-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brody M, et al. Perspectives in Nephrology and Hypertension. Critical role of the AV3V region in development and maintenance of experimental hypertension. In: Schmitt H, Meyers P, eds. New York, New York, USA: Wiley and Flammarion; 1978:76–84. [Google Scholar]
- 6.Lebrun CJ, Blume A, Herdegen T, Seifert K, Bravo R, Unger T. Angiotensin II induces a complex activation of transcription factors in the rat brain: expression of Fos, Jun and Krox proteins. Neuroscience. 1995;65(1):93–99. doi: 10.1016/0306-4522(94)00482-K. [DOI] [PubMed] [Google Scholar]
- 7.Bains JS, Potyok A, Ferguson AV. Angiotensin II actions in paraventricular nucleus: functional evidence for neurotransmitter role in efferents originating in subfornical organ. Brain Res. 1992;599(2):223–229. doi: 10.1016/0006-8993(92)90395-P. [DOI] [PubMed] [Google Scholar]
- 8.Young CN, et al. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest. 2012;122(11):3960–3964. doi: 10.1172/JCI64583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 10.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oyadomari S, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126(4):727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 12.Rutkowski DT, et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell. 2007;18(9):3681–3691. doi: 10.1091/mbc.E07-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95(2):210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- 14.Zimmerman MC, Sharma RV, Davisson RL. Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension. 2005;45(4):717–723. doi: 10.1161/01.HYP.0000153463.22621.5e. [DOI] [PubMed] [Google Scholar]
- 15.Yin JJ, et al. The role of autophagy in endoplasmic reticulum stress-induced pancreatic beta cell death. Autophagy. 2012;8(2):158–164. doi: 10.4161/auto.8.2.18807. [DOI] [PubMed] [Google Scholar]