

PMM2-CDG patients have phosphomannomutase (Pmm2) deficiency, with developmental and N-linked glycosylation defects attributed to depletion of mannose-1-phosphate and downstream lipid-linked oligosaccharides (LLOs). This, the first PMM2-CDG zebrafish model, shows, unexpectedly, that accumulation of the Pmm2 substrate mannose-6-phosphate explains LLO deficiency.
Abstract
Congenital disorder of glycosylation (PMM2-CDG) results from mutations in pmm2, which encodes the phosphomannomutase (Pmm) that converts mannose-6-phosphate (M6P) to mannose-1-phosphate (M1P). Patients have wide-spectrum clinical abnormalities associated with impaired protein N-glycosylation. Although it has been widely proposed that Pmm2 deficiency depletes M1P, a precursor of GDP-mannose, and consequently suppresses lipid-linked oligosaccharide (LLO) levels needed for N-glycosylation, these deficiencies have not been demonstrated in patients or any animal model. Here we report a morpholino-based PMM2-CDG model in zebrafish. Morphant embryos had developmental abnormalities consistent with PMM2-CDG patients, including craniofacial defects and impaired motility associated with altered motor neurogenesis within the spinal cord. Significantly, global N-linked glycosylation and LLO levels were reduced in pmm2 morphants. Although M1P and GDP-mannose were below reliable detection/quantification limits, Pmm2 depletion unexpectedly caused accumulation of M6P, shown earlier to promote LLO cleavage in vitro. In pmm2 morphants, the free glycan by-products of LLO cleavage increased nearly twofold. Suppression of the M6P-synthesizing enzyme mannose phosphate isomerase within the pmm2 background normalized M6P levels and certain aspects of the craniofacial phenotype and abrogated pmm2-dependent LLO cleavage. In summary, we report the first zebrafish model of PMM2-CDG and uncover novel cellular insights not possible with other systems, including an M6P accumulation mechanism for underglycosylation.
INTRODUCTION
The congenital disorders of glycosylation (CDGs) are a group of heterogeneous, hypomorphic, inherited diseases characterized by deficient N-glycosylation, typically detected by underglycosylation of serum glycoproteins (Jaeken and Matthijs, 2001; Marquardt and Freeze, 2001). Traditionally divided into two groups, the type I CDGs are classified by genetic defects at loci known to participate in the biosynthesis of the lipid-linked oligosaccharide (LLO) glucose3mannose9N-acetylglucosamine2-pyrophosphate-dolichol (G3M9Gn2-P-P-Dol), the glycan donor for N-glycosylation. This includes loci encoding proteins and enzymes involved in sugar and lipid metabolism, endoplasmic reticulum (ER)-localized glycosyltransferases, and proteins that facilitate utilization of substrates (Haeuptle and Hennet, 2009). First recognized in the clinic by Jaeken in the 1980s, the most common subtype of CDG is caused by deficiency of the metabolic enzyme phosphomannomutase 2 (Pmm2; Matthijs et al., 1997). This enzyme converts mannose-6-phosphate (M6P) to mannose-1-phosphate (M1P), the substrate needed for the synthesis of GDP-mannose and the eventual production of LLO. Designated PMM2-CDG (this subtype was formerly referred to as CDG-Ia), patients with this disease exhibit a constellation of abnormalities, including psychomotor and mental retardation, cerebellar atrophy, peripheral neuropathy, hypotonia, and ataxia (de Lonlay et al., 2001; Coman et al., 2008; Freeze et al., 2012). There are no current therapies for PMM2-CDG, and little is known about the relationship between the underglycosylation of proteins and the clinical features of the disease (Freeze, 2001, 2009).
Our limited understanding of type I CDG pathophysiology can be attributed to the paucity of suitable animal models for these diseases. Although many of the predicted CDG biochemical phenotypes have been confirmed by analyses of LLO and glycoprotein synthesis in cultured fibroblasts and leukocytes from patients, such culture systems fail to recapitulate the more complex physiological relationships that exist within the entire organism, yielding inconsistent results (Gao et al., 2005). These limitations have hampered evaluation of potential and actual therapeutic treatments for type I CDGs. For example, dietary mannose supplementation, which is expected to increase intracellular M6P concentrations and drive M1P synthesis by mass action, is clinically beneficial for MPI-CDG (also known as CDG-Ib) patients but not for PMM2-CDG patients. A mouse line completely lacking mannose phosphoisomerase (MPI), the enzyme responsible for the generation of M6P from fructose 6-phosphate (F6P), is an embryonic lethal (DeRossi et al., 2006). Attempts to generate viable mouse models for PMM2-CDG, the most common form of CDG, have also been particularly challenging. Complete knockout of Pmm2, or knock-in of the most common human PMM2-CDG allele, R137H, into this null background resulted in early embryonic lethality (Thiel et al., 2006). In contrast, knock-in of another common allele, F118L, resulted in only mild loss of enzymatic activity and no detectable phenotypes (Schneider et al., 2012). Compound heterozygotes with both the R137H and F118L alleles, however, survived to embryonic day 9.5 (Schneider et al., 2012). These embryos had reduced staining with wheat germ agglutinin, a plant lectin able to bind to sialic acid and N-acetylglucosamine residues and therefore capable of detecting several classes of glycoconjugates, including N-linked glycans. Strikingly, when the water given to the pregnant dams was supplemented with mannose, many of these embryos were born live, survived past weaning, and regained normal wheat germ agglutinin staining patterns. These data suggest that the R137H/F118L embryos had glycosylation deficiencies that were reversible by maternal mannose supplementation. Despite these findings, a tractable animal model that closely replicates the genetic, developmental, and glycosylation deficiencies documented for PMM2-CDG patients is still needed for a thorough biochemical and behavioral evaluation of the disease mechanisms and experimental treatments.
To overcome the present limitations of PMM2-CDG mouse models, we took advantage of the genetic and experimental attributes of zebrafish (Danio rerio), an excellent vertebrate system for both the study of embryonic development and the modeling of genetic diseases. We recently described a model of MPI-CDG (Chu et al., 2012), demonstrating the utility of this system for modeling CDG. Here we report that suppression of pmm2 expression using a morpholino-based approach resulted in morphant embryos that exhibit several cellular and molecular phenotypes consistent with the human disease, including altered craniofacial cartilage development, impaired motility, and reduced protein N-glycosylation. We also provide direct evidence for reduced LLO levels in the morphants. Surprisingly, although our analytical methods were too insensitive to reliably detect M1P and GDP-mannose in zebrafish, an increase in the level of the Pmm2 substrate M6P was consistently observed. Additional genetic and biochemical experiments revealed that loss of N-glycosylation in the pmm2 morphants was best explained by accelerated hydrolysis of LLO triggered by the accumulated M6P. Taken together, these results yield new insights into the cellular and molecular basis of type I CDGs, lend support to the concept of flux-based therapy, and further highlight the utility of the zebrafish system in the investigation of inherited metabolic disorders.
RESULTS
Pmm2 activity is shown to be significantly reduced in zebrafish embryos by using a morpholino-based strategy
To inhibit pmm2 expression, we tested two different antisense morpholinos (MOs) directed against zebrafish pmm2—one that inhibits mRNA translation (translation blocker [TB MO]) and one that inhibits mRNA splicing (splice blocker [SB MO]; Figure 1A). Over a 0–1.77 μM concentration range, the degree of knockdown with each morpholino was assessed by either reverse transcription (RT)-PCR (SB MO) or Pmm2 activity assays (SB and TB MOs). The SB MO caused a linear decrease in the amount of normally spliced pmm2 transcripts, with a maximal reduction at 0.52 μM (Figure 1B). Although a decrease in normally spliced transcript is often accompanied by a concomitant increase in an alternate splice product (either larger or smaller, depending on the junction targeted), we were unable to detect the larger intron-containing transcript after pmm2 inhibition. To assess whether the lack of this product after PCR amplification was due to the large amplicon size, we probed its presence with two additional sets of primers (see Materials and Methods for details) whose efficacies were previously validated using genomic DNA. In light of the fact that neither primer set amplified the alternate splice product, we conclude that this SB MO caused rapid elimination of the full-length pmm2 mRNA. Surprisingly and for reasons that are unclear, pmm2 transcript abundance increased with higher MO concentration (Figure 1B). To determine the effect of MO inhibition on protein expression, we measured Pmm2 activity in lysates using a coupled assay. Pmm2 activity was reduced to 33.4% of control with the SB MO, which paralleled the RT-PCR results, and to 58% of control with the TB MO. However, like the corresponding message, the enzymatic activity of Pmm2 could not be completely suppressed, and it rebounded at the highest SB MO concentration, suggesting the presence of compensatory mechanisms (Figure 1C).
FIGURE 1:
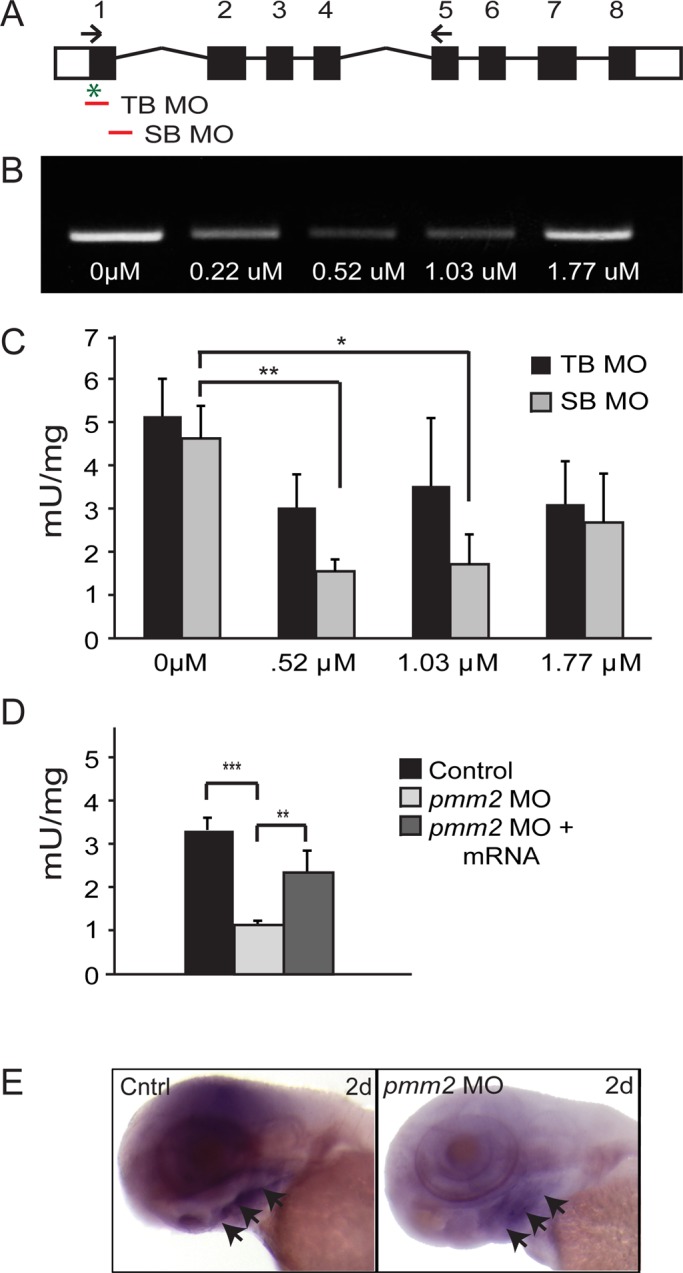
Injection of pmm2 directed antisense morpholinos into zebrafish embryos reduces pmm2 transcript abundance and activity. (A) Schematic representation of pmm2 gene. The positions of TB and SB MOs are indicated, as is the transcriptional start site (*). Arrows indicate the position of one set of primers used to assess transcript abundance (see Materials and Methods). (B) RT-PCR of 3-dpf embryos after injection of indicated concentrations of SB MO. (C) Pmm2 activity measurements of 3-dpf embryos after injection of either the TB or SB MO. n = 4 experiments. Throughout this article, *p < 0.05 and **p < 0.01 (Student's t test). (D) Measurement of Pmm2 and Mpi activity in 3-dpf control embryos, embryos injected with 0.52 μM SB, and mRNA-rescued morphants. n = 25 experiments. (E) In situ analysis of pmm2 expression in control and pmm2 morphant (MO) embryos. Arrowheads indicate ventral concentration of staining.
Based on these considerations, 0.52 μM SB MO was used for the remainder of the study. At this concentration, Pmm2 activity was stably reduced for the first 4 d of development (unpublished data). Coinjection of the SB MO and full-length pmm2 mRNA (whose sequence is nonhomologous to the SB MO) rescued Pmm2 activity to near-normal levels, demonstrating both the specificity of the SB MO and the efficacy of the injected mRNA (Figure 1D). In situ hybridization analyses of pmm2 revealed global expression throughout the heads of control embryos, with transcript concentration notably increased in ventral regions (Figure 1E; arrows highlight ventral concentration). Low levels of pmm2 expression were also noted in embryonic spinal cords (unpublished data). Consistent with decreased enzyme activity, both transcript patterns were clearly reduced in embryos injected with the pmm2 SB MO (Figure 1E).
pmm2 morphants exhibit alterations in craniofacial cartilage development
Motivated by the fact that PMM2-CDG patients exhibit craniofacial and skeletal malformations, we were prompted to analyze developing cartilage in pmm2 morphants in detail by staining with Alcian blue, a dye that binds acidic components of the extracellular matrix. pmm2 morphant embryos displayed several defects in cartilage morphogenesis, including protracted Meckel's (M) cartilages, misshapen palatoquadrate (PQ) and ceratohyal (CH) structures, and kinked pectoral fins (Figure 2A). Each of these phenotypes was rescued by coinjection of pmm2 mRNA, suggesting that they were specific to Pmm2 reduction and not potential off-target MO effects. Furthermore, simultaneous suppression of pmm2 and p53 did not ameliorate the craniofacial phenotypes (unpublished data), indicating that the pmm2 MO does not induce these abnormalities by promoting nonspecific apoptosis. This is an important consideration, as several studies have shown that one common off-target effect of morpholinos is the induction of p53-directed apoptosis (Ekker, 2000; Bill et al., 2009).
FIGURE 2:

pmm2 morphants have dysmorphic craniofacial cartilage. (A) Alcian blue stains of 4-dpf control and pmm2 morphant embryos revealed altered size and shape of Meckel's cartilage, as well as the palatoquadrate and ceratohyal cartilages. These defects were rescued by coinjection of pmm2 mRNA. n = 30–50 embryos per condition over three experiments. Arrow shows kinked pectoral fins in the pmm2 morphants. (B) The length of individual structures was measured and normalized to a standard length (SL), which was set as the distance between the eyes. Individual cartilage measurements are outlined in the fish schematic. n = 30–50 embryos per condition over three experiments. Here and throughout this article, ***p < 0.001. (C) The 4-dpf WT, pmm2 morphant (MO), and pmm2 mRNA–rescued embryos were dissected and the cartilages mounted flat. Analysis of these preparations showed that morphant chondrocytes were rounder and more underintercalated compared with WT chondrocytes. This was rescued by coinjection of pmm2 mRNA. n = 12–15 embryos per condition in three experiments. (D) Chondrocyte shape was measured in each of the affected structures by determining the ratio of cell short axis to long axis. The closer this number is to 1, the rounder is the cell. n = 12–15 embryos per condition in three experiments.
Measurements of the cartilage structures showed that they were not only misshapen, but also shorter than control cartilages (Figure 2B). In addition, the distance between the M and CH cartilages was reduced by 35% in the pmm2 morphants. With the exception of the length of the CH cartilage, coinjection of pmm2 mRNA significantly improved the size and shape of the craniofacial cartilages (Figure 2B). The lack of CH recovery and partial PQ recovery may be accounted for by the fact that these structures finish developing after the M cartilage and may therefore be outside the window of mRNA rescue.
Although analyses of control and morphant flat-mounted cartilages showed no significant differences in the total number of cells (unpublished data), they revealed alterations in the morphology of morphant chondrocytes (Figure 2C). Unlike control chondrocytes, which were oblong in shape and had converged to form a single line of cells, morphant cells remained round and were arranged in multicellular layers, yielding a “cobblestone”-like appearance. To quantitatively measure these differences in cellular shape, we calculated the ratio between the short and long axes of individual chondrocytes. The ratio between these axes was significantly larger in the morphants (Figure 2D), reflecting increased cellular roundness. Again, coinjection of pmm2 mRNA restored the typical oblong cellular morphology of the morphant chondrocytes. To ask whether the alterations in cell shape reflected an early defect in chondrocyte maturation, we stained control and morphant embryos with peanut agglutinin (PNA), which labels prechondrocytic mesenchymal cells, and type II collagen, one of the earliest markers of chondrocyte differentiation. PNA is a lectin that binds the terminal disaccharide Gal-β(1-3)-GalNAc in O-linked glycans, and is therefore not directly dependent on the mannose-derived products of Pmm2. No differences in the intensity, distribution, or timing of the expression of either marker were detected between control and morphant chondrocytes, suggesting that initial aspects of differentiation are unaffected by pmm2 knockdown (unpublished data). Collectively these findings indicate that reduced Pmm2 activity is associated with changes in craniofacial cartilage formation resulting from altered chondrocyte morphogenesis.
pmm2 morphants exhibit multiple motility defects
Because PMM2-CDG patients exhibit hypotonia, ataxia, and delayed motor development, we investigated whether pmm2 morphants demonstrated motor system defects. These analyses revealed multiple deficits at two developmental stages in morphant embryos when compared with controls. By 1 d postfertilization (dpf), normal zebrafish embryos exhibit two different motility behaviors: spontaneous movements, consisting of slow, alternating tail flexures, and elicited movements, in which embryos respond to external stimuli with several rapid tail flexures. Analyses of 1-dpf embryos revealed a slight but significant decrease in the number of spontaneous tail curls generated by pmm2 morphants (11.0 vs. 8.2; Figure 3A). In addition, pmm2 morphants exhibited reduced response to touch at 1 dpf. Although both produced a similar number of tail curls following stimulation, 88% of control embryos responded with a complete curl that made contact with the body. In contrast, the majority of morphant embryos (75%) only flexed the tip of their tail, never touching the body. By 3 dpf, deficits in touch responsiveness within the pmm2 morphants were further illustrated by both aberrant escape and swimming behaviors.
FIGURE 3:
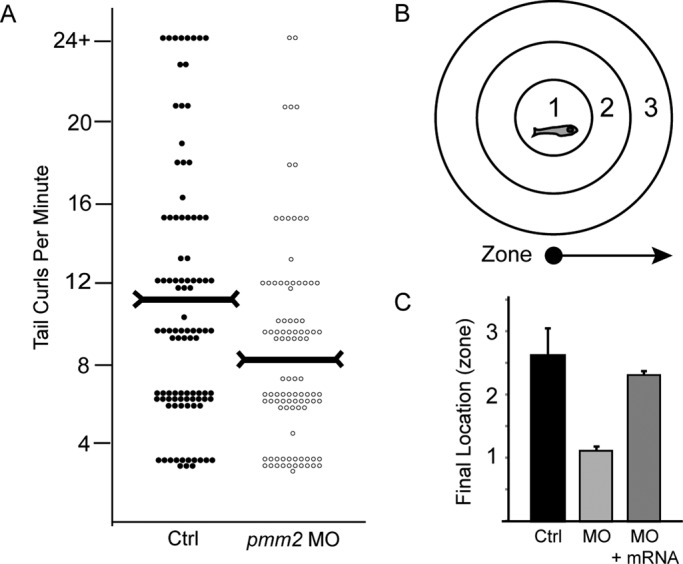
pmm2 morphants exhibit pronounced motility defects. (A) At 1 dpf the times that control and pmm2 morphant (MO) embryos spontaneously curled their tails in 1 min were counted and plotted. Each dot on the scatter plot represents one animal. Two hundred twenty-five embryos per condition were scored over five separate experiments. Horizontal markers indicate the median values. (B) At 3 dpf the swimming behaviors and “escape” responses of control, morphant, and pmm2 mRNA–rescued embryos were assessed by placing individual animals in the center of a Petri dish marked with three concentric rings. Embryos were lightly touched on the head and their behavior and final destination (zone 1, 2, or 3) recorded. (C) For each condition the average “final location” of 100 embryos from three experiments is graphed.
To quantify these behaviors, we placed 3-dpf embryos in the center of a plate marked with a series of concentric rings, which were used to assign swimming distances (Figure 3B). After stimulation, control embryos robustly swam away from the stimulus, ultimately crossing multiple rings. In contrast, morphant embryos typically swam in a circle toward the stimulus, remaining within the first ring. Both the early-stage (1 dpf) and late-stage (3 dpf) defects in elicited motility were significantly (in >50% of the embryos assayed) rescued by coinjection of pmm2 mRNA (Figure 3C). Taken together, these data suggest that reduction of pmm2 activity alters embryonic motility behaviors, possibly by affecting aspects of neural or muscular development.
Reduced motility of pmm2 morphants is associated with abnormal motoneuron development and spinal cord organization
Previous studies on zebrafish mutants with impaired glycosylation demonstrated altered migration, organization, and proliferation of multiple classes of motoneurons (Ohata et al., 2009; Song et al., 2010). In particular, the slytherin (srn) and towhead (twd) mutants, which carry distinct mutations in GDP-mannose-4,6-dehydratase (gmds)—an enzyme involved in synthesis of GDP-fucose—display increased motor neurogenesis and aberrant migration of motoneuron progenitors, respectively. To determine whether similar defects in neuronal development underlie the pmm2 motility phenotypes, we generated morphants within the hb9:GFP transgenic background, in which both primary and secondary motoneurons express green-fluorescent protein (GFP) under the control of the motoneuron-specific hb9 promoter (Flanagan-Steet et al., 2005).
Confocal analysis of control and morphant embryos at 1 and 3 dpf revealed an increase in the number of motoneurons within pmm2 morphant spinal cords, apparent both in the number of cell bodies and in the overall width of motoneuron occupancy within the spinal cord (Figure 4, A and B). Inhibition of Pmm2 activity did not obviously affect the developing skeletal muscle (as assessed with phalloidin staining) or motor axon trajectories and synaptic development. The increase in motoneuron number was rescued in >60% of the animals coinjected with pmm2 mRNA. Furthermore, morphological analysis of GFP-labeled cell bodies suggested that the increased motoneuron population was likely restricted to the secondary lineage. Although primary motoneurons, which are typically larger than secondary motoneurons, can be identified by both position and size, embryos were additionally stained with the secondary motoneuron marker zn-5. Analysis of zn-5 confirmed that increase in cell number primarily affected the population of secondary motoneurons (Figure 4, C and D).
FIGURE 4:

pmm2 morphant spinal cords contain increased numbers of secondary motoneurons. (A) Confocal images of control and pmm2 morphant (MO) embryos generated in the hb9:GFP background reveal increased numbers of motoneurons at 1 dpf (top) and 3 dpf (bottom) of development. Phalloidin staining of muscle fibers showed no differences in myotomal architecture between control and morphant embryos. White brackets indicate representative distances between the ventral spinal cord and the upper end of motoneuron occupancy. n = ∼30 embryos per condition. (B) Quantitation of the number of GFP-positive cell bodies per myotome at 1 dpf and width of motoneuron occupancy within both 1- and 3-dpf embryonic spinal cords demonstrated significant differences between control and pmm2 morphant (MO) embryos that were rescued by coinjection of pmm2 mRNA. n = ∼30 embryos per condition. (C) Whole-mount immunohistochemical stains of 3 dpf embryos using the secondary motoneuron marker zn5 show that the majority of neuronal increase occurs in the secondary motoneuron lineage of pmm2 morphant (MO) embryos. This increase is rescued by coinjection with pmm2 mRNA. Red brackets delineate the ventral edge of the spinal cord on upper limit of motoneuron occupancy. n = 22 embryos. (D) Schematic representation of increased numbers of motoneurons, which appears to primarily affect the secondary motoneuron (MN) population, represented as white cells, whereas the number of primary motoneurons (MN) is unaltered, represented as large green cells. The dotted arrow shows that the width of the spinal cord (SC) is similar between control (Cntrl), MO, and mRNA-rescued embryos.
Pmm2 deficiency in zebrafish results in global suppression of N-linked glycosylation
Hypoglycosylation of the serum glycoprotein transferrin is typically used in the clinical diagnosis of type I CDGs. In the absence of such a validated marker for zebrafish, we quantified the pool of N-glycans in protein fractions from entire animals by release with PNGase F, fluorophore-assisted carbohydrate electrophoresis (FACE) analysis, and normalization to protein content. Although no qualitative differences were detected in the profile of N-glycans, the size of the pool in five individual experiments was reduced on average to 86% of control after pmm2 knockdown (Figure 5A and Supplemental Figure S1A). These reductions of N-glycan levels were mitigated by rescue with pmm2 mRNA, arguing against an off-target effect of the pmm2 morpholino (Figure 5A). Thus it appears that N-glycan processing was not strongly affected by pmm2 knockdown—only the amount of glycan transferred to endogenous protein acceptors, which is highly reminiscent of PMM2-CDG patients.
FIGURE 5:

G3M9Gn2-P-P-Dol suppression and restoration in pmm2 morphants. G3M9Gn2-P-P-Dol and M6P were measured with the FACE technique from the same samples of zebrafish. Similar numbers of 4-dpf embryos (50–200, depending on need) were used for each comparison. All measurements were normalized to total protein, which did not vary significantly among the different genetically modified groups of fish. (A) Representative FACE gel for total N-glycans released from the proteins of yolk-containing zebrafish embryos. Total N-glycan amounts (normalized to control) are indicated above each lane. (B) Bar graph depicting the average percentage loss of total N-glycans in pmm2 morphants (with and without yolks) in two separate experiments (mean ± SEM), normalized to control embryos. Direct comparison of the control samples showed that yolk-free embryos had 49% of the N-glycan content of whole embryos. Related information is provided in Supplemental Figure S1. (C) Representative FACE images showing LLO glycans (M5Gn2 and G3M9Gn2 standards are indicated) and M6P from a single set of control fish, pmm2 morphants, and morphants rescued by coinjection of pmm2 mRNA. Vertical lines denote electronic removal of irrelevant lanes from the image. The portion below the M5Gn2-LLO standard on FACE gels generally contains mostly non-LLO species, but it is included here because this is where any accumulation of M3Gn2-M4Gn2-LLO is likely to appear. Related information is provided in Supplemental Figure S2. (D) G3M9Gn2-LLO and M6P measured in three independent experiments, mean ± SEM. Control levels were arbitrarily set as 100% (dashed line). (E) Top, schematic comparisons of hypotheses for M1P insufficiency (model A) and M6P accumulation (model B). Bottom, models A and B are indicated by light and dark gray bars, respectively, and contrasting double-morphant outcomes are highlighted with boxes.
Zebrafish yolk contains a large amount of N-glycosylated proteins (e.g., vitellogenin) that originate from the egg and therefore are not subject to hypoglycosylation due to injection of pmm2 morpholinos into embryos. This prompted us to determine whether the presence of yolk was masking a more pronounced change in morphant N-glycan content. As shown in Supplemental Figure S1B, N-linked glycans were readily observed in zebrafish eggs, and the quantities in embryos were greatly reduced by removal of yolk. Of importance, removal of yolk from the 4-dpf embryos showed that the N-glycan pool in pmm2 morphants was actually reduced on average to 54% of control. By comparison, in this experiment the N-glycan pool was reduced to 78% of control when yolks were present (Figure 5B). This demonstrated a robust masking effect of yolk-derived N-glycans. The qualitative profile of the N-glycans was unchanged in the morphants after yolk removal, again indicating that processing of embryonic N-linked glycoproteins was not substantially affected (Supplemental Figure S1B). Taken together, the loss of Pmm2 activity, broad developmental and motor abnormalities, including an unanticipated increase in motoneuron number, and deficiency of N-glycosylation indicate that the pmm2 morphants developed here represent a valid zebrafish model of PMM2-CDG disease.
LLO levels are suppressed in pmm2 morphants
It has been widely hypothesized that hypoglycosylation in PMM2-CDG patients is a result of reduced LLO concentrations, but no direct LLO analyses of patient biopsy material have been reported. Recent mass spectrometry data from PMM2-CDG patient fibroblasts grown in culture with physiological glucose indicate that the PMM2 deficiency creates a significant bottleneck for flux of mannosyl precursors (Sharma et al., 2011). However, in physiological glucose, PMM2-CDG fibroblasts also have normal steady-state levels of LLO (Gao et al., 2005) and have normal glycoform distributions for α1-antitrypsin and other N-linked glycoproteins (Marquardt et al., 1995, 1996; Dupre et al., 2000). These results leave open the question as to whether LLO levels in patients are truly reduced by PMM2 deficiency.
To begin to address this question, we used FACE to measure steady-state LLO levels in 4-dpf (larval stage) pmm2 morphants. Figure 5, C and D, shows that pmm2 morphants have 58% of control levels of G3M9Gn2-P-P-Dol. This amount is returned to normal after rescue by coinjecting pmm2 mRNA. Moreover, no relative accumulations of LLO intermediates were detected within the range M3Gn2-P-P-Dol to G2M9Gn2-P-P-Dol (Figure 5C and Supplemental Figure S2), which is the functional resolution established previously for the FACE system (Gao et al., 2005). These results show that LLO levels are suppressed in this animal model of pmm2 deficiency, with one possible explanation being inhibition of a step before the synthesis of M3Gn2-P-P-Dol.
Metabolic analysis of pmm2 morphants demonstrates an accumulation of mannose-6-phosphate
To elucidate the cause of the LLO deficiency, we subjected whole 4-dpf embryos to sugar analysis using FACE with reductive amination and fluorescence detection. An obvious caveat to this approach is that it measures sugars in total embryos, averaged over all the individual cell types, and is weighted toward those cells with higher sugar contents. Similarly, the embryos might contain unknown compounds able to interfere with the analysis. In preliminary experiments, we found that whole-embryo sugar phosphate contents were highly variable through 3 dpf but stabilized somewhat by 4 dpf. Consequently, we opted to analyze sugars in 4-dpf embryos.
An obvious explanation for LLO deficiency in pmm2 morphants is a reduction in the synthesis of M1P (which requires Pmm2 activity) and consequently lower concentrations of GDP-mannose that limit early mannose-dependent steps of LLO synthesis. FACE offers an advantage over other analytical methods in that sugar phosphates (M1P and M6P) and nucleotide sugars (like GDP-mannose) can be recovered from the same samples used for the earlier measurements of LLOs and N-glycans. However, we were unable to consistently detect either M1P or GDP-mannose in zebrafish by FACE due to the limited amounts of embryonic material (although mannosyl-1,6-bisphosphate, the cofactor for Pmm2, appeared unaffected). For this reason, the effects of Pmm2 insufficiency on M1P and GDP-mannose concentrations could not be reliably measured.
Curiously, FACE experiments showed that M6P levels were increased in pmm2 morphants (Figure 5C). FACE analyses of sugars isolated from the same three morphant and rescue samples used for the LLO analysis of Figure 5D were particularly informative. As shown in Figure 5D, pmm2 morphants exhibited an average increase of 1.9-fold in M6P levels, which was reduced by coinjection of pmm2 mRNA (Figure 5, C and D). This increase in M6P was independently confirmed by a high-performance liquid chromatography–based method able to directly assess levels of this sugar phosphate in an aqueous extract (Supplemental Table S1). The increase of M6P can be explained by the fact that M6P is normally consumed by Pmm2 for M1P synthesis and therefore might accumulate in pmm2 morphants. Of interest, in a recent study, we provided evidence that under certain conditions M6P could promote the hydrolysis of LLO both in permeabilized and in intact cells, causing release of the G3M9Gn2 glycan and Dol-P-P (Gao et al., 2011). The outcome was loss of LLO with no accumulation of truncated LLO biosynthetic intermediates. Because this result was similar to that observed in pmm2 morphants (Figure 5), we designed experiments to test the hypothesis that LLO depletion in the pmm2 morphants was caused by accumulated M6P, resulting in LLO hydrolysis.
LLO levels in pmm2 morphants are stabilized by manipulating the M6P-synthesizing enzyme mannose-6-phosphate isomerase
M6P can be formed from F6P in a reversible reaction catalyzed by Mpi (see Figure 5E). MPI-CDG patients have reduced Mpi activity, leading to an N-glycosylation deficiency similar to that in PMM2-CDG. Evidence suggests that the etiology of MPI-CDG is due to impaired conversion of F6P to M6P, leading to suppressed M1P concentrations (Panneerselvam and Freeze, 1996; Niehues et al., 1998). Moreover we recently demonstrated that zebrafish with suppressed Mpi activity had reduced levels of LLOs and N-glycans (Chu et al., 2012).
In light of these data, we hypothesized that the contribution of excess M6P to the biochemical phenotypes associated with Pmm2 deficiency could be tested by simultaneous knockdown of Pmm2 and Mpi (Figure 5E). In other words, if the LLO deficiencies in the pmm2 and mpi morphants were both due solely to lower concentrations of M1P and GDP-mannose, then we would expect a synergistic suppression of LLO levels in double morphants (Figure 5E, model A). Conversely, if higher concentrations of M6P caused the LLO deficiency in pmm2 morphants, we would predict offsetting effects in double morphants. As indicated in Figure 5E (model B), whereas mpi knockdown might cause LLO deficiency on its own due to M1P depletion, it might also counteract the effect of the pmm2 knockdown by inhibiting synthesis of M6P. According to model B, the net result of a simultaneous knockdown of both enzymes could be a loss of LLO no more severe than the loss due to either knockdown alone.
To address whether reducing Mpi levels would mitigate the increased levels of M6P detected in pmm2 morphants, we coinjected the pmm2 SB MO with the mpi translation blocking MO previously described (Chu et al., 2012). The effects on Mpi activity were assessed over a range of MO concentrations in control and pmm2 morphant embryos (Supplemental Figure S3). In an effort to lower but not eliminate Mpi levels, we ultimately chose to work with 0.02 μM MO, which reduced Mpi activity to 47 and 36% of wild type (WT) in the control and pmm2 morphant backgrounds, respectively (Figure 6A). Although this degree of suppression was less robust than that studied in Chu et al. (2012), which resulted in no greater than 20% mpi expression, it was nonetheless desirable, as it resulted in a rebalance of the normal 1:1.5 ratio of Pmm2:Mpi activity in the pmm2 morphants (Supplemental Figure S3). Although this concentration of the mpi MO on its own caused no developmental defects, they were clearly observed with higher degrees of knockdown (Chu et al., 2012). Of interest, whereas morpholino-based suppression of pmm2 and mpi (either separately or combined) resulted in decreases of mRNAs (unpublished data) and enzymatic activities (Figure 6A), individual inhibition of pmm2 expression also consistently resulted in a slight decrease in Mpi activity. However, a reciprocal decrease in Pmm2 activity was not observed when mpi was individually inhibited.
FIGURE 6:

Knockdown of mpi in the pmm2 background normalizes M6P levels, with no further loss of G3M9Gn2-LLO. (A) Pmm2 and Mpi enzyme activity for control embryos, pmm2 morphants, mpi morphants, and pmm2/mpi double morphants. (B) G3M9Gn2-P-P-Dol and M6P were measured in control embryos, pmm2 morphants, mpi morphants, and pmm2/mpi double morphants. Results are shown for four independent experiments, mean ± SEM, with control values set as 100%.
Knockdown of either pmm2 or mpi alone caused the expected losses of LLO (Figure 6B). It is striking that double morphants had LLO concentrations no lower than that of either individual morphant. In some individual experiments, LLO levels in the double morphants appeared to increase. In four sets of samples, sugar phosphate data obtained by FACE indicated that M6P increased ∼175% in the pmm2 morphant, was not significantly increased in the mpi morphant, and returned to control levels in the double morphant (Figure 6B). Consequently, this genetic approach supports the hypothesis that elevated M6P concentrations make a significant contribution to the LLO deficiency in pmm2 morphants, a phenotype that cannot be readily explained solely by a loss of M1P.
Free glycans, the products of M6P-dependent LLO cleavage, are increased in pmm2 morphants
Whether triggered in vitro with pure M6P (Gao et al., 2005) or in cultured cells by activating M6P signaling (Gao et al., 2011), the cleavage of LLO results in release of free glycans into the ER lumen. These free glycans then appear to undergo remodeling reactions within the secretory pathway, resulting in a heterogeneous mixture. We assessed free glycans in the same sample of pmm2 morphants used for LLO and sugar phosphate analysis (Figure 5). As shown in Figure 7A, total free glycans were increased approximately twofold in pmm2 morphants and returned to control levels after coinjection of pmm2 mRNA. However, there were no obvious qualitative differences in the free glycan pools, suggesting that remodeling reactions in the secretory pathway were unaffected in pmm2 morphants (Figure 7B). Consistent with a M6P-driven process (Figure 7, B and C), the increase in free glycan pool size was suppressed by coinjection of the pmm2 and mpi morpholinos.
FIGURE 7:
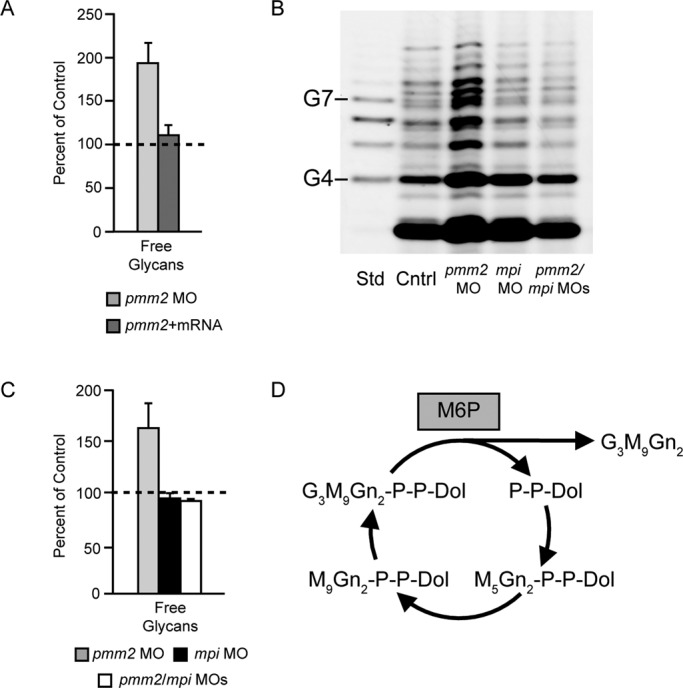
Free glycans in pmm2 morphants. Total free glycans were measured with FACE techniques from the same samples of zebrafish presented in Figures 5 and 6. (A) Free glycans were measured in control embryos, pmm2 morphants, and morphants rescued by coinjection of pmm2 mRNA. Results of three independent experiments, mean ± SEM. Control levels were arbitrarily set as 100%. (B) Representative FACE image showing total free glycans from control embryos, pmm2 morphants, mpi morphants, and pmm2/mpi double morphants. Glucose oligomer standards (G4–G7) are shown. As shown previously (Gao et al., 2011), G3M9Gn2 released from the LLO pool by M6P action is glycosidically degraded, resulting in the heterogeneous mixture of free glycans detected. The most abundant glycan detected near the bottom of the gel is likely to be M2Gn2, based on migration. (C) Results for four independent experiments as in B, mean ± SEM, with control set as 100%. (D) Schematic showing that dolichol pyrophosphate can be recycled for multiple rounds of LLO synthesis while free glycans are released by the action of M6P. This explains why the molar increase of free glycans in pmm2 morphants vastly exceeds the measured loss of LLO (see Discussion).
In previous studies regarding the M6P-dependent production of free glycans in mammalian cells, it was possible to demonstrate by selective permeabilization of plasma membranes that the released glycans were present in lumenal but not cytosolic compartments (Gao et al., 2011). This was not feasible in our zebrafish system, and, in principle, the results could be due to greater production of cytosolic free glycans generated by ER-associated degradation (ERAD) of glycoproteins that failed to fold due to impaired acquisition of N-glycans. However, this is unlikely for two reasons. First, knockdown of pmm2 did not induce mRNAs encoding GRP78/BiP or CHOP, both markers for ER stress, although pmm2 message itself was diminished to ∼20% of controls (unpublished data). Second, in deliberate attempts to increase ERAD by exposing mammalian cells to agents that impair folding, there were no changes in cytosolic free glycans (Gao et al., 2011). Consequently, the results of Figure 7 are best explained by increased production of lumenal free glycans from LLO. Taken together, our data with zebrafish pmm2 morphants strongly support a substrate-accumulation mechanism in which elevation of M6P increases LLO hydrolysis, expanding the lumenal free glycan pool and impairing protein N-glycosylation (Figure 7D).
Reducing Mpi levels in pmm2 morphants improves chondrocyte morphology
Although M6P and free glycan levels were normalized in mpi/pmm2 double morphants, these animals still had their respective enzyme levels reduced to one-third of normal (Figure 6A) and still had appreciable LLO deficiencies (Figure 6B). For these reasons we did not expect suppression of mpi to mitigate all of the phenotypic defects of pmm2 morphants, and indeed many of their phenotypes, including the motility deficits, were not significantly corrected by mpi knockdown. However, analysis of mpi/pmm2 double-morphant embryos demonstrated that there was substantial recovery of the chondrocyte morphology phenotype (Figure 8). Flat-mount preparations of Alcian blue–stained embryos revealed that although the size of individual cartilage structures was not significantly changed by mpi suppression, the round chondrocyte morphology characteristic of pmm2 deficiency was improved (Figure 8A). In each of the three structures analyzed, mpi/pmm2 double-morphant chondrocytes regained the columnar architecture typical of control chondrocytes (Figure 8B). Although occasionally thinner than control cells, double-morphant cells were clearly more elongated and less round than pmm2 chondrocytes.
FIGURE 8:

Inhibition of mpi in the pmm2 background improves craniofacial chondrocyte morphology. (A) Alcian blue stains of 4 dpf control, pmm2 morphant, mRNA-rescued, and pmm2/mpi double-morphant embryonic cartilage revealed significant improvement after mpi manipulation. Unlike pmm2 morphant chondrocytes, which were round and unorganized, reducing mpi in pmm2 morphants restored the elongated shape of chondrocytes within the Meckel's, ceratohyal, and palatoquadrate (not shown) cartilages. (B) Quantitative measurement of cell shape demonstrated significant recovery of cell elongation in pmm2 morphants after either introduction of pmm2 mRNA or reduction of mpi expression. Briefly, the long and short axes of individual chondrocytes were measured and their ratio calculated. A ratio of 1 indicates equivalence of these axes and a perfectly round cell. (C) Cellular organization was also partially improved in pmm2 morphants after mpi manipulation. This is quantitatively demonstrated in the number of cells spanning the width of the Meckel's and ceratohyal elements.
Although not fully restored, chondrocyte organization was also significantly improved after mpi suppression. Although mpi/pmm2 double-morphant chondrocytes were still present as multicellular layers, these regions were typically no more than two cells wide, a notable improvement from the three- to four-cell–wide structures observed in pmm2 morphants (Figure 8C). Therefore unlike pmm2 morphant cells, which exist in multilayered stacks, the chondrocytes of mpi/pmm2 double-morphant cartilages appear to have regained characteristics favoring the tendency to form a single cellular layer. It is noteworthy that, although additional aspects of the phenotypes (in particular, motility) were not significantly improved, they were also not exacerbated by mpi knockdown. The lack of complete phenotypic and biochemical correction in this double-morphant background is perhaps not surprising, considering the fact that both Pmm2 and Mpi activities are reduced to one-third their normal levels (Figure 6A), and these animals still have an appreciable LLO deficiency. In contrast, chondrocyte morphology defects appear to correlate with accumulation of free glycans rather than LLO deficiency.
DISCUSSION
PMM2-CDG, the most common and first identified subtype of CDG, clearly demonstrates that proper glycosylation is essential for normal human development and physiology. Abnormalities affecting the development and function of multiple organ systems are manifested by heritable deficiencies in the PMM2 enzyme. This enzyme converts M6P to M1P, the immediate precursor of GDP-mannose. GDP-mannose is required for the biosynthesis of at least four highly distinct classes of mannosyl conjugates (O-linked mannosyl glycans, C-linked mannose, glycosylphosphatidylinositol anchors, and N-linked glycans). Although many studies have demonstrated a clear association between PMM2 gene defects and aberrant N-glycosylation of the serum glycoprotein transferrin, the disease has been confounding for both basic scientists and clinicians with regard to its pathophysiology and treatment. Recent work by several groups has shown that bypassing defective metabolic steps or enhancing metabolic flux through depleted glycosylation pathways represents a promising mode of therapy for some type I CDGs (Freeze, 2009; Higashidani et al., 2009; Schroeder et al., 2011; Sharma et al., 2011). This concept is perhaps best supported by the clinical improvement of MPI-CDG patients who receive oral mannose supplementation. Uptake of mannose into cells and rapid conversion to M6P by hexokinase circumvents the need for Mpi to produce M6P from F6P. Our recent studies have demonstrated the same phenomenon in zebrafish: mpi morphants are rescued with mannose addition to their water (Chu et al., 2012).
In contrast, there is no clear clinical benefit for PMM2-CDG patients given mannose supplements (Mayatepek et al., 1997; Mayatepek and Kohlmuller, 1998). Although mannose-derived M6P in CDG-MPI cells has a direct metabolic path into N-glycans, in CDG-PMM2, the M6P is still dependent on impaired PMM2 activity for downstream use. Although this may help explain the lack of efficacy with mannose supplementation, it also highlights the metabolic complexity that underlies PMM2-CDG. For example, prenatal mannose supplementation was successful in overcoming embryonic lethality in pmm2 mice bearing a combination of disease alleles, suggesting that the timing of mannose supplementation may be critical (Schneider et al., 2012). In addition, other studies have provided support for therapeutic strategies for PMM2-CDG that either enhance mannose supplementation or are entirely independent of it. With cultured CDG-PMM2 fibroblasts, the drug metformin significantly improved the efficiency of mannose correction by increasing mannose uptake (Shang and Lehrman, 2004). Treatment of PMM2-CDG fibroblasts with zaragoic acid, which is believed to divert lipid precursors toward dolichol synthesis, was shown to rescue impaired N-glycosylation (Haeuptle et al., 2011). By a similar mechanism, high concentrations of ketoconazole were reported to counteract the effects of the glycosylation blocker tunicamycin (Harding et al., 2005). By lessening the demand on the LLO biosynthetic pathway, the translation attenuators diamide and disulfiram improved the quality of LLOs and the glycans transferred to protein (Shang et al., 2007). More recently, a compound that inhibited Mpi diverted mannose-derived M6P away from glycolytic catabolism and toward glycoconjugate production, providing a proof of principle for the strategy of enhancing glycosylation in PMM2-CDG cells by altering flux through key metabolic pathways (Higashidani et al., 2009; Sharma et al., 2011). The availability of valid, tractable animal models of PMM2-CDG would greatly aid the thorough evaluation of such pharmacological approaches and thus accelerate the search for useful clinical treatments.
Although there is no doubt that PMM2-CDG etiology involves loss of PMM2 expression and can result in hypoglycosylation of N-linked glycoproteins, the biochemical cause, like the search for a treatment, has remained enigmatic. In physiological (5 mM) glucose, cultured PMM2-CDG fibroblasts have normal LLO levels (Gao et al., 2005) and do not exhibit abnormal protein glycoforms detectable with immunoblots (Marquardt et al., 1995, 1996; Dupre et al., 2000). However, the PMM2 deficiency in these cells clearly causes a metabolic constriction that can be demonstrated in subphysiological (2.5 mM) glucose (Gao et al., 2005) or with physiological concentrations of stable isotopic glucose monitored by mass spectrometry (Sharma et al., 2011). Moreover, a recently validated marker for glycosylation deficiency, cell-surface ICAM-1, is diminished in PMM2-CDG fibroblasts cultured with physiological glucose and restored upon introduction of a functional Pmm2 gene (He et al., 2012). The picture is confounded further by chronic ER stress in these cells, which can alter LLO synthesis in several ways (Shang et al., 2002, 2007). The apparent contradictions in some of these results may originate from differences in the actual disease allele, donor age, and cell passage number. This emphasizes the need for a useful animal model amenable to multiple forms of experimentation, both to explore potential treatments and to provide mechanistic understandings of the systemic abnormalities of the disease.
The zebrafish model of Pmm2 deficiency reported here is reminiscent of human PMM2-CDG disease in several important ways: 1) Pmm2 activity is suppressed; 2) developmental abnormalities, including craniofacial and motility defects, are detected; and 3) global deficiency of N-glycosylation was identified. In addition, we directly demonstrated for the first time an LLO deficiency in Pmm2-depleted animal tissues, confirming a long-standing prediction for this genetic defect. Our biochemical analyses further established that the extent of protein underglycosylation in the PMM2-CDG zebrafish model was initially underestimated due to the abundance of yolk-derived glycoproteins. Along with the validation of this system as a CDG model, our observation that increased numbers of spinal motoneurons are associated with Pmm2 deficiency provides novel and unprecedented insight into the cellular basis for the neurological defects associated with PMM2-CDG. This insight will inform future studies geared at identifying the specific glycoproteins that are responsible for these neurological phenotypes. In light of the fact that >2200 N-linked sites were recently documented in zebrafish (Zielinska et al., 2012), the identification of these glycoproteins is beyond the scope of this study. Depletion of pmm2 in zebrafish resulted in cartilage defects that bear some resemblance to known mutants that affect proteoglycan glycosylation (Clément et al., 2008), reinforcing the necessity of proper glycosylation during cartilage development. Although they are phenotypically similar to other zebrafish mutants that affect matrix secretion via mutations in COPII subunits (Melville et al., 2011), we did not detect any alterations in collagen secretion or deposition or evidence of ER stress in the pmm2 morphants. This suggests that the mechanisms that account for the cartilage morphogenesis defects when N-glycosylation is impaired in zebrafish do not involve misfolding of glycoproteins or a block in their exit from the ER.
Although LLO levels were reduced in pmm2 morphants, the underlying mechanism revealed by our experiments was unexpected. Specifically, accumulation of the Pmm2 substrate M6P was detected and linked to LLO and N-glycosylation deficiency in a manner specific for the genetic lesion. The limits of sensitivity of our analytical methods prevented us from consistently assessing M1P or GDP-mannose. However, in the few experiments in which these molecules appeared detectable, they were not decreased by the pmm2 morpholino. The loss of LLO is therefore most easily explained by M6P promoting LLO hydrolysis, as reported previously in mammalian cells (Gao et al., 2011). This conclusion was supported by demonstrating both epistasis with mpi (the gene encoding the M6P-synthesizing enzyme mannose phosphate isomerase) and the Pmm2-dependent appearance of free glycans that are markers for LLO hydrolysis. Although our data show clear changes in the levels of key metabolic precursors within the pmm2 morphants, it is unclear whether these changes occur in a tissue-specific manner. In other words, it is possible that certain tissues or cell types such as neurons or chondrocytes are more susceptible to M6P accumulation and LLO hydrolysis than others. Furthermore, we cannot rule out that still other tissues in the embryo are subject to undergycosylation due to loss of M1P and GDP-mannose. Because our methods measured static metabolic levels, any alterations in substrate flux that did not reduce steady-state concentrations would also have gone undetected.
Our studies therefore suggest that two separate and potentially synergistic mechanisms of LLO loss and glycosylation deficiency, which may differ in significance, depending on the cell and tissue type and the point in development, should be considered in PMM2-CDG: absence of M1P, limiting the synthesis of mannosyl conjugates; and excess M6P, causing hydrolysis of LLO. The latter mechanism also raises the possibility of processed, lumenal free glycans acting as competitive inhibitors of vesicular lectin–dependent processes. As we showed earlier with permeabilized cells (Gao et al., 2005) and intact cells (Gao et al., 2011), the Dol-P-P released from LLO by M6P action can be recycled to make new LLO, whereas the corresponding free glycans accumulate (Figure 7D). LLO levels may either remain unchanged or decrease, depending on the rate of consumption (hydrolysis plus transfer to protein) and the existence of mutations or other factors that lower the rate of new synthesis. This concept predicts that the amount of free glycan generated may greatly exceed the apparent loss of LLO. Indeed, comparison of the total quantity of free glycan increased in Figure 7B with the LLO decreased in the pmm2 morphant samples revealed a remarkable ∼70-fold molar excess of free glycans. This amount does not include free glycans that may have been secreted or degraded and may be a significant underestimate. In some circumstances, the free glycan–generating potential of the M6P-dependent process described here may therefore be more significant than LLO depletion.
Our results help to explain the ineffectiveness of mannose supplementation for PMM2-CDG patients, a treatment that we suggest may ameliorate the loss M1P but at the same time exacerbate the M6P accumulation. The offsetting nature of these two mechanisms may also explain why further decreases in N-glycosylation of serum glycoproteins have not been detected in PMM2-CDG patients infused with mannose. In addition, substantial LLO loss attributed to M6P accumulation may not always be sufficient to impair N-glycosylation: in one study with mouse fibroblasts carrying a null allele for Mpi, high concentrations of M6P were measured and LLO levels were substantially reduced, but no change in overall protein N-glycosylation was detected (Higashidani et al., 2009). Manipulating metabolic flux is therefore likely to be a useful therapeutic approach, but it will require careful attention to conditions that offer beneficial effects while minimizing potential damage. With a useful animal model now in hand, these insights should enable specific, hypothesis-driven studies aimed at understanding the identified alterations in neurogenesis, as well as the roles of developmental time, tissue specificity, and enzyme concentration in the etiology of PMM2-CDG.
MATERIALS AND METHODS
Fish strains, maintenance, and husbandry
Wild-type zebrafish were obtained from the commercial source Fish2U (Gibsonton, FL) and maintained using standard protocols. Embryos were staged according to the criteria established by Kimmel et al. (1995). When necessary, pigment formation was inhibited by addition of 0.003% 1-phenyl-2-thiourea to embryonic growth medium. Although the data presented are derived primarily from experiments in F2U, all MO-generated phenotypes (e.g., craniofacial and motility defects) were confirmed in multiple genetic backgrounds (AB, TAB14, fli1a:EGFP). For analysis of neuronal phenotypes, morphants were generated in the hb9:GFP transgenic background. Handling and euthanasia of fish for all experiments were carried out in compliance with the University of Georgia's Institutional Animal Care and Use Committee (permit number A2011 8-144).
Antisense morpholino injection and mRNA rescue
Expression of pmm2 was inhibited by injection of antisense morpholinos (MO) into the yolks of one- to two-cell embryos. Translation-blocking (5′-GAATCCACGGTAGAGCCAGACATTG-3′) and splice-blocking (5′-TTCCGCATTAATTCACTCACTTGAC-3′) MOs were designed to target either the pmm2 5′ untranslated region or the pmm2 exon 1–intron 1 splice junction, respectively (Gene Tools, Eugene, OR). Optimal MO doses were determined by Pmm2 activity assays and RT-PCR analyses. Maximal inhibition (33% of control) of Pmm2 activity was achieved after injection of 0.5 nl of a 1 μM solution (0.52 μM final concentration based on a 1-μl estimate per egg) of the splice-blocking MO. The maximal inhibition achievable with the translation-blocking MO (60%) was found after injection of 0.5 nl of a 1 μM solution (0.52 μM final). To facilitate various recovery analyses, the full-length coding region of pmm2 was cloned by RT-PCR from mRNA isolated from 3-dpf embryos using the primers ttccaaatatcacaatgtctggc and tcagctgaaaaagagctccctgcag. The resulting product was cloned into the TA site of pCR2.1 (Life Technologies, Grand Island, NY) and subsequently excised by from vector-derived EcoRI and XbaI sites. This product was subcloned into pCSII. pCSII-pmm2 plasmid DNA was linearized with KpnI and full-length mRNA generated with Message Machine SP6 kit (Roche Applied Science, Indianapolis, IN.) Rescue experiments were performed by sequential injection of 0.52 μM splice-blocking MO and 200 pg of purified pmm2 mRNA into the yolks of one- to two-cell embryos. For mpi knockdown a translation-blocking MO was designed to the 5′ portion of the gene. This MO was validated using the described Mpi activity assay (see later discussion) over a range of MO concentrations (Supplemental Table S1). Because no phenotypic defects resulted in this concentration range, we did not examine the presence of off-target effects by mRNA rescue. However, our recent work demonstrated that all phenotypic effects caused by injection of higher mpi morpholino concentrations are rescued by coinjection of mpi mRNA (Chu et al., 2012).
RT-PCR analysis of pmm2 expression and Pmm2/Mpi activity assay
Total RNA was extracted from approximately 30 3-dpf embryos using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA was reverse transcribed to cDNA using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). RT-PCR analysis of pmm2 expression performed with primers flanking the first intron (tcacaatgtctggctctaccg and gagacattcagcattccattcc). Pmm2 transcript abundance was normalized to RPL4 transcripts. Using this method, we did not detect an accumulation of the larger PCR product expected if the intron was retained in an mRNA exported from the nucleus. This was assessed with two different primer pairs, including one yielding a smaller product to ensure ample amplification. The primer pairs were as follows: forward primer in exon 1, 5′-tcacaatgtctggctctaccg-3′, and one of two reverse primers—one in intron 1, 5′-cgacacaggtacaagtagtgttttg-3′ (product 552 base pairs), or one in exon 2, 5′-gagacattcagcattccattcc-3′ (product 2.7 kb with intron).
For analysis of Pmm2 enzyme activity, 50–100 embryos were collected at the indicated time points and the yolks manually removed. Embryos were lysed in 50 mM Na-4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, sonicated, and total protein quantitated using a microBCA protein assay kit (Pierce, Rockford, IL). Pmm2/Mpi enzyme activity assays were performed as previously described (Van Schaftingen and Jaeken, 1995). Briefly, Pmm2 activity is assayed by a coupled enzyme assay system that relies on the spectrophotometric detection of the reduction of NADP+ to NADPH using M1P as a substrate. Samples were measured 60 min after adding the substrate. Units were calculated as milliunits of enzyme activity/milligrams of protein based on the change in OD during the reaction time.
Histochemistry, immunohistochemistry, and whole-mount in situ analysis
To visualize craniofacial cartilage, we stained embryos with Alcian blue as described previously (Flanagan-Steet et al., 2009). Stained animals were photographed on an Olympus SZ16 stereoscope (Olympus, Tokyo, Japan) outfitted with a Retiga charge-coupled device camera (QImaging, Surrey, Canada). Morphometric measurements of Alcian blue–stained animals were performed using Photoshop (Adobe, San Jose, CA). To account for differences in animal size, all measurements were normalized to interlens distance. Secondary motoneuron identities were assessed by whole-mount immunohistochemistry using the zn-5 antibody (ZIRC, Eugene, OR). Immuno-whole mounts were performed as previously described (Flanagan-Steet et al., 2009). Confocal images were acquired on an Olympus FV100 laser-scanning microscope using a 40× (numerical aperture 1.2) water immersion objective. Whole-mount in situ hybridization for pmm2 was performed as previously described (Thisse and Thisse, 2008).
Motility assays
For assessment of early spontaneous and elicited motility, embryos were dechorionated at 22 h postfertilization (hpf) and allowed to age to 24 hpf. The number of spontaneous tail curls was then measured in a 60-s interval. Fifty fish were measured at a time from four separate injections. Elicited responses were measured at 24 hpf by touching embryos on the head near the otilith and recording the number and quality of subsequent tail responses. For assessment of later-stage elicited motility, 3-dpf embryos were placed in the center of a Petri dish marked by three concentric rings. Embryos were touched beside the otilith and their response recorded. Swimming distance of individual embryos was assigned as 1, 2, or 3, according to the number of rings crossed. The diameters of the zones were as follows: zone 1, 3 cm; zone 2, 6 cm; zone 3, 8 cm. Fifty fish were measured at a time from three separate injections.
Fluorophore-assisted carbohydrate electrophoresis analysis of zebrafish saccharides and glycoconjugates
In the lab of R.S., embryos were collected by centrifugation, excess water was removed, 1 ml of methanol (room temperature) was added to the tube, and the contents were disrupted by brief probe sonication (Branson Sonifier 150; low setting, 3× 5-s pulses). In some cases, the yolks were first removed from the embryos by gentle passage through a flame-polished Pasteur pipette. The suspension was transferred to a 15-ml conical tube, and an additional 10–12 ml of methanol was added. After thorough vortexing, the tubes were capped and sealed with parafilm, and sent by express shipping at room temperature to the lab of M.A.L. At that point, samples were dried and processed for FACE analyses as described (Gao and Lehrman, 2006; Gao et al., 2011). In brief, after removal of most lipids by extraction with chloroform:methanol (2:1), aqueous saccharides were recovered by extraction with water, and LLOs were then obtained by extraction with chloroform:methanol:water (10:10:3), with N-linked glycoproteins remaining in the residual material. Ion exchange fractionation of the aqueous saccharides yielded free sugars and free glycans in an uncharged fraction, sugar monophosphates in a weakly anionic fraction, and both nucleotide-sugars and sugar 1,6-bisphosphates in a more strongly anionic fraction. Phosphate esters of the 1-position of monosaccharides and oligosaccharides were released with weak acid (a condition that retained 6-phosphate esters), and N-glycans were released with peptide N-glycosidase F. All monosaccharide species were then conjugated with 2-aminoacridone and separated on a monosaccharide profiling gel, and all oligosaccharide species were conjugated with 7-amino-1,3-naphthalenedisulfonic acid and detected with an oligosaccharide profiling gel. Where appropriate, standards were included consisting of monosaccharide mixtures, glucose oligomers, or LLO-derived glycans. Fluorescent saccharide conjugates were detected with a Bio-Rad Fluor-S scanner and quantified with Quantity-One software. Loading of various quantities of standards established that detection was linear, with a sensitivity of ∼2 pmol of saccharide. For quantitation of free glycan experiments, only the portions in the size range delimited by the G4 and G7 standards were used.
Supplementary Material
Acknowledgments
This study was funded by the National Institutes of Health-American Recovery and Reinvestment Act (RC1 HD064159, awarded jointly to H.H.F, K.C.S., M.A.L., and R.S.), the National Institutes of Health (R01-GM038545 to M.A.L., R01-GM086524 to R.S., and R01-AA018886 to K.C.S.), the Welch Foundation (I-1168 to M.A.L.), and The Rocket Fund (to H.H.F).
Abbreviations used:
- CDG
congenital disorder of glycosylation
- CH
ceratohyal
- dpf
day postfertilization
- Dol
dolichol
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- FACE
fluorophore-assisted carbohydrate electrophoresis
- F6P
fructose-6-phosphate
- G3M9Gn2-P-P-Dol, glucose3mannose9N-acetylglucosamine2-pyrophosphate-dolichol
- hpf
hour postfertilization
- LLO
lipid-linked oligosaccharide
- M1P
mannose-1-phosphate
- M6P
mannose-6-phosphate
- MO
morpholino
- Pmm2
phosphomannomutase 2
- PQ
palatoquadrate
- SB
splice blocker
- TB
translation blocker
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-05-0411) on September 5, 2012.
*Joint lead authors.
REFERENCES
- Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6:69–77. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Mir A, Gao N, Rosa S, Monson C, Sharma V, Steet R, Freeze HH, Lehrman MA, Sadler KC. A zebrafish model of congenital disorders of glycosylation with phosphomannose isomerase deficiency reveals an early opportunity for corrective mannose supplementation. Dis Model Mech. 2012 doi: 10.1242/dmm.010116. DOI: 10.1242/dmm.010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément A, Wiweger M, von der Hardt S, Rusch MA, Selleck SB, Chien CB, Roehl HH. Regulation of zebrafish skeletogenesis by ext2/dackel and papst1/pinscher. PLoS Genet. 2008;4:e1000136. doi: 10.1371/journal.pgen.1000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coman D, Irving M, Kannu P, Jaeken J, Savarirayan R. The skeletal manifestations of the congenital disorders of glycosylation. Clin Genet. 2008;73:507–515. doi: 10.1111/j.1399-0004.2008.01015.x. [DOI] [PubMed] [Google Scholar]
- de Lonlay P, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001;38:14–19. doi: 10.1136/jmg.38.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRossi C, Bode L, Eklund EA, Zhang F, Davis JA, Westphal V, Wang L, Borowsky AD, Freeze HH. Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J Biol Chem. 2006;281:5916–5927. doi: 10.1074/jbc.M511982200. [DOI] [PubMed] [Google Scholar]
- Dupre T, Barnier A, de Lonlay P, Cormier-Daire V, Durand G, Codogno P, Seta N. Defect in N-glycosylation of proteins is tissue-dependent in congenital disorders of glycosylation Ia. Glycobiology. 2000;10:1277–1281. doi: 10.1093/glycob/10.12.1277. [DOI] [PubMed] [Google Scholar]
- Ekker SC. Morphants: a new systematic vertebrate functional genomics approach. Yeast. 2000;17:302–306. doi: 10.1002/1097-0061(200012)17:4<302::AID-YEA53>3.0.CO;2-#. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan-Steet H, Fox MA, Meyer D, Sanes JR. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development. 2005;132:4471–4481. doi: 10.1242/dev.02044. [DOI] [PubMed] [Google Scholar]
- Flanagan-Steet H, Sias C, Steet R. Altered chondrocyte differentiation and extracellular matrix homeostasis in a zebrafish model for mucolipidosis II. Am J Pathol. 2009;175:2063–2075. doi: 10.2353/ajpath.2009.090210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH. Update and perspectives on congenital disorders of glycosylation. Glycobiology. 2001;11:129R–143R. [PubMed] [Google Scholar]
- Freeze HH. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim Biophys Acta. 2009;1792:835–840. doi: 10.1016/j.bbadis.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012;11:453–466. doi: 10.1016/S1474-4422(12)70040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, Lehrman MA. Non-radioactive analysis of lipid-linked oligosaccharide compositions by fluorophore-assisted carbohydrate electrophoresis. Methods Enzymol. 2006;415:3–20. doi: 10.1016/S0076-6879(06)15001-6. [DOI] [PubMed] [Google Scholar]
- Gao N, Shang J, Lehrman MA. Analysis of glycosylation in CDG-Ia fibroblasts by fluorophore-assisted carbohydrate electrophoresis: implications for extracellular glucose and intracellular mannose 6-phosphate. J Biol Chem. 2005;280:17901–17909. doi: 10.1074/jbc.M500510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, et al. Mannose-6-phosphate regulates destruction of lipid-linked oligosaccharides. Mol Biol Cell. 2011;22:2994–3009. doi: 10.1091/mbc.E11-04-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- Haeuptle MA, Welti M, Troxler H, Hulsmeier AJ, Imbach T, Hennet T. Improvement of dolichol-linked oligosaccharide biosynthesis by the squalene synthase inhibitor zaragozic acid. J Biol Chem. 2011;286:6085–6091. doi: 10.1074/jbc.M110.165795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Khersonsky S, Marciniak S, Scheuner D, Kaufman RJ, Javitt N, Chang YT, Ron D. Bioactive small molecules reveal antagonism between the integrated stress response and sterol-regulated gene expression. Cell Metab. 2005;2:361–371. doi: 10.1016/j.cmet.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Ng BG, Losfeld ME, Zhu W, Freeze HH. Identification of intercellular cell adhesion molecule 1 (ICAM-1) as a hypo-glycosylation marker in congenital disorders of glycosylation cells. J Biol Chem. 2012;287:18210–18217. doi: 10.1074/jbc.M112.355677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashidani A, Bode L, Nishikawa A, Freeze HH. Exogenous mannose does not raise steady state mannose-6-phosphate pools of normal or N-glycosylation-deficient human fibroblasts. Mol Genet Metab. 2009;96:268–272. doi: 10.1016/j.ymgme.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeken J, Matthijs G. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet. 2001;2:129–151. doi: 10.1146/annurev.genom.2.1.129. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Freeze H. Congenital disorders of glycosylation: glycosylation defects in man and biological models for their study. Biol Chem. 2001;382:161–177. doi: 10.1515/BC.2001.024. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ullrich K, Niehues R, Koch HG, Harms E. Carbohydrate-deficient glycoprotein syndrome type I: determination of the oligosaccharide structure of newly synthesized glycoproteins by analysis of calnexin binding. J Inherit Metab Dis. 1996;19:246–250. doi: 10.1007/BF01799441. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ullrich K, Zimmer P, Hasilik A, Deufel T, Harms E. Carbohydrate-deficient glycoprotein syndrome (CDGS)—glycosylation, folding and intracellular transport of newly synthesized glycoproteins. Eur J Cell Biol. 1995;66:268–273. [PubMed] [Google Scholar]
- Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ, Van Schaftingen E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome) Nat Genet. 1997;16:88–92. doi: 10.1038/ng0597-88. [DOI] [PubMed] [Google Scholar]
- Mayatepek E, Kohlmuller D. Mannose supplementation in carbohydrate-deficient glycoprotein syndrome type I and phosphomannomutase deficiency. Eur J Pediatr. 1998;157:605–606. doi: 10.1007/s004310050889. [DOI] [PubMed] [Google Scholar]
- Mayatepek E, Schroder M, Kohlmuller D, Bieger WP, Nutzenadel W. Continuous mannose infusion in carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr. 1997;86:1138–1140. doi: 10.1111/j.1651-2227.1997.tb14825.x. [DOI] [PubMed] [Google Scholar]
- Melville DB, Montero-Balaguer M, Levic DS, Bradley K, Smith JR, Hatzopoulos AK, Knapik EW. The feelgood mutation in zebrafish dysregulates COPII-dependent secretion of select extracellular matrix proteins in skeletal morphogenesis. Dis Mech Model. 2011;4:763–776. doi: 10.1242/dmm.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehues R, et al. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414–1420. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata S, Kinoshita S, Aoki R, Tanaka H, Wada H, Tsuruoka-Kinoshita S, Tsuboi T, Watabe S, Okamoto H. Neuroepithelial cells require fucosylated glycans to guide the migration of vagus motor neuron progenitors in the developing zebrafish hindbrain. Development. 2009;136:1653–1663. doi: 10.1242/dev.033290. [DOI] [PubMed] [Google Scholar]
- Panneerselvam K, Freeze HH. Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J Clin Invest. 1996;97:1478–1487. doi: 10.1172/JCI118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Thiel C, Rindermann J, DeRossi C, Popovici D, Hoffmann GF, Grone HJ, Korner C. Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat Med. 2012;18:71–73. doi: 10.1038/nm.2548. [DOI] [PubMed] [Google Scholar]
- Schroeder AS, Kappler M, Bonfert M, Borggraefe I, Schoen C, Reiter K. Seizures and stupor during intravenous mannose therapy in a patient with CDG syndrome type 1b (MPI-CDG) J Inherit Metab Dis. 2011 doi: 10.1007/s10545-010-9252-x. DOI: 10.1007/s10545-010-9252-x. [DOI] [PubMed] [Google Scholar]
- Shang J, Lehrman MA. Metformin-stimulated mannose transport in dermal fibroblasts. J Biol Chem. 2004;279:9703–9712. doi: 10.1074/jbc.M310837200. [DOI] [PubMed] [Google Scholar]
- Shang J, Gao N, Kaufman RJ, Ron D, Harding HP, Lehrman MA. Translation attenuation by PERK balances ER glycoprotein synthesis with lipid-linked oligosaccharide flux. J Cell Biol. 2007;176:605–616. doi: 10.1083/jcb.200607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Korner C, Freeze H, Lehrman MA. Extension of lipid-linked oligosaccharides is a high-priority aspect of the unfolded protein response: endoplasmic reticulum stress in type I congenital disorder of glycosylation fibroblasts. Glycobiology. 2002;12:307–317. doi: 10.1093/glycob/12.5.307. [DOI] [PubMed] [Google Scholar]
- Sharma V, Ichikawa M, He P, Scott DA, Bravo Y, Dahl R, Ng BG, Cosford ND, Freeze HH. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase-deficient fibroblasts. J Biol Chem. 2011;286:39431–39438. doi: 10.1074/jbc.M111.285502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Willer JR, Scherer PC, Panzer JA, Kugath A, Skordalakes E, Gregg RG, Willer GB, Balice-Gordon RJ. Neural and synaptic defects in slytherin, a zebrafish model for human congenital disorders of glycosylation. PLoS One. 2010;5:e13743. doi: 10.1371/journal.pone.0013743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel C, Lubke T, Matthijs G, von Figura K, Korner C. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol Cell Biol. 2006;26:5615–5620. doi: 10.1128/MCB.02391-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Van Schaftingen E, Jaeken J. Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett. 1995;377:318–320. doi: 10.1016/0014-5793(95)01357-1. [DOI] [PubMed] [Google Scholar]
- Zielinska DF, Gnad F, Schropp K, Wisniewski JR, Mann M. Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol Cell. 2012;46:542–548. doi: 10.1016/j.molcel.2012.04.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.